Transcription Factors in Cancer Development and Therapy



Abstract
1. Introduction
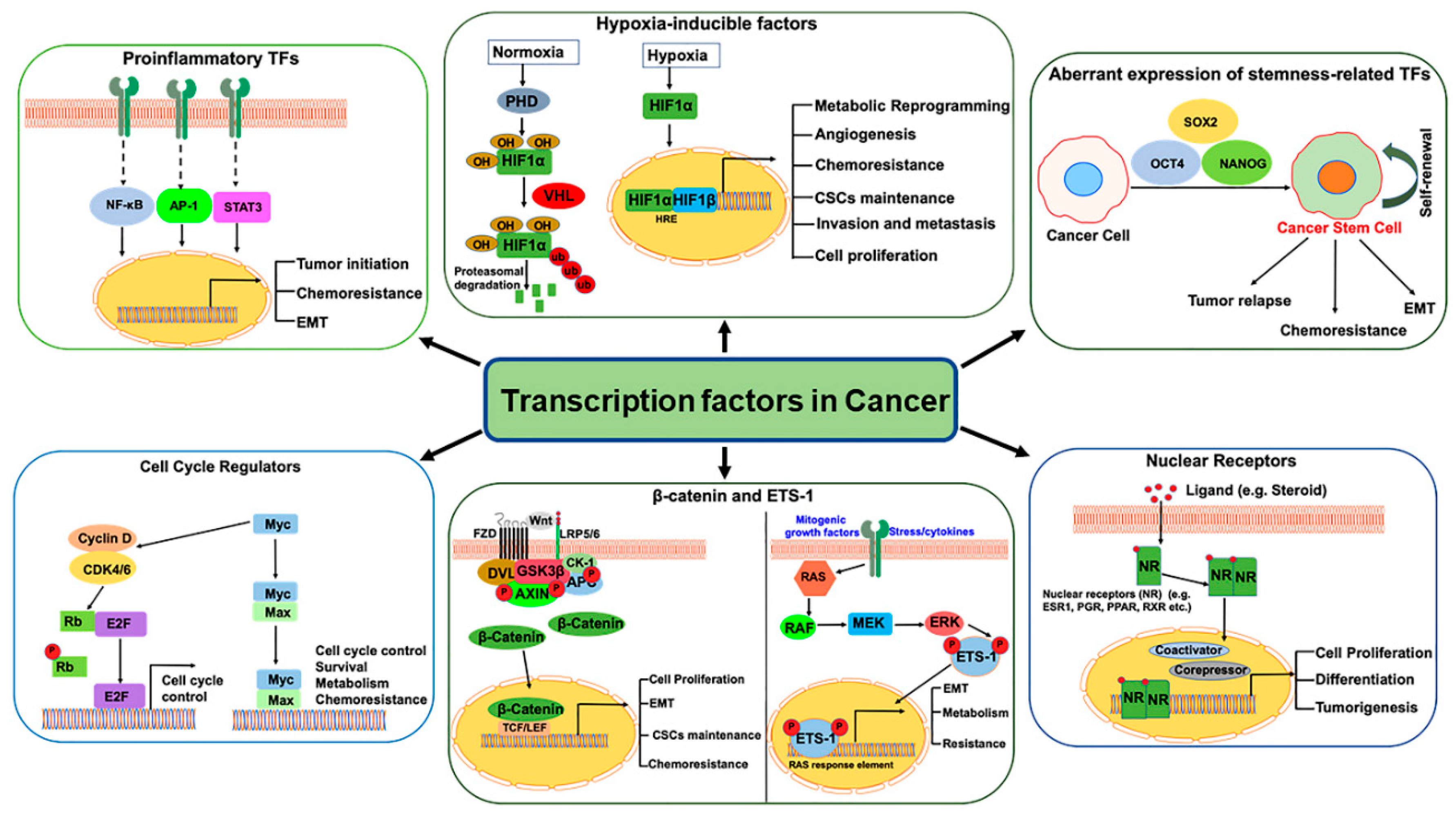
2. Pro-Inflammatory Transcription Factors
2.1. Nuclear Factor-kappa B (NF-kB)
2.2. Activator Protein 1 (AP-1)
2.3. Signal Transducer and Activator of Transcription 3 (STAT3)
3. Hypoxic Tumor Microenvironment in Cancer
3.1. Hypoxia-Inducible Factors (HIFs)
3.2. HIFs in Cancer Stem Cells (CSCs)
4. TFs Regulating Cell Proliferation, Invasion, and Metastasis
4.1. Myc
4.2. E2F
4.3. ETS1
4.4. β-Catenin
5. Nuclear Receptors (NRs)
6. Chemoresistance and TFs
7. Targeting TFs in Cancers
7.1. STAT3 and NF-κB Inhibitors
7.2. Myc Inhibitors
7.3. HIF1 Inhibitors
7.4. E2F Inhibitors
7.5. Wnt/β-Catenin Inhibitors
8. Targeting Druggable Nuclear Receptors
8.1. Estrogen Receptor α Inhibitors
8.2. Androgen Receptor Inhibitors
8.3. Glucocorticoid Receptor (GR) Inhibitors
8.4. Progesterone Receptor (PR) Inhibitors
8.5. Retinoic Acid Receptor Inhibitors
8.6. Peroxisome Proliferator-Activated Receptor (PPAR) Inhibitors
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.H. Targeting Transcription Factors for Cancer Treatment. Molecules 2018, 23, 1479. [Google Scholar] [CrossRef]
- Sun, W.; Yang, J. Functional mechanisms for human tumor suppressors. J. Cancer 2010, 1, 136–140. [Google Scholar] [CrossRef]
- Sionov, R.V.; Haupt, Y. The cellular response to p53: The decision between life and death. Oncogene 1999, 18, 6145–6157. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Yori, J.L.; Johnson, E.; Zhou, G.; Jain, M.K.; Keri, R.A. Kruppel-like factor 4 inhibits epithelial-to-mesenchymal transition through regulation of E-cadherin gene expression. J. Biol. Chem. 2010, 285, 16854–16863. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.N.; Abou-Kheir, W.; Yin, J.J.; Fang, L.; Hynes, P.; Casey, O.; Hu, D.; Wan, Y.; Seng, V.; Sheppard-Tillman, H.; et al. Critical and reciprocal regulation of KLF4 and SLUG in transforming growth factor beta-initiated prostate cancer epithelial-mesenchymal transition. Mol. Cell Biol. 2012, 32, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, Z.; Zhang, X.; Yang, S.; Lin, X.; Yang, X.; Lin, X.; Shi, J.; Wang, S.; Zhao, W.; et al. Klf4 reduces stemness phenotype, triggers mesenchymal-epithelial transition (MET)-like molecular changes, and prevents tumor progression in nasopharygeal carcinoma. Oncotarget 2017, 8, 93924–93941. [Google Scholar] [CrossRef]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef]
- Eppert, K.; Scherer, S.W.; Ozcelik, H.; Pirone, R.; Hoodless, P.; Kim, H.; Tsui, L.C.; Bapat, B.; Gallinger, S.; Andrulis, I.L.; et al. MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell 1996, 86, 543–552. [Google Scholar] [CrossRef]
- Maesawa, C.; Tamura, G.; Nishizuka, S.; Iwaya, T.; Ogasawara, S.; Ishida, K.; Sakata, K.; Sato, N.; Ikeda, K.; Kimura, Y.; et al. MAD-related genes on 18q21.1, Smad2 and Smad4, are altered infrequently in esophageal squamous cell carcinoma. Jpn. J. Cancer Res. 1997, 88, 340–343. [Google Scholar] [CrossRef]
- Silberstein, G.B.; Van Horn, K.; Strickland, P.; Roberts, C.T., Jr.; Daniel, C.W. Altered expression of the WT1 wilms tumor suppressor gene in human breast cancer. Proc. Natl. Acad. Sci. USA 1997, 94, 8132–8137. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Shi, Q.; Wang, W. Double agents: Genes with both oncogenic and tumor-suppressor functions. Oncogenesis 2018, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, L.J.; Al-Attar, R.; Storey, K.B. Transcriptional regulation of metabolism in disease: From transcription factors to epigenetics. PeerJ 2018, 6, e5062. [Google Scholar] [CrossRef]
- Gulumian, M. The role of oxidative stress in diseases caused by mineral dusts and fibres: Current status and future of prophylaxis and treatment. Mol. Cell Biochem. 1999, 196, 69–77. [Google Scholar] [CrossRef]
- Burd, E.M. Human papillomavirus and cervical cancer. Clin. Microbiol. Rev. 2003, 16, 1–17. [Google Scholar] [CrossRef]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Wang, D.J.; Ratnam, N.M.; Byrd, J.C.; Guttridge, D.C. NF-kappaB functions in tumor initiation by suppressing the surveillance of both innate and adaptive immune cells. Cell Rep. 2014, 9, 90–103. [Google Scholar] [CrossRef]
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Liu, Z.G.; Hsu, H.; Goeddel, D.V.; Karin, M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell 1996, 87, 565–576. [Google Scholar] [CrossRef]
- Chen, C.; Edelstein, L.C.; Gelinas, C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol. Cell Biol. 2000, 20, 2687–2695. [Google Scholar] [CrossRef]
- Cui, X.; Shen, D.; Kong, C.; Zhang, Z.; Zeng, Y.; Lin, X.; Liu, X. NF-kappaB suppresses apoptosis and promotes bladder cancer cell proliferation by upregulating survivin expression in vitro and in vivo. Sci. Rep. 2017, 7, 40723. [Google Scholar] [CrossRef]
- James, M.A.; Lee, J.H.; Klingelhutz, A.J. Human papillomavirus type 16 E6 activates NF-kappaB, induces cIAP-2 expression, and protects against apoptosis in a PDZ binding motif-dependent manner. J. Virol. 2006, 80, 5301–5307. [Google Scholar] [CrossRef]
- Hinz, M.; Krappmann, D.; Eichten, A.; Heder, A.; Scheidereit, C.; Strauss, M. NF-kappaB function in growth control: Regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol. Cell Biol. 1999, 19, 2690–2698. [Google Scholar] [CrossRef]
- Lee, W.; Mitchell, P.; Tjian, R. Purified transcription factor AP-1 interacts with TPA-inducible enhancer elements. Cell 1987, 49, 741–752. [Google Scholar] [CrossRef]
- Angel, P.; Karin, M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1991, 1072, 129–157. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef]
- Qiao, Y.; He, H.; Jonsson, P.; Sinha, I.; Zhao, C.; Dahlman-Wright, K. AP-1 is a key regulator of proinflammatory cytokine TNFalpha-mediated triple-negative breast cancer progression. J. Biol. Chem. 2016, 291, 18309. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, O.; Schwitalla, S.; et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef]
- Liu, Y.; Li, P.K.; Li, C.; Lin, J. Inhibition of STAT3 signaling blocks the anti-apoptotic activity of IL-6 in human liver cancer cells. J. Biol. Chem. 2010, 285, 27429–27439. [Google Scholar] [CrossRef]
- Bronte-Tinkew, D.M.; Terebiznik, M.; Franco, A.; Ang, M.; Ahn, D.; Mimuro, H.; Sasakawa, C.; Ropeleski, M.J.; Peek, R.M., Jr.; Jones, N.L. Helicobacter pylori cytotoxin-associated gene A activates the signal transducer and activator of transcription 3 pathway in vitro and in vivo. Cancer Res. 2009, 69, 632–639. [Google Scholar] [CrossRef]
- Choudhari, S.R.; Khan, M.A.; Harris, G.; Picker, D.; Jacob, G.S.; Block, T.; Shailubhai, K. Deactivation of Akt and STAT3 signaling promotes apoptosis, inhibits proliferation, and enhances the sensitivity of hepatocellular carcinoma cells to an anticancer agent, Atiprimod. Mol. Cancer Ther. 2007, 6, 112–121. [Google Scholar] [CrossRef]
- Sun, S.; Steinberg, B.M. PTEN is a negative regulator of STAT3 activation in human papillomavirus-infected cells. J. Gen. Virol. 2002, 83, 1651–1658. [Google Scholar] [CrossRef]
- Migone, T.S.; Lin, J.X.; Cereseto, A.; Mulloy, J.C.; O’Shea, J.J.; Franchini, G.; Leonard, W.J. Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I. Science 1995, 269, 79–81. [Google Scholar] [CrossRef]
- Muromoto, R.; Ikeda, O.; Okabe, K.; Togi, S.; Kamitani, S.; Fujimuro, M.; Harada, S.; Oritani, K.; Matsuda, T. Epstein-Barr virus-derived EBNA2 regulates STAT3 activation. Biochem. Biophys. Res. Commun. 2009, 378, 439–443. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Contino, G.; Deshpande, V.; Tzatsos, A.; Conrad, C.; Benes, C.H.; Levy, D.E.; Settleman, J.; Engelman, J.A.; Bardeesy, N. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011, 71, 5020–5029. [Google Scholar] [CrossRef]
- Eales, K.L.; Hollinshead, K.E.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef] [PubMed]
- Hockel, M.; Schlenger, K.; Aral, B.; Mitze, M.; Schaffer, U.; Vaupel, P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996, 56, 4509–4515. [Google Scholar]
- Walenta, S.; Wetterling, M.; Lehrke, M.; Schwickert, G.; Sundfor, K.; Rofstad, E.K.; Mueller-Klieser, W. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 2000, 60, 916–921. [Google Scholar]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Harrison, L. Tumor hypoxia: Causative factors, compensatory mechanisms, and cellular response. Oncologist 2004, 9 (Suppl. S5), 4–9. [Google Scholar] [CrossRef]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Z.; Moran, S.M.; Hogenesch, J.B.; Wartman, L.; Bradfield, C.A. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr. 1998, 7, 205–213. [Google Scholar] [PubMed]
