Skipping Exon-v6 from CD44v6-Containing Isoforms Influences Chemotherapy Response and Self-Renewal Capacity of Gastric Cancer Cells


Abstract
1. Introduction
2. Results
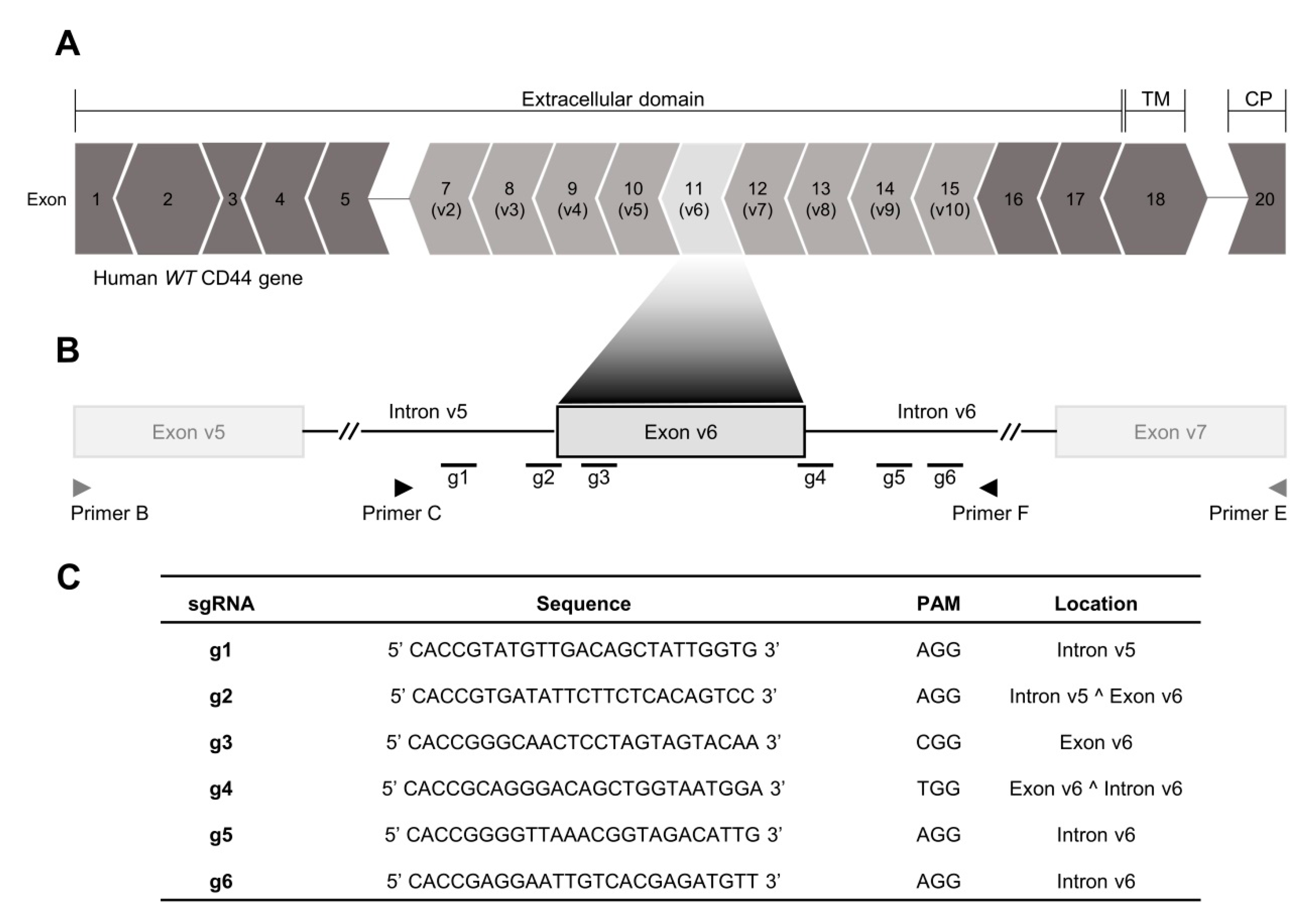
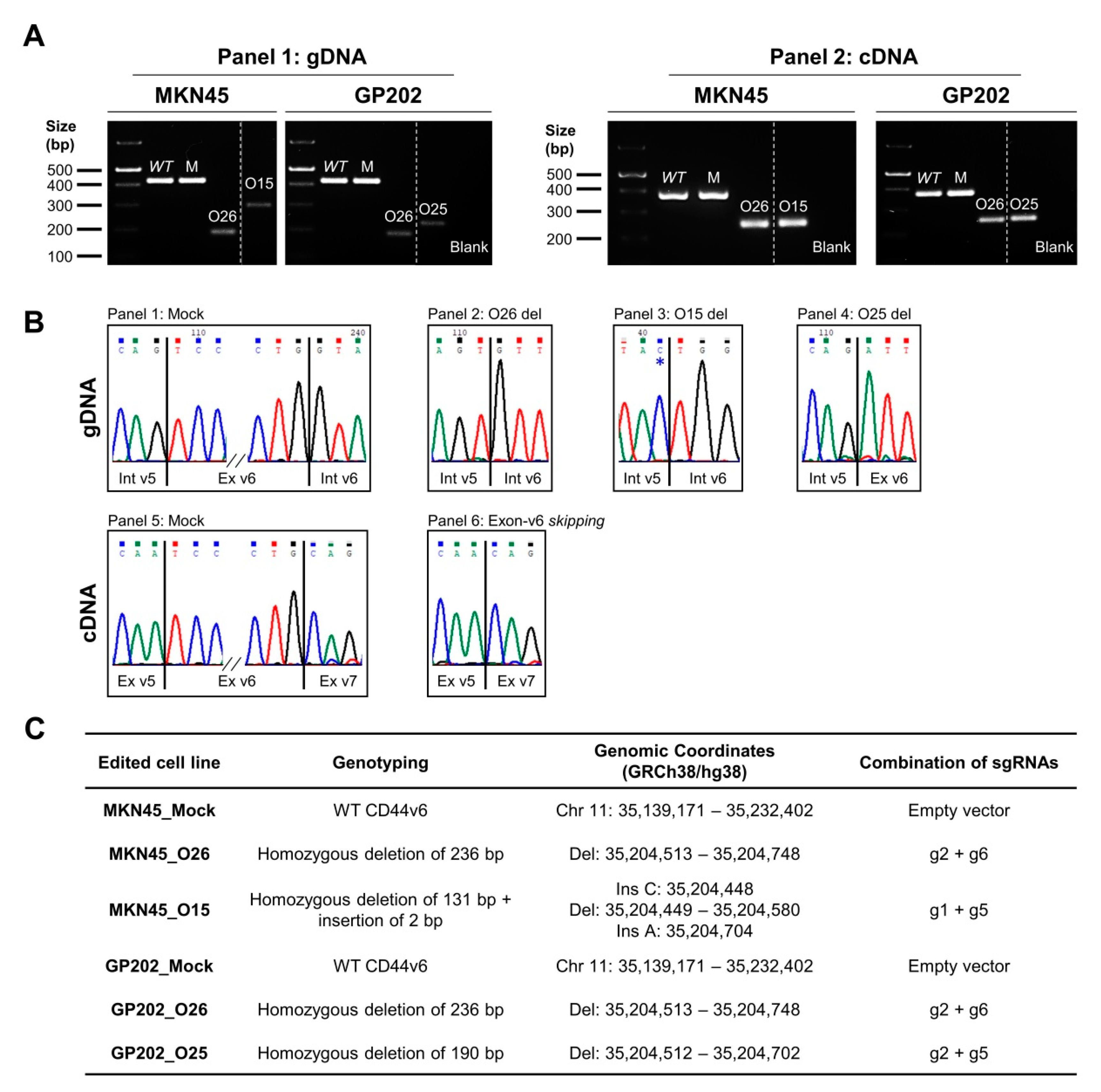
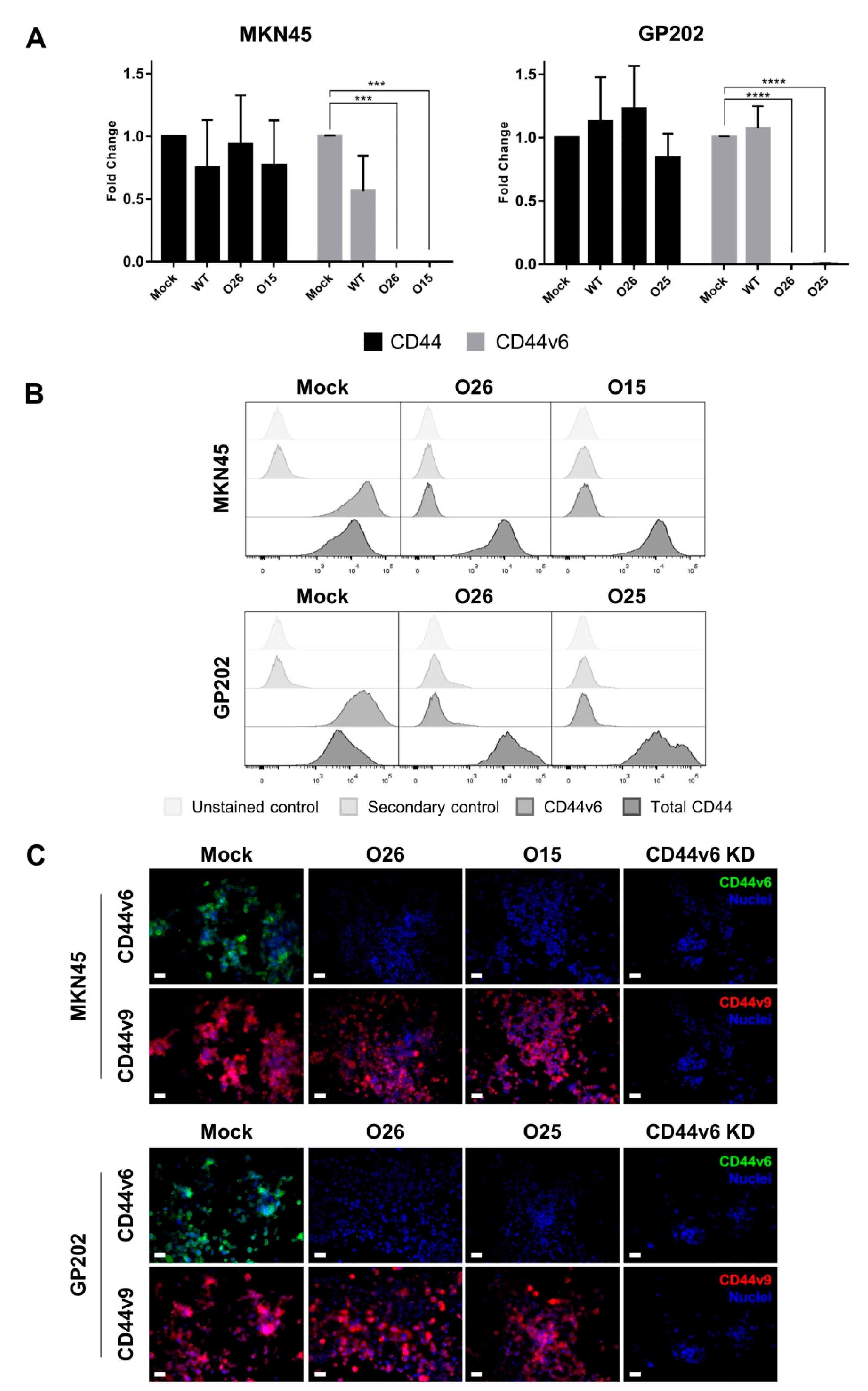
2.1. CRISPR/Cas9 Editing at Exon-v6 Splice-Sites Generates CD44v Isoforms Lacking Exon-v6
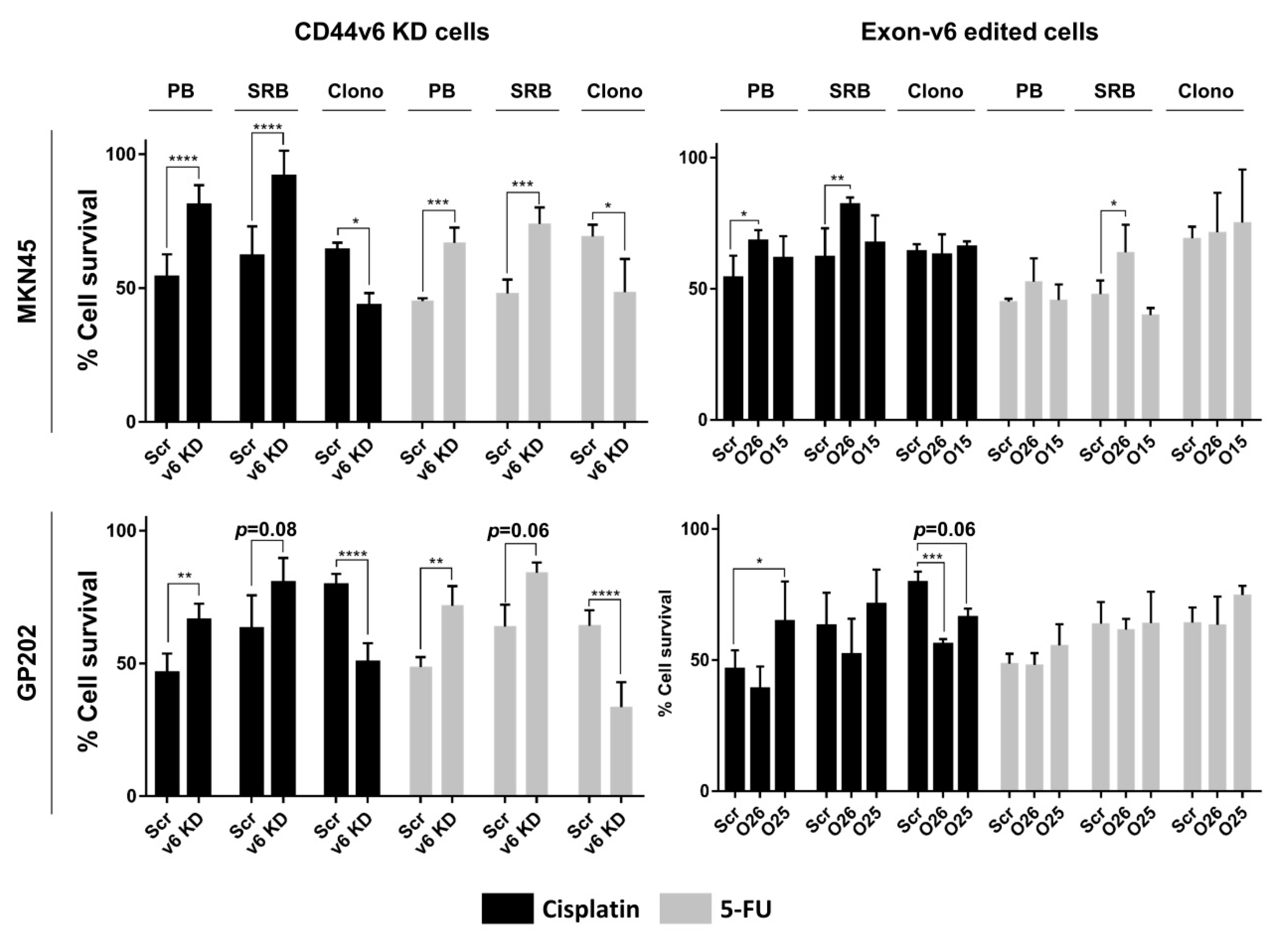
2.2. CD44v6-Containing Isoforms and Exon-v6 Itself Are Positive Modulators of Cisplatin Response
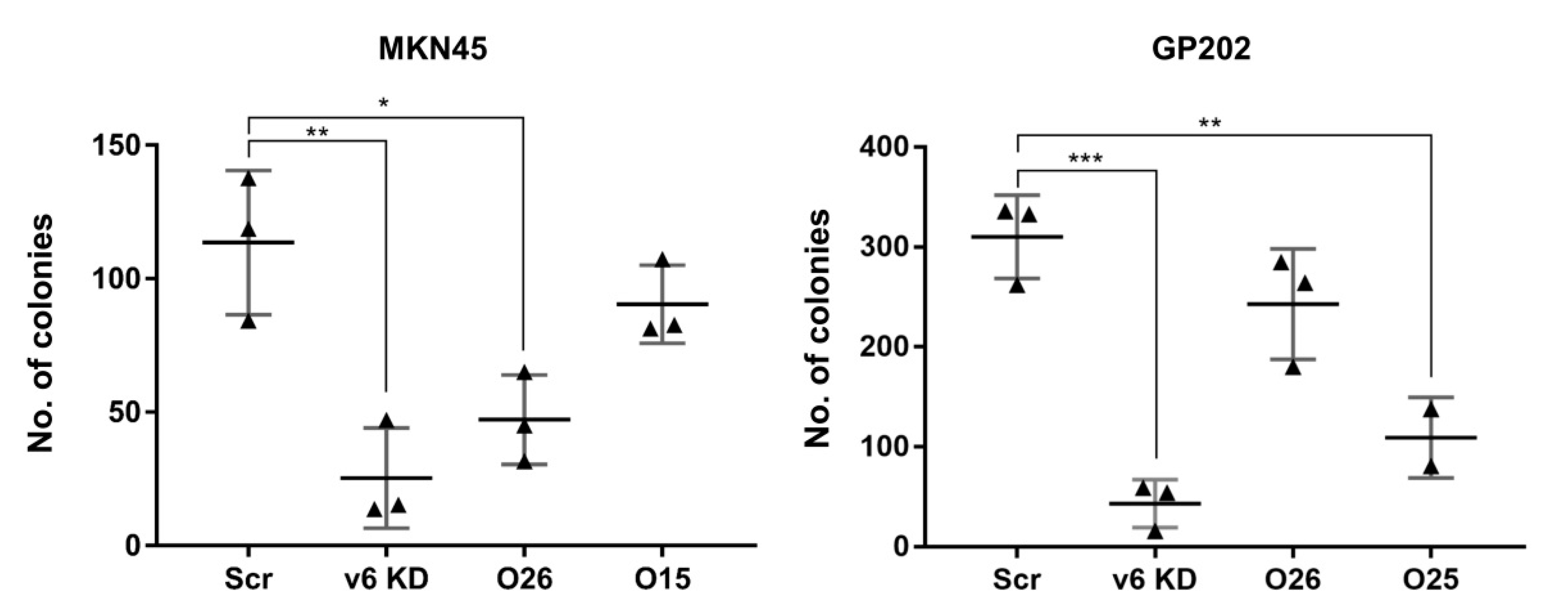
2.3. Exon-v6 Depletion Decreases Self-Renewal Capacity of GC Cells to the Same Extent as Knocking-Down CD44v6-Containing Isoforms
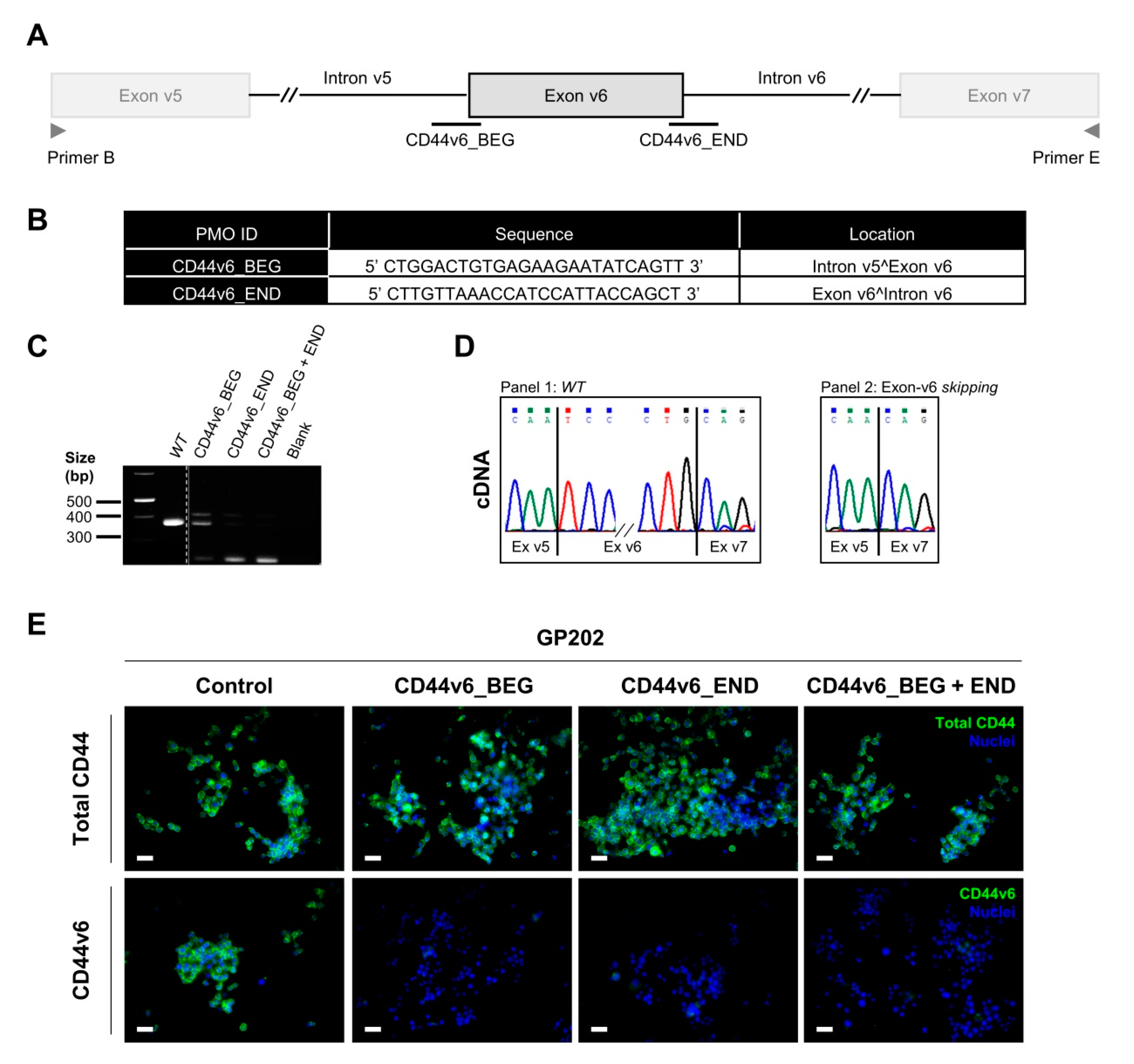
2.4. Transient Exon-v6 Skipping Using Phosphorodiamidate Morpholino Oligomers (PMOs)
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Generation of a Permanent Exon-v6 Skipping Model by CRISPR/Cas9
4.3. Genotyping Analysis of CRISPR/Cas9 Skipping Models
4.4. CD44/CD44v Expression Analysis
4.5. Inhibition of CD44v6 Expression by siRNA
4.6. Cell Survival Assays
4.7. Transient Exon-v6 Skipping Using Morpholinos