- Unwith, S.; Zhao, H.; Hennah, L.; Ma, D. The potential role of HIF on tumour progression and dissemination. Int. J. Cancer 2015, 136, 2491–2503. [Google Scholar] [CrossRef]
- Firth, J.D.; Ebert, B.L.; Pugh, C.W.; Ratcliffe, P.J. Oxygen-regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase A genes: Similarities with the erythropoietin 3′ enhancer. Proc. Natl. Acad. Sci. USA 1994, 91, 6496–6500. [Google Scholar] [CrossRef]
- Chen, C.; Pore, N.; Behrooz, A.; Ismail-Beigi, F.; Maity, A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J. Biol. Chem. 2001, 276, 9519–9525. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.M.; Tian, R.F.; Liu, W.P.; Fei, Z.; Long, Q.F.; Wang, X.A.; Zhang, X. The expression and significance of HIF-1alpha and GLUT-3 in glioma. Brain Res. 2009, 1304, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar]
- Yu, M.; Yongzhi, H.; Chen, S.; Luo, X.; Lin, Y.; Zhou, Y.; Jin, H.; Hou, B.; Deng, Y.; Tu, L.; et al. The prognostic value of GLUT1 in cancers: A systematic review and meta-analysis. Oncotarget 2017, 8, 43356–43367. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, M.; McSheehy, P.M.; Griffiths, J.R.; Bashford, C.L. Causes and consequences of tumour acidity and implications for treatment. Mol. Med. Today 2000, 6, 15–19. [Google Scholar] [CrossRef]
- Fukumura, D.; Xu, L.; Chen, Y.; Gohongi, T.; Seed, B.; Jain, R.K. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res. 2001, 61, 6020–6024. [Google Scholar]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Gerweck, L.E. Tumor pH: Implications for treatment and novel drug design. Semin. Radiat. Oncol. 1998, 8, 176–182. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Das, B.; Tsuchida, R.; Malkin, D.; Koren, G.; Baruchel, S.; Yeger, H. Hypoxia enhances tumor stemness by increasing the invasive and tumorigenic side population fraction. Stem. Cells 2008, 26, 1818–1830. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Kreso, A.; Jamieson, C.H. Cancer stem cells and self-renewal. Clin. Cancer Res. 2010, 16, 3113–3120. [Google Scholar] [CrossRef] [PubMed]
- Hepburn, A.C.; Steele, R.E.; Veeratterapillay, R.; Wilson, L.; Kounatidou, E.E.; Barnard, A.; Berry, P.; Cassidy, J.R.; Moad, M.; El-Sherif, A.; et al. The induction of core pluripotency master regulators in cancers defines poor clinical outcomes and treatment resistance. Oncogene 2019, 38, 4412–4424. [Google Scholar] [CrossRef] [PubMed]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Schoning, J.P.; Monteiro, M.; Gu, W. Drug resistance and cancer stem cells: The shared but distinct roles of hypoxia-inducible factors HIF1alpha and HIF2alpha. Clin. Exp. Pharm. Physiol. 2017, 44, 153–161. [Google Scholar] [CrossRef]
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 2009, 28, 3949–3959. [Google Scholar] [CrossRef]
- Kitajima, S.; Lee, K.L.; Hikasa, H.; Sun, W.; Huang, R.Y.; Yang, H.; Matsunaga, S.; Yamaguchi, T.; Araki, M.; Kato, H.; et al. Hypoxia-inducible factor-1alpha promotes cell survival during ammonia stress response in ovarian cancer stem-like cells. Oncotarget 2017, 8, 114481–114494. [Google Scholar] [CrossRef]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2alpha regulates Oct-4: Effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006, 20, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Manzano, V.; Rodriguez-Jimenez, F.J.; Acena-Bonilla, J.L.; Fustero-Lardies, S.; Erceg, S.; Dopazo, J.; Montaner, D.; Stojkovic, M.; Sanchez-Puelles, J.M. FM19G11, a new hypoxia-inducible factor (HIF) modulator, affects stem cell differentiation status. J. Biol. Chem. 2010, 285, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Bres, V.; Yoshida, T.; Pickle, L.; Jones, K.A. SKIP interacts with c-Myc and Menin to promote HIV-1 Tat transactivation. Mol. Cell 2009, 36, 75–87. [Google Scholar] [CrossRef]
- Eberhardy, S.R.; Farnham, P.J. c-Myc mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J. Biol. Chem. 2001, 276, 48562–48571. [Google Scholar] [CrossRef]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef]
- Hermeking, H.; Rago, C.; Schuhmacher, M.; Li, Q.; Barrett, J.F.; Obaya, A.J.; O’Connell, B.C.; Mateyak, M.K.; Tam, W.; Kohlhuber, F.; et al. Identification of CDK4 as a target of c-MYC. Proc. Natl. Acad. Sci. USA 2000, 97, 2229–2234. [Google Scholar] [CrossRef]
- Bouchard, C.; Dittrich, O.; Kiermaier, A.; Dohmann, K.; Menkel, A.; Eilers, M.; Luscher, B. Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: Myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev. 2001, 15, 2042–2047. [Google Scholar] [CrossRef]
- Claassen, G.F.; Hann, S.R. A role for transcriptional repression of p21CIP1 by c-Myc in overcoming transforming growth factor beta -induced cell-cycle arrest. Proc. Natl. Acad. Sci. USA 2000, 97, 9498–9503. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Shen, J.; Wu, M.; Arsura, M.; FitzGerald, M.; Suldan, Z.; Kim, D.W.; Hofmann, C.S.; Pianetti, S.; Romieu-Mourez, R.; et al. Repression of transcription of the p27(Kip1) cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene 2001, 20, 1688–1702. [Google Scholar] [CrossRef] [PubMed]
- Poli, V.; Fagnocchi, L.; Fasciani, A.; Cherubini, A.; Mazzoleni, S.; Ferrillo, S.; Miluzio, A.; Gaudioso, G.; Vaira, V.; Turdo, A.; et al. MYC-driven epigenetic reprogramming favors the onset of tumorigenesis by inducing a stem cell-like state. Nat. Commun. 2018, 9, 1024. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Cheryan, V.T.; Xu, L.; Rishi, A.K.; Reddy, K.B. Myc mediates cancer stem-like cells and EMT changes in triple negative breast cancers cells. PLoS ONE 2017, 12, e0183578. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Zeller, K.I.; Wang, Y.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell Biol. 2004, 24, 5923–5936. [Google Scholar] [CrossRef]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Lukas, J.; Bartkova, J.; Rohde, M.; Strauss, M.; Bartek, J. Cyclin D1 is dispensable for G1 control in retinoblastoma gene-deficient cells independently of cdk4 activity. Mol. Cell Biol. 1995, 15, 2600–2611. [Google Scholar] [CrossRef]
- Trimarchi, J.M.; Lees, J.A. Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 2002, 3, 11–20. [Google Scholar] [CrossRef]
- Di Fiore, R.; D’Anneo, A.; Tesoriere, G.; Vento, R. RB1 in cancer: Different mechanisms of RB1 inactivation and alterations of pRb pathway in tumorigenesis. J. Cell Physiol. 2013, 228, 1676–1687. [Google Scholar] [CrossRef] [PubMed]
- Azechi, H.; Nishida, N.; Fukuda, Y.; Nishimura, T.; Minata, M.; Katsuma, H.; Kuno, M.; Ito, T.; Komeda, T.; Kita, R.; et al. Disruption of the p16/cyclin D1/retinoblastoma protein pathway in the majority of human hepatocellular carcinomas. Oncology 2001, 60, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Pascale, R.M.; Simile, M.M.; De Miglio, M.R.; Muroni, M.R.; Calvisi, D.F.; Asara, G.; Casabona, D.; Frau, M.; Seddaiu, M.A.; Feo, F. Cell cycle deregulation in liver lesions of rats with and without genetic predisposition to hepatocarcinogenesis. Hepatology 2002, 35, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, T.; Bijsmans, I.T.; Van de Vijver, K.K.; Bekaert, S.; Oosting, J.; Van Criekinge, W.; van Engeland, M.; Sieben, N.L. E2Fs mediate a fundamental cell-cycle deregulation in high-grade serous ovarian carcinomas. J. Pathol. 2009, 217, 14–20. [Google Scholar] [CrossRef]
- Lan, W.; Bian, B.; Xia, Y.; Dou, S.; Gayet, O.; Bigonnet, M.; Santofimia-Castano, P.; Cong, M.; Peng, L.; Dusetti, N.; et al. E2F signature is predictive for the pancreatic adenocarcinoma clinical outcome and sensitivity to E2F inhibitors, but not for the response to cytotoxic-based treatments. Sci. Rep. 2018, 8, 8330. [Google Scholar] [CrossRef]
- Iaquinta, P.J.; Lees, J.A. Life and death decisions by the E2F transcription factors. Curr. Opin. Cell Biol. 2007, 19, 649–657. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; Gonzalez-Baron, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef]
- Hsing, M.; Wang, Y.; Rennie, P.S.; Cox, M.E.; Cherkasov, A. ETS transcription factors as emerging drug targets in cancer. Med. Res. Rev. 2020, 40, 413–430. [Google Scholar] [CrossRef]
- Sizemore, G.M.; Pitarresi, J.R.; Balakrishnan, S.; Ostrowski, M.C. The ETS family of oncogenic transcription factors in solid tumours. Nat. Rev. Cancer 2017, 17, 337–351. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.S.; Hauser, C.A.; Henkel, G.; Colman, M.S.; Van Beveren, C.; Stacey, K.J.; Hume, D.A.; Maki, R.A.; Ostrowski, M.C. Ras-mediated phosphorylation of a conserved threonine residue enhances the transactivation activities of c-Ets1 and c-Ets2. Mol. Cell Biol. 1996, 16, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Wasylyk, B.; Wasylyk, C.; Flores, P.; Begue, A.; Leprince, D.; Stehelin, D. The c-ets proto-oncogenes encode transcription factors that cooperate with c-Fos and c-Jun for transcriptional activation. Nature 1990, 346, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Watabe, T.; Yoshida, K.; Shindoh, M.; Kaya, M.; Fujikawa, K.; Sato, H.; Seiki, M.; Ishii, S.; Fujinaga, K. The Ets-1 and Ets-2 transcription factors activate the promoters for invasion-associated urokinase and collagenase genes in response to epidermal growth factor. Int. J. Cancer 1998, 77, 128–137. [Google Scholar] [CrossRef]
- Westermarck, J.; Seth, A.; Kahari, V.M. Differential regulation of interstitial collagenase (MMP-1) gene expression by ETS transcription factors. Oncogene 1997, 14, 2651–2660. [Google Scholar] [CrossRef] [PubMed]
- Hashiya, N.; Jo, N.; Aoki, M.; Matsumoto, K.; Nakamura, T.; Sato, Y.; Ogata, N.; Ogihara, T.; Kaneda, Y.; Morishita, R. In vivo evidence of angiogenesis induced by transcription factor Ets-1: Ets-1 is located upstream of angiogenesis cascade. Circulation 2004, 109, 3035–3041. [Google Scholar] [CrossRef]
- Furlan, A.; Vercamer, C.; Heliot, L.; Wernert, N.; Desbiens, X.; Pourtier, A. Ets-1 drives breast cancer cell angiogenic potential and interactions between breast cancer and endothelial cells. Int. J. Oncol. 2019, 54, 29–40. [Google Scholar] [CrossRef]
- Khanna, A.; Mahalingam, K.; Chakrabarti, D.; Periyasamy, G. Ets-1 expression and gemcitabine chemoresistance in pancreatic cancer cells. Cell Mol. Biol. Lett. 2011, 16, 101–113. [Google Scholar] [CrossRef]
- Shao, Z.; Li, Y.; Dai, W.; Jia, H.; Zhang, Y.; Jiang, Q.; Chai, Y.; Li, X.; Sun, H.; Yang, R.; et al. ETS-1 induces Sorafenib-resistance in hepatocellular carcinoma cells via regulating transcription factor activity of PXR. Pharm. Res. 2018, 135, 188–200. [Google Scholar] [CrossRef]
- Khatun, S.; Fujimoto, J.; Toyoki, H.; Tamaya, T. Clinical implications of expression of ETS-1 in relation to angiogenesis in ovarian cancers. Cancer Sci. 2003, 94, 769–773. [Google Scholar] [CrossRef]
- Yalim-Camci, I.; Balcik-Ercin, P.; Cetin, M.; Odabas, G.; Tokay, N.; Sayan, A.E.; Yagci, T. ETS1 is coexpressed with ZEB2 and mediates ZEB2-induced epithelial-mesenchymal transition in human tumors. Mol. Carcinog. 2019, 58, 1068–1081. [Google Scholar] [CrossRef] [PubMed]
- Gluck, C.; Glathar, A.; Tsompana, M.; Nowak, N.; Garrett-Sinha, L.A.; Buck, M.J.; Sinha, S. Molecular dissection of the oncogenic role of ETS1 in the mesenchymal subtypes of head and neck squamous cell carcinoma. PLoS Genet. 2019, 15, e1008250. [Google Scholar] [CrossRef] [PubMed]
- Verschoor, M.L.; Wilson, L.A.; Verschoor, C.P.; Singh, G. Ets-1 regulates energy metabolism in cancer cells. PLoS ONE 2010, 5, e13565. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.A.; Gemin, A.; Espiritu, R.; Singh, G. ets-1 is transcriptionally up-regulated by H2O2 via an antioxidant response element. FASEB J. 2005, 19, 2085–2087. [Google Scholar] [CrossRef] [PubMed]
- Verschoor, M.L.; Singh, G. Ets-1 regulates intracellular glutathione levels: Key target for resistant ovarian cancer. Mol. Cancer 2013, 12, 138. [Google Scholar] [CrossRef]
- Ozawa, M.; Baribault, H.; Kemler, R. The cytoplasmic domain of the cell adhesion molecule uvomorulin associates with three independent proteins structurally related in different species. EMBO J. 1989, 8, 1711–1717. [Google Scholar] [CrossRef]
- Gregorieff, A.; Clevers, H. Wnt signaling in the intestinal epithelium: From endoderm to cancer. Genes Dev. 2005, 19, 877–890. [Google Scholar] [CrossRef]
- Niehrs, C.; Acebron, S.P. Mitotic and mitogenic Wnt signalling. EMBO J. 2012, 31, 2705–2713. [Google Scholar] [CrossRef]
- Chen, S.; Guttridge, D.C.; You, Z.; Zhang, Z.; Fribley, A.; Mayo, M.W.; Kitajewski, J.; Wang, C.Y. Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J. Cell Biol. 2001, 152, 87–96. [Google Scholar] [CrossRef]
- Huang, M.; Wang, Y.; Sun, D.; Zhu, H.; Yin, Y.; Zhang, W.; Yang, S.; Quan, L.; Bai, J.; Wang, S.; et al. Identification of genes regulated by Wnt/beta-catenin pathway and involved in apoptosis via microarray analysis. BMC Cancer 2006, 6, 221. [Google Scholar] [CrossRef][Green Version]
- Schmalhofer, O.; Brabletz, S.; Brabletz, T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009, 28, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillo, E.; de Barrios, O.; Siles, L.; Cuatrecasas, M.; Castells, A.; Postigo, A. beta-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc. Natl. Acad. Sci. USA 2011, 108, 19204–19209. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillo, E.; Lazaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Guo, Y.; Wang, X.; Zhao, H.; Ji, Z.; Cheng, C.; Li, L.; Fang, Y.; Xu, D.; Zhu, H.H.; et al. WNT/beta-Catenin Directs Self-Renewal Symmetric Cell Division of hTERT(high) Prostate Cancer Stem Cells. Cancer Res. 2017, 77, 2534–2547. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Cheung, A.K.; Ko, J.M.; Phoon, Y.P.; Chiu, P.M.; Lo, P.H.; Waterman, M.L.; Lung, M.L. Physiological beta-catenin signaling controls self-renewal networks and generation of stem-like cells from nasopharyngeal carcinoma. BMC Cell Biol. 2013, 14, 44. [Google Scholar] [CrossRef] [PubMed]
- Pandit, H.; Li, Y.; Li, X.; Zhang, W.; Li, S.; Martin, R.C.G. Enrichment of cancer stem cells via beta-catenin contributing to the tumorigenesis of hepatocellular carcinoma. BMC Cancer 2018, 18, 783. [Google Scholar] [CrossRef]
- Zhang, X.; Zheng, X.; Lou, Y.; Wang, H.; Xu, J.; Zhang, Y.; Han, B. beta-catenin inhibitors suppress cells proliferation and promote cells apoptosis in PC9 lung cancer stem cells. Int. J. Clin. Exp. Pathol. 2017, 10, 11968–11978. [Google Scholar]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schutz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef]
- Thomas, C.; Gustafsson, J.A. Progesterone receptor-estrogen receptor crosstalk: A novel insight. Trends Endocrinol. Metab. 2015, 26, 453–454. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.G.; Dickler, M.N. Endocrine resistance in hormone-responsive breast cancer: Mechanisms and therapeutic strategies. Endocr. Relat. Cancer 2016, 23, R337–R352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Giuliano, M.; Trivedi, M.V.; Schiff, R.; Osborne, C.K. Metastasis dormancy in estrogen receptor-positive breast cancer. Clin. Cancer Res. 2013, 19, 6389–6397. [Google Scholar] [CrossRef] [PubMed]
- Nagaich, A.K.; Rayasam, G.V.; Martinez, E.D.; Becker, M.; Qiu, Y.; Johnson, T.A.; Elbi, C.; Fletcher, T.M.; John, S.; Hager, G.L. Subnuclear trafficking and gene targeting by steroid receptors. Ann. N. Y. Acad. Sci. 2004, 1024, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Liao, L.; Redmond, A.; Young, L.; Yuan, Y.; Chen, H.; O’Malley, B.W.; Xu, J. The AIB1 oncogene promotes breast cancer metastasis by activation of PEA3-mediated matrix metalloproteinase 2 (MMP2) and MMP9 expression. Mol. Cell Biol. 2008, 28, 5937–5950. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Liu, Z.; Chen, H.; Xu, J. The steroid receptor coactivator-1 regulates twist expression and promotes breast cancer metastasis. Cancer Res. 2009, 69, 3819–3827. [Google Scholar] [CrossRef]
- Daniel, A.R.; Hagan, C.R.; Lange, C.A. Progesterone receptor action: Defining a role in breast cancer. Expert Rev. Endocrinol. Metab. 2011, 6, 359–369. [Google Scholar] [CrossRef]
- Kerbel, R.S.; Kobayashi, H.; Graham, C.H. Intrinsic or acquired drug resistance and metastasis: Are they linked phenotypes? J. Cell Biochem. 1994, 56, 37–47. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- Bushweller, J.H. Targeting transcription factors in cancer-from undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef]
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: Results of a phase 1b trial. J. Immunother. Cancer 2018, 6, 119. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Khor, T.O.; Shu, L.; Su, Z.Y.; Fuentes, F.; Lee, J.H.; Kong, A.N. Plants vs. cancer: A review on natural phytochemicals in preventing and treating cancers and their druggability. Anticancer Agents Med. Chem. 2012, 12, 1281–1305. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.R.; Dollard, C.; Winston, F.; Beck, S.; Trowsdale, J.; Dawid, I.B. The bromodomain: A conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res. 1992, 20, 2603. [Google Scholar] [CrossRef]
- Chiang, C.M. Brd4 engagement from chromatin targeting to transcriptional regulation: Selective contact with acetylated histone H3 and H4. F1000 Biol. Rep. 2009, 1, 98. [Google Scholar] [CrossRef]