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smyth, E.C.; Verheij, M.; Allum, W.; Cunningham, D.; Cervantes, A.; Arnold, D.; Committee, E.G. Gastric cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Goodison, S.; Urquidi, V.; Tarin, D. CD44 cell adhesion molecules. Mol. Pathol. 1999, 52, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Orian-Rousseau, V. CD44, a therapeutic target for metastasising tumours. Eur. J. Cancer 2010, 46, 1271–1277. [Google Scholar] [CrossRef]
- Zoller, M. CD44, Hyaluronan, the Hematopoietic Stem Cell, and Leukemia-Initiating Cells. Front. Immunol. 2015, 6, 235. [Google Scholar] [CrossRef] [PubMed]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Hasenauer, S.; Malinger, D.; Koschut, D.; Pace, G.; Matzke, A.; von Au, A.; Orian-Rousseau, V. Internalization of Met requires the co-receptor CD44v6 and its link to ERM proteins. PLoS ONE 2013, 8, 62357. [Google Scholar] [CrossRef]
- Xie, J.W.; Chen, P.C.; Zheng, C.H.; Li, P.; Wang, J.B.; Lin, J.X.; Lu, J.; Chen, Q.Y.; Cao, L.L.; Lin, M.; et al. Evaluation of the prognostic value and functional roles of CD44v6 in gastric cancer. J. Cancer Res. Clin. Oncol. 2015, 141, 1809–1817. [Google Scholar] [CrossRef]
- Xu, Y.Y.; Guo, M.; Yang, L.Q.; Zhou, F.; Yu, C.; Wang, A.; Pang, T.H.; Wu, H.Y.; Zou, X.P.; Zhang, W.J.; et al. Regulation of CD44v6 expression in gastric carcinoma by the IL-6/STAT3 signaling pathway and its clinical significance. Oncotarget 2017, 8, 45848–45861. [Google Scholar] [CrossRef]
- Hu, B.; Luo, W.; Hu, R.T.; Zhou, Y.; Qin, S.Y.; Jiang, H.X. Meta-Analysis of Prognostic and Clinical Significance of CD44v6 in Esophageal Cancer. Medicine 2015, 94, 1238. [Google Scholar] [CrossRef]
- Ma, L.; Dong, L.; Chang, P. CD44v6 engages in colorectal cancer progression. Cell Death Dis. 2019, 10, 30. [Google Scholar] [CrossRef]
- Jiang, H.; Patel, P.H.; Kohlmaier, A.; Grenley, M.O.; McEwen, D.G.; Edgar, B.A. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell 2009, 137, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Qiao, G.L.; Song, L.N.; Deng, Z.F.; Chen, Y.; Ma, L.J. Prognostic value of CD44v6 expression in breast cancer: A meta-analysis. OncoTargets Ther. 2018, 11, 5451–5457. [Google Scholar] [CrossRef] [PubMed]
- Matzke-Ogi, A.; Jannasch, K.; Shatirishvili, M.; Fuchs, B.; Chiblak, S.; Morton, J.; Tawk, B.; Lindner, T.; Sansom, O.; Alves, F.; et al. Inhibition of Tumor Growth and Metastasis in Pancreatic Cancer Models by Interference With CD44v6 Signaling. Gastroenterology 2016, 150, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Reeder, J.A.; Gotley, D.C.; Walsh, M.D.; Fawcett, J.; Antalis, T.M. Expression of antisense CD44 variant 6 inhibits colorectal tumor metastasis and tumor growth in a wound environment. Cancer Res. 1998, 58, 3719–3726. [Google Scholar]