- Ba, M.; Long, H.; Yan, Z.; Wang, S.; Wu, Y.; Tu, Y.; Gong, Y.; Cui, S. BRD4 promotes gastric cancer progression through the transcriptional and epigenetic regulation of c-MYC. J. Cell Biochem. 2018, 119, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, D.; Tao, D.; Xiang, W.; Xiao, X.; Wang, M.; Wang, L.; Luo, G.; Li, Y.; Zeng, F.; et al. BRD4 Regulates EZH2 Transcription through Upregulation of C-MYC and Represents a Novel Therapeutic Target in Bladder Cancer. Mol. Cancer Ther. 2016, 15, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Coleman, D.J.; Gao, L.; Schwartzman, J.; Korkola, J.E.; Sampson, D.; Derrick, D.S.; Urrutia, J.; Balter, A.; Burchard, J.; King, C.J.; et al. Maintenance of MYC expression promotes de novo resistance to BET bromodomain inhibition in castration-resistant prostate cancer. Sci. Rep. 2019, 9, 3823. [Google Scholar] [CrossRef]
- Shi, J.; Wang, Y.; Zeng, L.; Wu, Y.; Deng, J.; Zhang, Q.; Lin, Y.; Li, J.; Kang, T.; Tao, M.; et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 2014, 25, 210–225. [Google Scholar] [CrossRef]
- Lu, L.; Chen, Z.; Lin, X.; Tian, L.; Su, Q.; An, P.; Li, W.; Wu, Y.; Du, J.; Shan, H.; et al. Inhibition of BRD4 suppresses the malignancy of breast cancer cells via regulation of Snail. Cell Death Differ. 2020, 27, 255–268. [Google Scholar] [CrossRef]
- Dauch, D.; Rudalska, R.; Cossa, G.; Nault, J.C.; Kang, T.W.; Wuestefeld, T.; Hohmeyer, A.; Imbeaud, S.; Yevsa, T.; Hoenicke, L.; et al. A MYC-aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med. 2016, 22, 744–753. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, T.; Zhou, H.; Feng, B.; Chen, Y.; Zhi, Y.; Wang, R. A Novel Aurora-A Inhibitor (MLN8237) Synergistically Enhances the Antitumor Activity of Sorafenib in Hepatocellular Carcinoma. Mol. Ther. Nucleic Acids 2018, 13, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Bubna, A.K. Vorinostat-An Overview. Indian J. Derm. 2015, 60, 419. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Jeong, J.W.; Park, J.A.; Lee, J.W.; Seo, J.H.; Jung, B.K.; Bae, M.K.; Kim, K.W. Regulation of the HIF-1alpha stability by histone deacetylases. Oncol. Rep. 2007, 17, 647–651. [Google Scholar] [PubMed]
- Sapra, P.; Kraft, P.; Pastorino, F.; Ribatti, D.; Dumble, M.; Mehlig, M.; Wang, M.; Ponzoni, M.; Greenberger, L.M.; Horak, I.D. Potent and sustained inhibition of HIF-1alpha and downstream genes by a polyethyleneglycol-SN38 conjugate, EZN-2208, results in anti-angiogenic effects. Angiogenesis 2011, 14, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Norris, R.E.; Shusterman, S.; Gore, L.; Muscal, J.A.; Macy, M.E.; Fox, E.; Berkowitz, N.; Buchbinder, A.; Bagatell, R. Phase 1 evaluation of EZN-2208, a polyethylene glycol conjugate of SN38, in children adolescents and young adults with relapsed or refractory solid tumors. Pediatr. Blood Cancer 2014, 61, 1792–1797. [Google Scholar] [CrossRef]
- Wu, J.; Contratto, M.; Shanbhogue, K.P.; Manji, G.A.; O’Neil, B.H.; Noonan, A.; Tudor, R.; Lee, R. Evaluation of a locked nucleic acid form of antisense oligo targeting HIF-1alpha in advanced hepatocellular carcinoma. World J. Clin. Oncol. 2019, 10, 149–160. [Google Scholar] [CrossRef]
- Zhang, H.; Qian, D.Z.; Tan, Y.S.; Lee, K.; Gao, P.; Ren, Y.R.; Rey, S.; Hammers, H.; Chang, D.; Pili, R.; et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc. Natl. Acad. Sci. USA 2008, 105, 19579–19586. [Google Scholar] [CrossRef]
- Sage, J. Hope in sight for retinoblastoma. Nat. Med. 2007, 13, 30–31. [Google Scholar] [CrossRef]
- La Thangue, N.B. The yin and yang of E2F-1: Balancing life and death. Nat. Cell Biol. 2003, 5, 587–589. [Google Scholar] [CrossRef]
- Kent, L.N.; Leone, G. The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 2019, 19, 326–338. [Google Scholar] [CrossRef]
- Bisi, J.E.; Sorrentino, J.A.; Jordan, J.L.; Darr, D.D.; Roberts, P.J.; Tavares, F.X.; Strum, J.C. Preclinical development of G1T38: A novel, potent and selective inhibitor of cyclin dependent kinases 4/6 for use as an oral antineoplastic in patients with CDK4/6 sensitive tumors. Oncotarget 2017, 8, 42343–42358. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Semenov, M.; Tamai, K.; Zeng, X. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: Arrows point the way. Development 2004, 131, 1663–1677. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.M.; Cancilla, B.; Yeung, V.P.; Cattaruzza, F.; Chartier, C.; Murriel, C.L.; Cain, J.; Tam, R.; Cheng, C.Y.; Evans, J.W.; et al. WNT antagonists exhibit unique combinatorial antitumor activity with taxanes by potentiating mitotic cell death. Sci. Adv. 2017, 3, e1700090. [Google Scholar] [CrossRef] [PubMed]
- Le, P.N.; McDermott, J.D.; Jimeno, A. Targeting the Wnt pathway in human cancers: Therapeutic targeting with a focus on OMP-54F28. Pharm. Ther. 2015, 146, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Takemaru, K.I.; Moon, R.T. The transcriptional coactivator CBP interacts with beta-catenin to activate gene expression. J. Cell Biol. 2000, 149, 249–254. [Google Scholar] [CrossRef]
- Dejmek, J.; Dejmek, A.; Safholm, A.; Sjolander, A.; Andersson, T. Wnt-5a protein expression in primary dukes B colon cancers identifies a subgroup of patients with good prognosis. Cancer Res. 2005, 65, 9142–9146. [Google Scholar] [CrossRef]
- Jonsson, M.; Dejmek, J.; Bendahl, P.O.; Andersson, T. Loss of Wnt-5a protein is associated with early relapse in invasive ductal breast carcinomas. Cancer Res. 2002, 62, 409–416. [Google Scholar]
- Liang, H.; Chen, Q.; Coles, A.H.; Anderson, S.J.; Pihan, G.; Bradley, A.; Gerstein, R.; Jurecic, R.; Jones, S.N. Wnt5a inhibits B cell proliferation and functions as a tumor suppressor in hematopoietic tissue. Cancer Cell 2003, 4, 349–360. [Google Scholar] [CrossRef]
- Prasad, C.P.; Manchanda, M.; Mohapatra, P.; Andersson, T. WNT5A as a therapeutic target in breast cancer. Cancer Metastasis Rev. 2018, 37, 767–778. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef]
- Dhiman, V.K.; Bolt, M.J.; White, K.P. Nuclear receptors in cancer-uncovering new and evolving roles through genomic analysis. Nat. Rev. Genet. 2018, 19, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.; Tarulli, G.; Portman, N.; Hickey, T.E.; Tilley, W.D.; Palmieri, C. Pushing estrogen receptor around in breast cancer. Endocr. Relat. Cancer 2016, 23, T227–T241. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Brown, M. Molecular determinants for the tissue specificity of SERMs. Science 2002, 295, 2465–2468. [Google Scholar] [CrossRef] [PubMed]
- Wakeling, A.E.; Dukes, M.; Bowler, J. A potent specific pure antiestrogen with clinical potential. Cancer Res. 1991, 51, 3867–3873. [Google Scholar] [PubMed]
- Fu, X.; Jeselsohn, R.; Pereira, R.; Hollingsworth, E.F.; Creighton, C.J.; Li, F.; Shea, M.; Nardone, A.; De Angelis, C.; Heiser, L.M.; et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E6600–E6609. [Google Scholar] [CrossRef]
- Bi, M.; Zhang, Z.; Jiang, Y.Z.; Xue, P.; Wang, H.; Lai, Z.; Fu, X.; De Angelis, C.; Gong, Y.; Gao, Z.; et al. Enhancer reprogramming driven by high-order assemblies of transcription factors promotes phenotypic plasticity and breast cancer endocrine resistance. Nat. Cell Biol. 2020, 22, 701–715. [Google Scholar] [CrossRef]
- Provinciali, N.; Suen, C.; Dunn, B.K.; DeCensi, A. Raloxifene hydrochloride for breast cancer risk reduction in postmenopausal women. Expert Rev. Clin. Pharm. 2016, 9, 1263–1272. [Google Scholar] [CrossRef]
- Komm, B.S.; Mirkin, S.; Jenkins, S.N. Development of conjugated estrogens/bazedoxifene, the first tissue selective estrogen complex (TSEC) for management of menopausal hot flashes and postmenopausal bone loss. Steroids 2014, 90, 71–81. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martin, M.; et al. Phase III Randomized Study of Ribociclib and Fulvestrant in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: MONALEESA-3. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 2465–2472. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [CrossRef]
- Turner, N.C.; Slamon, D.J.; Ro, J.; Bondarenko, I.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2018, 379, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Guichard, S.M.; Curwen, J.; Bihani, T.; D’Cruz, C.M.; Yates, J.W.; Grondine, M.; Howard, Z.; Davies, B.R.; Bigley, G.; Klinowska, T.; et al. AZD2014, an Inhibitor of mTORC1 and mTORC2, Is Highly Effective in ER+ Breast Cancer When Administered Using Intermittent or Continuous Schedules. Mol. Cancer Ther. 2015, 14, 2508–2518. [Google Scholar] [CrossRef] [PubMed]
- Xiong, R.; Zhao, J.; Gutgesell, L.M.; Wang, Y.; Lee, S.; Karumudi, B.; Zhao, H.; Lu, Y.; Tonetti, D.A.; Thatcher, G.R. Novel Selective Estrogen Receptor Downregulators (SERDs) Developed against Treatment-Resistant Breast Cancer. J. Med. Chem. 2017, 60, 1325–1342. [Google Scholar] [CrossRef] [PubMed]