- Stroomer, J.W.; Roos, J.C.; Sproll, M.; Quak, J.J.; Heider, K.H.; Wilhelm, B.J.; Castelijns, J.A.; Meyer, R.; Kwakkelstein, M.O.; Snow, G.B.; et al. Safety and biodistribution of 99mTechnetium-labeled anti-CD44v6 monoclonal antibody BIWA 1 in head and neck cancer patients. Clin. Cancer Res. 2000, 6, 3046–3055. [Google Scholar]
- Tijink, B.M.; Buter, J.; de Bree, R.; Giaccone, G.; Lang, M.S.; Staab, A.; Leemans, C.R.; van Dongen, G.A. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin. Cancer Res. 2006, 12, 6064–6072. [Google Scholar] [CrossRef]
- Da Cunha, C.B.; Oliveira, C.; Wen, X.; Gomes, B.; Sousa, S.; Suriano, G.; Grellier, M.; Huntsman, D.G.; Carneiro, F.; Granja, P.L.; et al. De novo expression of CD44 variants in sporadic and hereditary gastric cancer. Lab. Investig. 2010, 90, 1604–1614. [Google Scholar] [CrossRef]
- Pereira, C.; Ferreira, D.; Mendes, N.; Granja, P.L.; Almeida, G.M.; Oliveira, C. Expression of CD44v6-Containing Isoforms Influences Cisplatin Response in Gastric Cancer Cells. Cancers 2020, 12, 858. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, K.; Hackert, T.; Zoller, M. CD44/CD44v6 a Reliable Companion in Cancer-Initiating Cell Maintenance and Tumor Progression. Front. Cell Dev. Biol. 2018, 6, 97. [Google Scholar] [CrossRef]