- Wardell, S.E.; Nelson, E.R.; Chao, C.A.; Alley, H.M.; McDonnell, D.P. Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader. Endocr. Relat. Cancer 2015, 22, 713–724. [Google Scholar] [CrossRef]
- Harte, M.T.; Gorski, J.J.; Savage, K.I.; Purcell, J.W.; Barros, E.M.; Burn, P.M.; McFarlane, C.; Mullan, P.B.; Kennedy, R.D.; Perkins, N.D.; et al. NF-kappaB is a critical mediator of BRCA1-induced chemoresistance. Oncogene 2014, 33, 713–723. [Google Scholar] [CrossRef]
- Kwon, O.; Kim, K.A.; Kim, S.O.; Ha, R.; Oh, W.K.; Kim, M.S.; Kim, H.S.; Kim, G.D.; Kim, J.W.; Jung, M.; et al. NF-kappaB inhibition increases chemosensitivity to trichostatin A-induced cell death of Ki-Ras-transformed human prostate epithelial cells. Carcinogenesis 2006, 27, 2258–2268. [Google Scholar] [CrossRef]
- Bentires-Alj, M.; Barbu, V.; Fillet, M.; Chariot, A.; Relic, B.; Jacobs, N.; Gielen, J.; Merville, M.P.; Bours, V. NF-kappaB transcription factor induces drug resistance through MDR1 expression in cancer cells. Oncogene 2003, 22, 90–97. [Google Scholar] [CrossRef]
- Fan, F.; Bashari, M.H.; Morelli, E.; Tonon, G.; Malvestiti, S.; Vallet, S.; Jarahian, M.; Seckinger, A.; Hose, D.; Bakiri, L.; et al. The AP-1 transcription factor JunB is essential for multiple myeloma cell proliferation and drug resistance in the bone marrow microenvironment. Leukemia 2017, 31, 1570–1581. [Google Scholar] [CrossRef]
- Yan, D.; Yan, X.; Dai, X.; Chen, L.; Sun, L.; Li, T.; He, F.; Lian, J.; Cai, W. Activation of AKT/AP1/FoxM1 signaling confers sorafenib resistance to liver cancer cells. Oncol. Rep. 2019, 42, 785–796. [Google Scholar] [CrossRef]
- Wang, Y.; Wan, G.H.; Wu, Y.M.; Wang, H.S.; Wang, H.F.; Zhang, G.; Lu, L.L.; Li, Z.Q.; Chan, K.Y.; Zhou, Y.; et al. AP-1 confers resistance to anti-cancer therapy by activating XIAP. Oncotarget 2018, 9, 14124–14137. [Google Scholar] [CrossRef][Green Version]
- Han, Z.; Feng, J.; Hong, Z.; Chen, L.; Li, W.; Liao, S.; Wang, X.; Ji, T.; Wang, S.; Ma, D.; et al. Silencing of the STAT3 signaling pathway reverses the inherent and induced chemoresistance of human ovarian cancer cells. Biochem. Biophys. Res. Commun. 2013, 435, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Ren, C.; Wang, J.; Wang, S.; Yang, L.; Han, X.; Chen, Y.; Tong, G.; Yang, G. The crosstalk between STAT3 and p53/RAS signaling controls cancer cell metastasis and cisplatin resistance via the Slug/MAPK/PI3K/AKT-mediated regulation of EMT and autophagy. Oncogenesis 2019, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Liu, K.; Shao, Y.; Feng, X.; Ji, Z.; Chang, B.; Wang, Y.; Xu, L.; Yang, G. ID1 confers cancer cell chemoresistance through STAT3/ATF6-mediated induction of autophagy. Cell Death Dis. 2020, 11, 137. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, Y.T.; Liu, P.F.; Li, J.Y.; Liu, L.F.; Kuo, S.Y.; Hsieh, C.W.; Lee, C.H.; Wu, C.H.; Hsiao, M.; Chang, H.T.; et al. Kinome-Wide siRNA Screening Identifies Src-Enhanced Resistance of Chemotherapeutic Drugs in Triple-Negative Breast Cancer Cells. Front. Pharm. 2018, 9, 1285. [Google Scholar] [CrossRef] [PubMed]
- Sowa, T.; Menju, T.; Chen-Yoshikawa, T.F.; Takahashi, K.; Nishikawa, S.; Nakanishi, T.; Shikuma, K.; Motoyama, H.; Hijiya, K.; Aoyama, A.; et al. Hypoxia-inducible factor 1 promotes chemoresistance of lung cancer by inducing carbonic anhydrase IX expression. Cancer Med. 2017, 6, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Liu, F.; Han, L.; Zhao, L.; Chen, J.; Olopade, O.I.; He, M.; Wei, M. HIF-2alpha promotes conversion to a stem cell phenotype and induces chemoresistance in breast cancer cells by activating Wnt and Notch pathways. J. Exp. Clin. Cancer Res. 2018, 37, 256. [Google Scholar] [CrossRef]
- Van Oosterwijk, J.G.; Buelow, D.R.; Drenberg, C.D.; Vasilyeva, A.; Li, L.; Shi, L.; Wang, Y.D.; Finkelstein, D.; Shurtleff, S.A.; Janke, L.J.; et al. Hypoxia-induced upregulation of BMX kinase mediates therapeutic resistance in acute myeloid leukemia. J. Clin. Invest. 2018, 128, 369–380. [Google Scholar] [CrossRef]
- Zhao, C.X.; Luo, C.L.; Wu, X.H. Hypoxia promotes 786-O cells invasiveness and resistance to sorafenib via HIF-2alpha/COX-2. Med. Oncol. 2015, 32, 419. [Google Scholar] [CrossRef]
- Zhao, D.; Zhai, B.; He, C.; Tan, G.; Jiang, X.; Pan, S.; Dong, X.; Wei, Z.; Ma, L.; Qiao, H.; et al. Upregulation of HIF-2alpha induced by sorafenib contributes to the resistance by activating the TGF-alpha/EGFR pathway in hepatocellular carcinoma cells. Cell. Signal. 2014, 26, 1030–1039. [Google Scholar] [CrossRef]
- Lee, K.M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sanchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647. [Google Scholar] [CrossRef]
- Zhang, H.L.; Wang, P.; Lu, M.Z.; Zhang, S.D.; Zheng, L. c-Myc maintains the self-renewal and chemoresistance properties of colon cancer stem cells. Oncol. Lett. 2019, 17, 4487–4493. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Zhang, Z.; Luo, K.; Zheng, G.; Lu, M.; Song, Y.; Liu, H.; Qiu, H.; He, Z. TCRP1 transcriptionally regulated by c-Myc confers cancer chemoresistance in tongue and lung cancer. Sci. Rep. 2017, 7, 3744. [Google Scholar] [CrossRef]
- Pan, X.N.; Chen, J.J.; Wang, L.X.; Xiao, R.Z.; Liu, L.L.; Fang, Z.G.; Liu, Q.; Long, Z.J.; Lin, D.J. Inhibition of c-Myc overcomes cytotoxic drug resistance in acute myeloid leukemia cells by promoting differentiation. PLoS ONE 2014, 9, e105381. [Google Scholar] [CrossRef]
- Kato, T.; Fujita, Y.; Nakane, K.; Kojima, T.; Nozawa, Y.; Deguchi, T.; Ito, M. ETS1 promotes chemoresistance and invasion of paclitaxel-resistant, hormone-refractory PC3 prostate cancer cells by up-regulating MDR1 and MMP9 expression. Biochem. Biophys. Res. Commun. 2012, 417, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.A.; Yamamoto, H.; Singh, G. Role of the transcription factor Ets-1 in cisplatin resistance. Mol. Cancer Ther. 2004, 3, 823–832. [Google Scholar] [PubMed]
- Wei, J.; Zhou, Y.; Jiang, G.Q.; Xiao, D. Silencing of ETS1 reverses adriamycin resistance in MCF-7/ADR cells via downregulation of MDR1. Cancer Cell Int. 2014, 14, 22. [Google Scholar] [CrossRef] [PubMed]
- Ruan, X.; Liu, A.; Zhong, M.; Wei, J.; Zhang, W.; Rong, Y.; Liu, W.; Li, M.; Qing, X.; Chen, G.; et al. Silencing LGR6 Attenuates Stemness and Chemoresistance via Inhibiting Wnt/beta-Catenin Signaling in Ovarian Cancer. Mol. Ther. Oncolytics 2019, 14, 94–106. [Google Scholar] [CrossRef]
- Sinnberg, T.; Menzel, M.; Ewerth, D.; Sauer, B.; Schwarz, M.; Schaller, M.; Garbe, C.; Schittek, B. beta-Catenin signaling increases during melanoma progression and promotes tumor cell survival and chemoresistance. PLoS ONE 2011, 6, e23429. [Google Scholar] [CrossRef]
- Liu, Y.; Ye, X.; Zhang, J.B.; Ouyang, H.; Shen, Z.; Wu, Y.; Wang, W.; Wu, J.; Tao, S.; Yang, X.; et al. PROX1 promotes hepatocellular carcinoma proliferation and sorafenib resistance by enhancing beta-catenin expression and nuclear translocation. Oncogene 2015, 34, 5524–5535. [Google Scholar] [CrossRef]
- Deng, L.; Sun, J.; Chen, X.; Liu, L.; Wu, D. Nek2 augments sorafenib resistance by regulating the ubiquitination and localization of beta-catenin in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 316. [Google Scholar] [CrossRef]
- He, L.; Zhu, H.; Zhou, S.; Wu, T.; Wu, H.; Yang, H.; Mao, H.; SekharKathera, C.; Janardhan, A.; Edick, A.M.; et al. Wnt pathway is involved in 5-FU drug resistance of colorectal cancer cells. Exp. Mol. Med. 2018, 50, 101. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.S.; Shieh, G.S.; Wang, C.T.; Su, B.H.; Su, Y.C.; Chen, Y.C.; Su, W.C.; Wu, P.; Yang, W.H.; Shiau, A.L.; et al. Chemotherapeutics-induced Oct4 expression contributes to drug resistance and tumor recurrence in bladder cancer. Oncotarget 2017, 8, 30844–30858. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Hsu, H.S.; Chen, Y.W.; Tsai, T.H.; How, C.K.; Wang, C.Y.; Hung, S.C.; Chang, Y.L.; Tsai, M.L.; Lee, Y.Y.; et al. Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS ONE 2008, 3, e2637. [Google Scholar] [CrossRef] [PubMed]
- Bora-Singhal, N.; Mohankumar, D.; Saha, B.; Colin, C.M.; Lee, J.Y.; Martin, M.W.; Zheng, X.; Coppola, D.; Chellappan, S. Novel HDAC11 inhibitors suppress lung adenocarcinoma stem cell self-renewal and overcome drug resistance by suppressing Sox2. Sci. Rep. 2020, 10, 4722. [Google Scholar] [CrossRef]
- Lai, S.C.; Su, Y.T.; Chi, C.C.; Kuo, Y.C.; Lee, K.F.; Wu, Y.C.; Lan, P.C.; Yang, M.H.; Chang, T.S.; Huang, Y.H. DNMT3b/OCT4 expression confers sorafenib resistance and poor prognosis of hepatocellular carcinoma through IL-6/STAT3 regulation. J. Exp. Clin. Cancer Res. 2019, 38, 474. [Google Scholar] [CrossRef] [PubMed]