- Riethoven, J.J. Regulatory regions in DNA: Promoters, enhancers, silencers, and insulators. Methods Mol. Biol. 2010, 674, 33–42. [Google Scholar] [CrossRef]
- Perri, F.; Longo, F.; Giuliano, M.; Sabbatino, F.; Favia, G.; Ionna, F.; Addeo, R.; Della Vittoria Scarpati, G.; Di Lorenzo, G.; Pisconti, S. Epigenetic control of gene expression: Potential implications for cancer treatment. Crit. Rev. Oncol. Hematol. 2017, 111, 166–172. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, J.T.; Farrington, W.J.; Tollefsbol, T.O. An overview of epigenetic assays. Mol. Biotechnol. 2008, 38, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Zavros, Y. Initiation and Maintenance of Gastric Cancer: A Focus on CD44 Variant Isoforms and Cancer Stem Cells. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.; Shiina, M.; Li, J.J. Hyaluronan-CD44 interaction promotes oncogenic signaling, microRNA functions, chemoresistance, and radiation resistance in cancer stem cells leading to tumor progression. Adv. Cancer Res. 2014, 123, 255–275. [Google Scholar] [CrossRef]
- Ishiwata, T.; Matsuda, Y.; Yoshimura, H.; Sasaki, N.; Ishiwata, S.; Ishikawa, N.; Takubo, K.; Arai, T.; Aida, J. Pancreatic cancer stem cells: Features and detection methods. Pathol. Oncol. Res. 2018, 24, 797–805. [Google Scholar] [CrossRef]
- Kozovska, Z.; Gabrisova, V.; Kucerova, L. Colon cancer: Cancer stem cells markers, drug resistance and treatment. Biomed Pharmacother. 2014, 68, 911–916. [Google Scholar] [CrossRef]
- Yan, Y.; Zuo, X.; Wei, D. Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl. Med. 2015, 4, 1033–1043. [Google Scholar] [CrossRef]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef]
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef]
- Clarke, M.F. Clinical and Therapeutic Implications of Cancer Stem Cells. N. Engl. J. Med. 2019, 380, 2237–2245. [Google Scholar] [CrossRef]
- Miyoshi, S.; Tsugawa, H.; Matsuzaki, J.; Hirata, K.; Mori, H.; Saya, H.; Kanai, T.; Suzuki, H. Inhibiting xCT Improves 5-Fluorouracil Resistance of Gastric Cancer Induced by CD44 Variant 9 Expression. Anticancer Res. 2018, 38, 6163–6170. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Iwasaki, R.; Kawata, N.; Kawakami, R.; Kumagai, K.; Migita, T.; Sano, T.; Yamaguchi, K.; Seimiya, H. In silico chemical screening identifies epidermal growth factor receptor as a therapeutic target of drug-tolerant CD44v9-positive gastric cancer cells. Br. J. Cancer 2019, 121, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, Y.; Zhao, J. CRISPR editing in biological and biomedical investigation. J. Cell. Physiol. 2018, 233, 3875–3891. [Google Scholar] [CrossRef] [PubMed]