- Huser, L.; Sachindra, S.; Granados, K.; Federico, A.; Larribere, L.; Novak, D.; Umansky, V.; Altevogt, P.; Utikal, J. SOX2-mediated upregulation of CD24 promotes adaptive resistance toward targeted therapy in melanoma. Int. J. Cancer 2018, 143, 3131–3142. [Google Scholar] [CrossRef]
- Lu, Y.; Zhu, H.; Shan, H.; Lu, J.; Chang, X.; Li, X.; Lu, J.; Fan, X.; Zhu, S.; Wang, Y.; et al. Knockdown of Oct4 and Nanog expression inhibits the stemness of pancreatic cancer cells. Cancer Lett. 2013, 340, 113–123. [Google Scholar] [CrossRef]
- Wong, Y.N.; Ferraldeschi, R.; Attard, G.; de Bono, J. Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nat. Rev. Clin. Oncol. 2014, 11, 365–376. [Google Scholar] [CrossRef]
- Zhao, L.; Zhou, S.; Gustafsson, J.A. Nuclear Receptors: Recent Drug Discovery for Cancer Therapies. Endocr. Rev. 2019, 40, 1207–1249. [Google Scholar] [CrossRef]
- Rathkopf, D.E.; Antonarakis, E.S.; Shore, N.D.; Tutrone, R.F.; Alumkal, J.J.; Ryan, C.J.; Saleh, M.; Hauke, R.J.; Bandekar, R.; Maneval, E.C.; et al. Safety and Antitumor Activity of Apalutamide (ARN-509) in Metastatic Castration-Resistant Prostate Cancer with and without Prior Abiraterone Acetate and Prednisone. Clin. Cancer Res. 2017, 23, 3544–3551. [Google Scholar] [CrossRef]
- Bastos, D.A.; Antonarakis, E.S. Darolutamide for Castration-Resistant Prostate Cancer. Onco Targets Ther. 2019, 12, 8769–8777. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2019, 380, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Nevedomskaya, E.; Baumgart, S.J.; Haendler, B. Recent Advances in Prostate Cancer Treatment and Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1359. [Google Scholar] [CrossRef] [PubMed]
- Srinath, R.; Dobs, A. Enobosarm (GTx-024, S-22): A potential treatment for cachexia. Future Oncol. 2014, 10, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Meissner, R.S.; Gentile, M.A.; Chisamore, M.J.; Opas, E.E.; Scafonas, A.; Cusick, T.E.; Gambone, C.; Pennypacker, B.; Hodor, P.; et al. Identification of an anabolic selective androgen receptor modulator that actively induces death of androgen-independent prostate cancer cells. J. Steroid Biochem. Mol. Biol. 2014, 143, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.T.; Wang, L.H. New dimension of glucocorticoids in cancer treatment. Steroids 2016, 111, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Sundahl, N.; Bridelance, J.; Libert, C.; De Bosscher, K.; Beck, I.M. Selective glucocorticoid receptor modulation: New directions with non-steroidal scaffolds. Pharm. Ther. 2015, 152, 28–41. [Google Scholar] [CrossRef]
- Sundahl, N.; Clarisse, D.; Bracke, M.; Offner, F.; Berghe, W.V.; Beck, I.M. Selective glucocorticoid receptor-activating adjuvant therapy in cancer treatments. Oncoscience 2016, 3, 188–202. [Google Scholar] [CrossRef]
- Orqueda, A.J.; Dansey, M.V.; Espanol, A.; Veleiro, A.S.; Bal de Kier Joffe, E.; Sales, M.E.; Burton, G.; Pecci, A. The rigid steroid 21-hydroxy-6,19-epoxyprogesterone (21OH-6,19OP) is a dissociated glucocorticoid receptor modulator potentially useful as a novel coadjuvant in breast cancer chemotherapy. Biochem. Pharm. 2014, 89, 526–535. [Google Scholar] [CrossRef]
- Haridas, V.; Xu, Z.X.; Kitchen, D.; Jiang, A.; Michels, P.; Gutterman, J.U. The anticancer plant triterpenoid, avicin D, regulates glucocorticoid receptor signaling: Implications for cellular metabolism. PLoS ONE 2011, 6, e28037. [Google Scholar] [CrossRef]
- Lesovaya, E.; Yemelyanov, A.; Swart, A.C.; Swart, P.; Haegeman, G.; Budunova, I. Discovery of Compound A--a selective activator of the glucocorticoid receptor with anti-inflammatory and anti-cancer activity. Oncotarget 2015, 6, 30730–30744. [Google Scholar] [CrossRef] [PubMed]
- Hunt, H.J.; Belanoff, J.K.; Walters, I.; Gourdet, B.; Thomas, J.; Barton, N.; Unitt, J.; Phillips, T.; Swift, D.; Eaton, E. Identification of the Clinical Candidate (R)-(1-(4-Fluorophenyl)-6-((1-methyl-1H-pyrazol-4-yl)sulfonyl)-4,4a,5,6,7,8-hexah ydro-1H-pyrazolo[3,4-g]isoquinolin-4a-yl)(4-(trifluoromethyl)pyridin-2-yl)methano ne (CORT125134): A Selective Glucocorticoid Receptor (GR) Antagonist. J. Med. Chem. 2017, 60, 3405–3421. [Google Scholar] [CrossRef] [PubMed]
- Rew, Y.; Du, X.; Eksterowicz, J.; Zhou, H.; Jahchan, N.; Zhu, L.; Yan, X.; Kawai, H.; McGee, L.R.; Medina, J.C.; et al. Discovery of a Potent and Selective Steroidal Glucocorticoid Receptor Antagonist (ORIC-101). J. Med. Chem. 2018, 61, 7767–7784. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J.; Shao, J.; Gao, Y.; Xu, J.; Yu, S.; Liu, Z.; Jia, L. The unique pharmacological characteristics of mifepristone (RU486): From terminating pregnancy to preventing cancer metastasis. Med. Res. Rev. 2014, 34, 979–1000. [Google Scholar] [CrossRef] [PubMed]
- Benagiano, G.; Bastianelli, C.; Farris, M. Selective progesterone receptor modulators 3: Use in oncology, endocrinology and psychiatry. Expert Opin. Pharm. 2008, 9, 2487–2496. [Google Scholar] [CrossRef] [PubMed]
- Wiehle, R.; Lantvit, D.; Yamada, T.; Christov, K. CDB-4124, a progesterone receptor modulator, inhibits mammary carcinogenesis by suppressing cell proliferation and inducing apoptosis. Cancer Prev. Res. 2011, 4, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Stringer-Reasor, E.M.; Baker, G.M.; Skor, M.N.; Kocherginsky, M.; Lengyel, E.; Fleming, G.F.; Conzen, S.D. Glucocorticoid receptor activation inhibits chemotherapy-induced cell death in high-grade serous ovarian carcinoma. Gynecol. Oncol. 2015, 138, 656–662. [Google Scholar] [CrossRef]
- Carroll, J.S.; Hickey, T.E.; Tarulli, G.A.; Williams, M.; Tilley, W.D. Deciphering the divergent roles of progestogens in breast cancer. Nat. Rev. Cancer 2017, 17, 54–64. [Google Scholar] [CrossRef]
- Mohammed, H.; Russell, I.A.; Stark, R.; Rueda, O.M.; Hickey, T.E.; Tarulli, G.A.; Serandour, A.A.; Birrell, S.N.; Bruna, A.; Saadi, A.; et al. Progesterone receptor modulates ERalpha action in breast cancer. Nature 2015, 523, 313–317. [Google Scholar] [CrossRef]
- Niederreither, K.; Dolle, P. Retinoic acid in development: Towards an integrated view. Nat. Rev. Genet. 2008, 9, 541–553. [Google Scholar] [CrossRef]
- Solt, L.A.; Burris, T.P. Action of RORs and their ligands in (patho)physiology. Trends Endocrinol. Metab. 2012, 23, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Benomar, Y.; Staels, B. Retinoid X receptors: Common heterodimerization partners with distinct functions. Trends Endocrinol. Metab. 2010, 21, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Milanesi, N.; D’Erme, A.M.; Gola, M. Nail improvement during alitretinoin treatment: Three case reports and review of the literature. Clin. Exp. Derm. 2015, 40, 533–536. [Google Scholar] [CrossRef]
- Cheng, C.; Michaels, J.; Scheinfeld, N. Alitretinoin: A comprehensive review. Expert Opin. Investig. Drugs 2008, 17, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Guo, Y.; Zhang, P.; Cao, X.; Luan, Y. Preventive and Therapeutic Effects of the Retinoid X Receptor Agonist Bexarotene on Tumors. Curr. Drug Metab. 2016, 17, 118–128. [Google Scholar] [CrossRef]
- Rodon, J.; Jacobs, C.D.; Chu, Q.; Rowinsky, E.K.; Lopez-Anaya, A.; Takimoto, C.H.; Wakelee, H.A. A phase I pharmacokinetic study of bexarotene with paclitaxel and carboplatin in patients with advanced non-small cell lung cancer (NSCLC). Cancer Chemother. Pharm. 2012, 69, 825–834. [Google Scholar] [CrossRef]
- Shinagawa, K.; Yanada, M.; Sakura, T.; Ueda, Y.; Sawa, M.; Miyatake, J.; Dobashi, N.; Kojima, M.; Hatta, Y.; Emi, N.; et al. Tamibarotene as maintenance therapy for acute promyelocytic leukemia: Results from a randomized controlled trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 3729–3735. [Google Scholar] [CrossRef][Green Version]
- Wang, X.; Dasari, S.; Nowakowski, G.S.; Lazaridis, K.N.; Wieben, E.D.; Kadin, M.E.; Feldman, A.L.; Boddicker, R.L. Retinoic acid receptor alpha drives cell cycle progression and is associated with increased sensitivity to retinoids in T-cell lymphoma. Oncotarget 2017, 8, 26245–26255. [Google Scholar] [CrossRef]
- Alonso, S.; Hernandez, D.; Chang, Y.T.; Gocke, C.B.; McCray, M.; Varadhan, R.; Matsui, W.H.; Jones, R.J.; Ghiaur, G. Hedgehog and retinoid signaling alters multiple myeloma microenvironment and generates bortezomib resistance. J. Clin. Invest. 2016, 126, 4460–4468. [Google Scholar] [CrossRef]
- Liby, K.; Royce, D.B.; Risingsong, R.; Williams, C.R.; Wood, M.D.; Chandraratna, R.A.; Sporn, M.B. A new rexinoid, NRX194204, prevents carcinogenesis in both the lung and mammary gland. Clin. Cancer Res. 2007, 13, 6237–6243. [Google Scholar] [CrossRef]
- Clarke, E.; Jarvis, C.I.; Goncalves, M.B.; Kalindjian, S.B.; Adams, D.R.; Brown, J.T.; Shiers, J.J.; Taddei, D.M.A.; Ravier, E.; Barlow, S.; et al. Design and synthesis of a potent, highly selective, orally bioavailable, retinoic acid receptor alpha agonist. Bioorg. Med. Chem. 2018, 26, 798–814. [Google Scholar] [CrossRef]