- Brokowski, C.; Adli, M. CRISPR Ethics: Moral Considerations for Applications of a Powerful Tool. J. Mol. Biol. 2019, 431, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Gartner, F.; David, L.; Seruca, R.; Machado, J.C.; Sobrinho-Simoes, M. Establishment and characterization of two cell lines derived from human diffuse gastric carcinomas xenografted in nude mice. Virchows Arch. 1996, 428, 91–98. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Morrisey Lab Protocol: Generating Large (>1kb) Genomic Deletions Using CRISPRs. Available online: https://www.med.upenn.edu/genetics/tcmf/protocols.shtml. (accessed on 2 July 2020).
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, 67. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Orientation | In-Text Name | Binding Site | Primer Sequence (5′–3′) | Melting Temperature |
|---|---|---|---|---|
| Forward | A | pSpCas9(BB)_Bbs I | 5′-GGGCCTATTTCCCATGATTCCTT-3′ | Tm = 68.5 °C |
| B | CD44 Exon 10 (v5) | 5′-ATGTAGACAGAAATGGCACCAC-3′ | Tm = 62.7 °C | |
| C | CD44 Intron 10 | 5′-ATCAGTGGCCTGTTTCCTTG-3′ | Tm = 64.0 °C | |
| Reverse | D | pSpCas9(BB)_Bbs I | 5′-GACTCGGTGCCACTTTTTCAA-3′ | Tm = 66.2 °C |
| E | CD44 Exon 12 (v7) | 5′-CCATCCTTCTTCCTGCTTGATG-3′ | Tm = 66.8 °C | |
| F | CD44 Intron 11 | 5′-TTTGGCTCTGTGTGAACTGC-3′ | Tm = 64.1 °C |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lobo, S.; Pereira, C.; Oliveira, C.; Almeida, G.M. Skipping Exon-v6 from CD44v6-Containing Isoforms Influences Chemotherapy Response and Self-Renewal Capacity of Gastric Cancer Cells. Cancers 2020, 12, 2378. https://doi.org/10.3390/cancers12092378
Lobo S, Pereira C, Oliveira C, Almeida GM. Skipping Exon-v6 from CD44v6-Containing Isoforms Influences Chemotherapy Response and Self-Renewal Capacity of Gastric Cancer Cells. Cancers. 2020; 12(9):2378. https://doi.org/10.3390/cancers12092378
Chicago/Turabian StyleLobo, Silvana, Carla Pereira, Carla Oliveira, and Gabriela M. Almeida. 2020. "Skipping Exon-v6 from CD44v6-Containing Isoforms Influences Chemotherapy Response and Self-Renewal Capacity of Gastric Cancer Cells" Cancers 12, no. 9: 2378. https://doi.org/10.3390/cancers12092378
APA StyleLobo, S., Pereira, C., Oliveira, C., & Almeida, G. M. (2020). Skipping Exon-v6 from CD44v6-Containing Isoforms Influences Chemotherapy Response and Self-Renewal Capacity of Gastric Cancer Cells. Cancers, 12(9), 2378. https://doi.org/10.3390/cancers12092378