- Peters, J.M.; Shah, Y.M.; Gonzalez, F.J. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat. Rev. Cancer 2012, 12, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, E.; Romei, C.; Biagini, A.; Sabini, E.; Agate, L.; Mazzeo, S.; Materazzi, G.; Sellari-Franceschini, S.; Ribechini, A.; Torregrossa, L.; et al. Anaplastic thyroid carcinoma: From clinicopathology to genetics and advanced therapies. Nat. Rev. Endocrinol. 2017, 13, 644–660. [Google Scholar] [CrossRef] [PubMed]
- Smallridge, R.C.; Copland, J.A.; Brose, M.S.; Wadsworth, J.T.; Houvras, Y.; Menefee, M.E.; Bible, K.C.; Shah, M.H.; Gramza, A.W.; Klopper, J.P.; et al. Efatutazone, an oral PPAR-gamma agonist, in combination with paclitaxel in anaplastic thyroid cancer: Results of a multicenter phase 1 trial. J. Clin. Endocrinol. Metab. 2013, 98, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Keith, R.L.; Blatchford, P.J.; Kittelson, J.; Minna, J.D.; Kelly, K.; Massion, P.P.; Franklin, W.A.; Mao, J.; Wilson, D.O.; Merrick, D.T.; et al. Oral iloprost improves endobronchial dysplasia in former smokers. Cancer Prev. Res. 2011, 4, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Morales, C.P.; Souza, R.F.; Spechler, S.J. Hallmarks of cancer progression in Barrett’s oesophagus. Lancet 2002, 360, 1587–1589. [Google Scholar] [CrossRef]
- Bernstein, J.M.; Bernstein, C.R.; West, C.M.; Homer, J.J. Molecular and cellular processes underlying the hallmarks of head and neck cancer. Eur. Arch. Otorhinolaryngol. 2013, 270, 2585–2593. [Google Scholar] [CrossRef]
- Stahl, M.; Kohrman, N.; Gore, S.D.; Kim, T.K.; Zeidan, A.M.; Prebet, T. Epigenetics in Cancer: A Hematological Perspective. PLoS Genet. 2016, 12, e1006193. [Google Scholar] [CrossRef]
- Datta, D.; Aftabuddin, M.; Gupta, D.K.; Raha, S.; Sen, P. Human Prostate Cancer Hallmarks Map. Sci. Rep. 2016, 6, 30691. [Google Scholar] [CrossRef]
- Noroxe, D.S.; Poulsen, H.S.; Lassen, U. Hallmarks of glioblastoma: A systematic review. Esmo Open 2016, 1, e000144. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wang, E.; Milazzo, J.P.; Wang, Z.; Kinney, J.B.; Vakoc, C.R. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol. 2015, 33, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Koehler, A.N. Transcription Factor Inhibition: Lessons Learned and Emerging Targets. Trends Mol. Med. 2020, 26, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Transcription Factors | Chemo-Resistance Mechanism | Cancer Type | Drug | Reference |
|---|---|---|---|---|
| NF-κB | p50 subunit of NF-κB associate with BRCA1 on the promoter of genes encoding anti-apoptotic proteins to promote BRC1-mediated resistance to DNA damage | Breast cancer | Etoposide and Camptothecin | [185] |
| p65/RelA activation in 267B1/K-ras overexpressing tumorigenic cells promote chemo-resistance | Prostate cancer | Trichostatin A | [186] | |
| NF-κB regulates MDR1 gene expression | Colon cancer | Daunomycin | [187] | |
| AP1 | The AP-1 family member JunB promoted growth and dexamethasone-resistance in multiple myeloma cells. These were reversed by the silencing of JunB. | Multiple myeloma cell | Dexamethasone | [188] |
| c-Jun upregulated FoxM1 in sorafenib-resistant liver cancer cells. Knocking down c-jun expression reversed this, resulting in enhanced sensitivity of cells to sorafenib. | Liver cancer | Sorafenib | [189] | |
| Activation of AP-1 expression induced transcription of XIAP conferring resistance to chemotherapeutics | Breast and liver cancer cells | HDAC inhibitor, JNJ-2648158 | [190] | |
| STAT3 | Expression of pSTAT3 was higher in cisplatin-resistant cells and silencing of STAT3 increased chemotherapy sensitivity | Ovarian cancer | Cisplatin | [191] |
| Activation of STAT3 in association with p53/RAS pathway controls metastasis and cisplatin-resistance. This also involves Slug, MAPK and PI3K/AKT axes. | Ovarian cancer | Cisplatin | [192] | |
| ID1 mediates resistance via STAT3-mediated induction of ATF6 transcription to induce autophagy | Ovarian cancer | Paclitaxel and cisplatin | [193] | |
| Involvement of Src/STAT3 signaling pathway in chemotherapy resistance | Triple negative breast cancer | Camptothecin, Doxorubicin | [194] | |
| HIF-1 | HIF-1 induced carbonic anhydrase IX expression. Inhibition of carbonic anhydrase IX restored chemo-sensitivity against vinorelbine | Lung cancer | Vinorelbine | [195] |
| HIF-2α overexpression increased the expression of stem cell markers (c-Myc, OCT4, Nanog) and Paclitaxel-resistance. These were accomplished via activation of Wnt and Notch pathways. | Breast cancer | Paclitaxel | [196] | |
| Treatment of breast cancer cell lines with Paclitaxel or Gemcitabine increased HIF activity. This in turn enriched the cancer stem cell population and increased the expression of multidrug resistance 1 (MDR1) | Breast cancer | Paclitaxel or Gemcitabine | [70] | |
| HIF-dependent BMX kinase upregulation resulted in therapeutic resistance through a compensatory pro-survival signaling mechanism | Acute myeloid leukemia | Sorafenib | [197] | |
| HIF2 and COX2 activated Snail and downregulated E-cadherin expression to promote invasion and resistance to sorafenib | Renal cancer | Sorafenib | [198] | |
| HIF-2α activated TGF-α/EGFR pathway to promote proliferation and sorafenib-resistance, which was antagonized by HIF-2α siRNA | Hepatocellular carcinoma | Sorafenib | [199] | |
| MYC | MYC cooperated with MCL1 to increase mitochondrial oxidative phosphorylation and ROS to maintain CSCs and promote chemo-resistance | Triple negative breast cancer | Paclitaxel | [200] |
| c-Myc promoted the self-renewal, tumorigenicity, invasion and drug-resistance of colon CSCs | Colon cancer | 5-Fluorouracil, Oxaliplatin, FOLFOX | [201] | |
| c-Myc upregulated tongue cancer resistance-associated protein 1 (TCRP1) to promote chemoresistance, and this axis acted as a negative biomarker of prognosis | Tongue and lung cancer | Cisplatin | [202] | |
| Overexpression of c-Myc promoted resistance to chemotherapeutic drugs, increased colony formation and inhibited cell differentiation | Acute Myeloid Leukemia Cells | Cytarabine (Ara-C), Daunomycin, Doxorubicin | [203] | |
| ETS1 | ETS1 upregulated MDR1 and MMP9 expressions to promote paclitaxel-resistance and invasion | Hormone-refractory prostate cancer | Paclitaxel | [204] |
| ETS1 promoted cisplatin-resistance by transcriptional activation of genes involved in reducing cisplatin toxicity, including metallothioneins and DNA repair enzymes | Ovarian cancer | Cisplatin | [205] | |
| ETS1 binding to Pregnane X receptor (PXR) increases the transcriptional activity of PXR, leading to sorafenib-resistance through induction of multi-drug resistance genes | Hepatocellular carcinoma | Sorafenib | [109] | |
| ETS1 increased MDR1 expression, and siRNA-mediated silencing of ETS1 reduced MDR1 expression and effectively reversed drug-resistance | Adriamycin-resistant breast cancer cells | Adriamycin | [206] | |
| β-catenin/TCF | Leucine-rich-repeat-containing G protein-coupled receptor (LGR) promoted stemness and chemo-resistance via activating Wnt/β-catenin signaling pathway | Ovarian cancer | Cisplatin and Paclitaxel | [207] |
| β-catenin promoted survival of metastatic melanoma cells and not benign melanocytes or primary, non-invasive melanoma cells. Downregulation of β-catenin sensitized metastatic melanoma cells towards chemotherapy | Metastatic melanoma cells | Temozolomide, Cisplatin and Doxorubicin | [208] | |
| Prospero-related homeobox 1 (PROX1) upregulated β-catenin transcription and nuclear translocation to activate the Wnt/β-catenin pathway in HCC, which lead to high proliferation and sorafenib-resistance | Hepatocellular carcinoma | Sorafenib | [209] | |
| Nek2 stabilized β-catenin, increased its nuclear translocation, and activated the transcription of its downstream target genes, to promote sorafenib-resistance | Hepatocellular carcinoma | Sorafenib | [210] | |
| Suppression of checkpoint kinase 1 (CHK1) pathway by Wnt/β-catenin in p53 wild-type colorectal cancer cells, promoted drug-resistance | Colorectal cancer | 5-Fluorouracil | [211] | |
| Transcription factors related to stemness (Oct-4, Sox-2, Nanog) | Chemotherapy-induced Oct-4 expression promoted acquired resistance in cancer | Bladder cancer | Cisplatin | [212] |
| High expression of Oct-4 in CD133+CSCs maintained self-renewal and drug-resistance in lung cancer. Knocking down Oct-4 expression in CD133+CSCs significantly inhibited tumor invasion and colony formation, and increased apoptosis | Lung cancer | Cisplatin, Etoposide, Doxorubicin, and Paclitaxel | [213] | |
| HDAC11-mediated increase in expression of Sox2 is required for the maintenance of CSCs to promote drug resistance | Lung adenocarcinoma | Cisplatin, Erlotinib and Gefitinib | [214] | |
| IL-6/p-STAT3 activation increased the expression of DNMT3b/OCT4 which conferred early recurrence and poor prognosis in HCC | Hepatocellular carcinoma | Sorafenib | [215] | |
| The expression of Sox2 and CD24 were upregulated in targeted-therapy resistant melanoma cells, which was mediated by activated STAT3. Activation of STAT3, Sox2 and CD24 promoted adaptive-resistance to BRAF inhibitors. | Melanoma | BRAF inhibitors (Vemurafenib, plx8394 and pIx7904) | [216] | |
| Increased expression of Oct-4 and Nanog play important roles in the proliferation, migration, invasion and chemoresistance of pancreatic CSCs, and might serve as important prognostic biomarkers and therapeutic targets for pancreatic cancer. | Pancreatic cancer | Gemcitabine | [217] |
| TFs | Therapeutics | Phase | Cancer Type | Status | Trial Identifier |
|---|---|---|---|---|---|
| STAT3 | OPB-31121 | Phase I | Advanced solid tumors | Completed | NCT00955812 |
| OPB-51602 | Phase I | Advanced cancers | Completed | NCT01423903 | |
| AZD9150 (IONIS-STAT3Rx) | Phase I/II | Advanced cancers, Diffuse Large B Cell Lymphoma, Lymphoma | Completed | NCT01563302 | |
| WP1066 | Phase I | Metastatic malignant neoplasm in the brain, metastatic melanoma, recurrent brain neoplasm, recurrent glioblastoma, recurrent malignant glioma | Recruiting | NCT01904123 | |
| TTI-101 | Phase I | Breast cancer, head and neck squamous cell carcinoma, Non-small cell lung cancer (NSCLC), HCC, colorectal cancer, gastric adenocarcinoma, melanoma, advanced cancer | Recruiting | NCT03195699 | |
| NF-κB | Imx-110 | Phase I/II | Advanced solid tumors, pancreatic cancer, breast Cancer, ovarian cancer | Recruiting | NCT03382340 |
| BR-DIM | Phase I | Nonmetastatic hormone-refractory prostate cancer | Completed | NCT00305747 | |
| Curcumin | Phase II | Breast cancer | Completed | NCT01740323 | |
| HIF-1 | Digoxin | Phase II | Breast cancer | Completed | NCT01763931 |
| Topotecan | Phase I | Refractory advanced solid neoplasms expressing HIF-1α | Completed | NCT00117013 | |
| PX-478 | Phase I | Advanced solid tumors, lymphoma | Completed | NCT00522652 | |
| EZN-2208 | Phase I | Refractory solid tumors (in combination with Bevacizumab) | Completed | NCT01251926 | |
| CRLX101 | Phase II | Recurrent platinum-resistant ovarian cancer, Fallopian tube cancer, primary peritoneal cancer (in combination with Bevacizumab) | Completed | NCT01652079 | |
| RO7070179 | Phase I | Hepatocellular carcinoma | Completed | NCT02564614 | |
| Vorinostat | Phase I | Advanced Breast Cancer (in combination with Capecitabine) | Completed | NCT00719875 | |
| PT2385 | Phase I | Advanced clear cell renal cell carcinoma, kidney cancer (alone or in combination with nivolumab or cabozantinib) | Active | NCT02293980 | |
| Myc | BMS-986158 | Phase I | Pediatric solid tumors, lymphoma, or brain tumor | Recruiting | NCT03936465 |
| GSK525762 | Phase I/II | Relapsed, refractory hematological malignancies | Completed | NCT01943851 | |
| MLN8237 | Phase II | Histologically confirmed or clinically suspected metastatic neuroendocrine Prostate cancer | Completed | NCT01799278 | |
| RO6870810 | Phase I | Relapsed/refractory acute myeloid leukemia (AML) | Completed | NCT02308761 | |
| Wnt/Beta-catenin | PRI-724 | Phase I | Pancreatic adenocarcinoma, which is locally advanced, metastatic, or otherwise inoperable. These patients are candidates for second-line therapy after failing FOLFIRINOX as first-line therapy (in combination with gemcitabine) | Completed | NCT01764477 |
| CWP232291 | Phase I/II | Relapsed or refractory AML (in combination with Cytarabine) | Active | NCT03055286 | |
| Vantictumab | Phase I | Previously untreated stage IV pancreatic cancer (in combination with nab-paclitaxel and gemcitabine) | Completed | NCT02005315 | |
| Ipafricept (OMP-54F28) | Phase I | Solid tumors | Completed | NCT01608867 | |
| Ipafricept (OMP-54F28) | Phase I | Previously untreated stage IV pancreatic cancer (in combination with nab-paclitaxel and gemcitabine) | Completed | NCT02050178 | |
| Foxy-5 | Phase II | Resected colon cancer patients treated with FOLFOX chemotherapy regimen | Recruiting | NCT03883802 | |
| E2F | Ribociclib | Phase I | Recurrent Glioblastoma or anaplastic Glioma | Unknown | NCT02345824 |
| Palbociclib | Phase I/II | Advanced KRAS mutant NSCLC (in combination with MEK inhibitor Binimetinib) | Recruiting | NCT03170206 | |
| Abemaciclib | Phase II | Chemo-refractory, Rb wild-type extensive Small-cell lung cancer | Recruiting | NCT04010357 | |
| G1T38 | Phase I/II | EGRF mutation-positive metastatic NSCLC (in combination with Osimertinib) | Active | NCT03455829 |
| Nuclear Receptors | Therapeutics | Phase | Cancer Type | Status | Trial Identifier |
|---|---|---|---|---|---|
| ER | Bazedoxifene | Phase II | Ductal Breast Carcinoma In Situ | Recruiting | NCT02694809 |
| Z-Endoxifen Hydrochloride | Phase I | Breast cancer | Active | NCT01327781 | |
| Lasofoxifene | Phase II | Locally Advanced or Metastatic breast cancer | Recruiting | NCT03781063 | |
| Acolbifene Hydrochloride | Phase II | Breast cancer | Completed | NCT00853996 | |
| Goserelin | Phase I | Patients with Advanced ER+ (Her2 Negative) Breast cancer | Active | NCT02586675 | |
| Elacestrant (RAD-1901) | Phase III | ER+/HER2- advanced breast cancer (EMERALD) | Recruiting | NCT03778931 | |
| AZD9496 | Phase I | Postmenopausal women with ER+ HER2− primary breast cancer | Completed | NCT03236974 | |
| AR | Darolutamide (BAY1841788, ODM-201) | Phase III | Non-metastatic CRPC (nmCRPC) | Active | NCT02200614 |
| Darolutamide | Phase III | Metastatic Hormone Sensitive Prostate Cancer (mHSPC) (in combination with ADT and Docetaxel) | Active | NCT02799602 | |
| TRC253 | Phase I/II | mCRPC and prostate adenocarcinoma | Active | NCT02987829 | |
| TAS3681 | Phase I | mCRPC (multinational study) | Recruiting | NCT02566772 | |
| RAD140 | Phase I | Hormone receptor positive malignant neoplasm of breast | Active | NCT03088527 | |
| Proxalutamide (GT0918) | Phase II | mCRPC | Recruiting | NCT03899467 | |
| Seviteronel (VT-464) | Phase II | CRPC | Completed | NCT02012920 | |
| Enobosarm | Phase II | AR+ metastatic TNBC | Active | NCT02971761 | |
| GR | Relacorilant (CORT125134) | Phase I/II | Solid tumors in combination with nab-paclitaxel | Active | NCT02762981 |
| ORIC-101 | Phase I | Advanced or metastatic solid tumors (in combination with nab-paclitaxel) | Recruiting | NCT03928314 | |
| PR | Onapristone | Phase I/II | Prostate cancer, Androgen-independent Prostate cancer (in combination with abiraterone) | Unknown | NCT02049190 |
| Telapristone acetate (CDB-4124) | Phase II | Stage 1A, 1B and 2 breast cancer | Active | NCT01800422 | |
| Mifepristone | Not Applicable | Breast cancers with ratios of PRA/PRB higher than 1.5 and PR higher than 50% | Active | NCT02651844 | |
| RAR/RXR | IRX4204 | Phase I | Previously treated advanced NSCLC (in combination with erlotinib) | Recruiting | NCT02991651 |
| 9-cis-UAB-30 | Phase I | Early-Stage Breast Carcinoma and Invasive Breast Carcinoma | Recruiting | NCT02876640 | |
| Tamibarotene (SY-1425/Am80) | Phase II | Acute Myeloid Leukemia (as a monotherapy or in combination with azacytidine) | Active | NCT02807558 | |
| Tretinoin | Phase I | Acute Myelogenous Leukemia (in combination with Tranylcypromine) | Active | NCT02273102 | |
| PPAR | Efatutazone (inolitazone, CS-7017, RS-5444) (PPARγ agonist) | Phase II | Previously treated myxoid liposarcoma that cannot be removed by surgery | Active | NCT02249949 |
| Pioglitazone Hydrochloride (PPARγ agonist) | Phase II | Head and Neck Cancer, Oral Leukoplakia | Completed | NCT00099021 | |
| Iloprost (PPARα/δ agonist) | Phase II | Lung cancer, Precancerous Condition | Completed | NCT00084409 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vishnoi, K.; Viswakarma, N.; Rana, A.; Rana, B. Transcription Factors in Cancer Development and Therapy. Cancers 2020, 12, 2296. https://doi.org/10.3390/cancers12082296
Vishnoi K, Viswakarma N, Rana A, Rana B. Transcription Factors in Cancer Development and Therapy. Cancers. 2020; 12(8):2296. https://doi.org/10.3390/cancers12082296
Chicago/Turabian StyleVishnoi, Kanchan, Navin Viswakarma, Ajay Rana, and Basabi Rana. 2020. "Transcription Factors in Cancer Development and Therapy" Cancers 12, no. 8: 2296. https://doi.org/10.3390/cancers12082296
APA StyleVishnoi, K., Viswakarma, N., Rana, A., & Rana, B. (2020). Transcription Factors in Cancer Development and Therapy. Cancers, 12(8), 2296. https://doi.org/10.3390/cancers12082296