Clonal Hematopoiesis in Liquid Biopsy: From Biological Noise to Valuable Clinical Implications




Abstract

1. Introduction
2. Circulating Cell-Free DNA (cfDNA) and Circulating Tumor DNA (ctDNA) in Liquid Biopsy
3. Clonal Hematopoiesis
3.1. Definition of Clonal Hematopoiesis (CH)
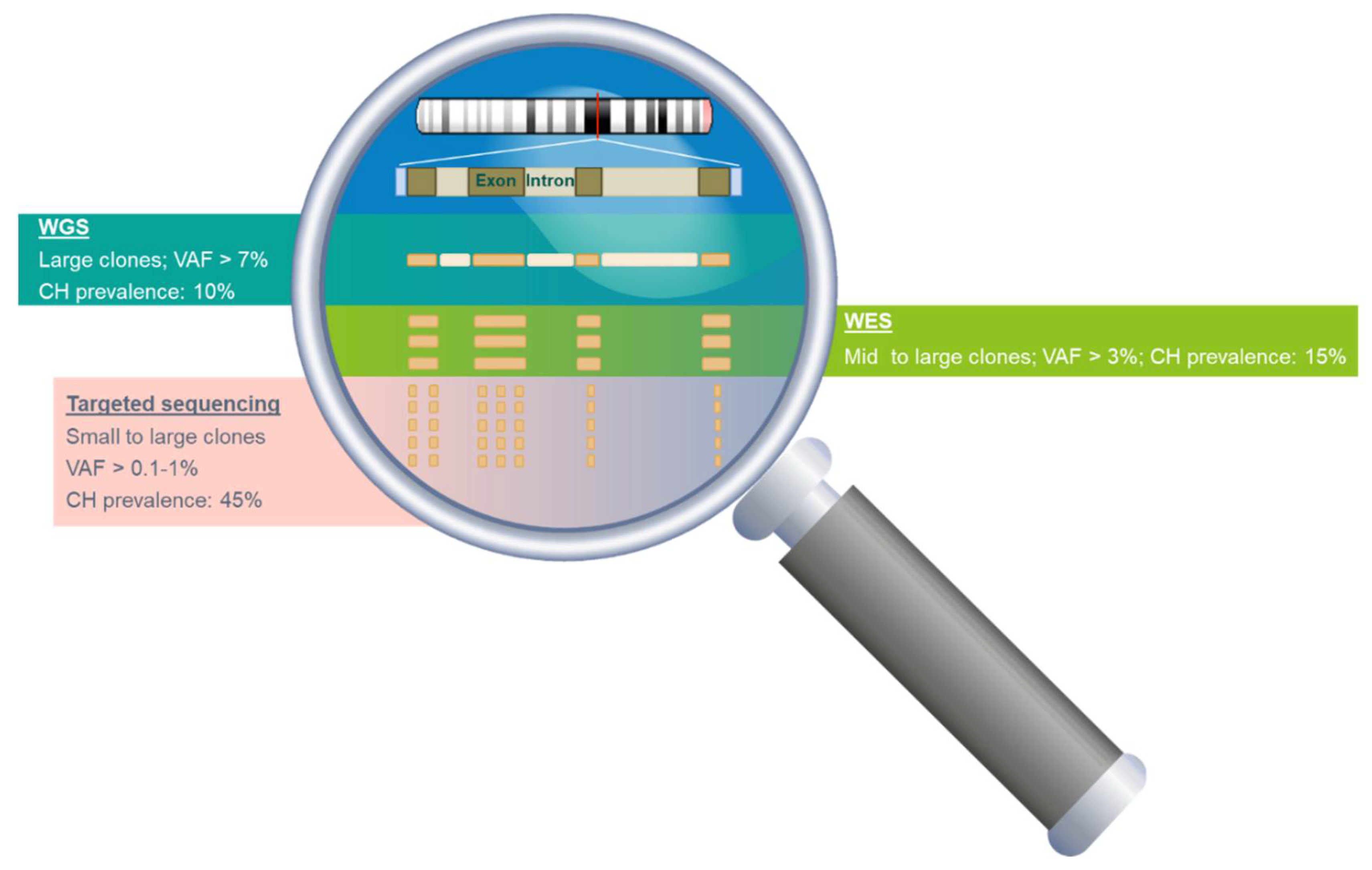
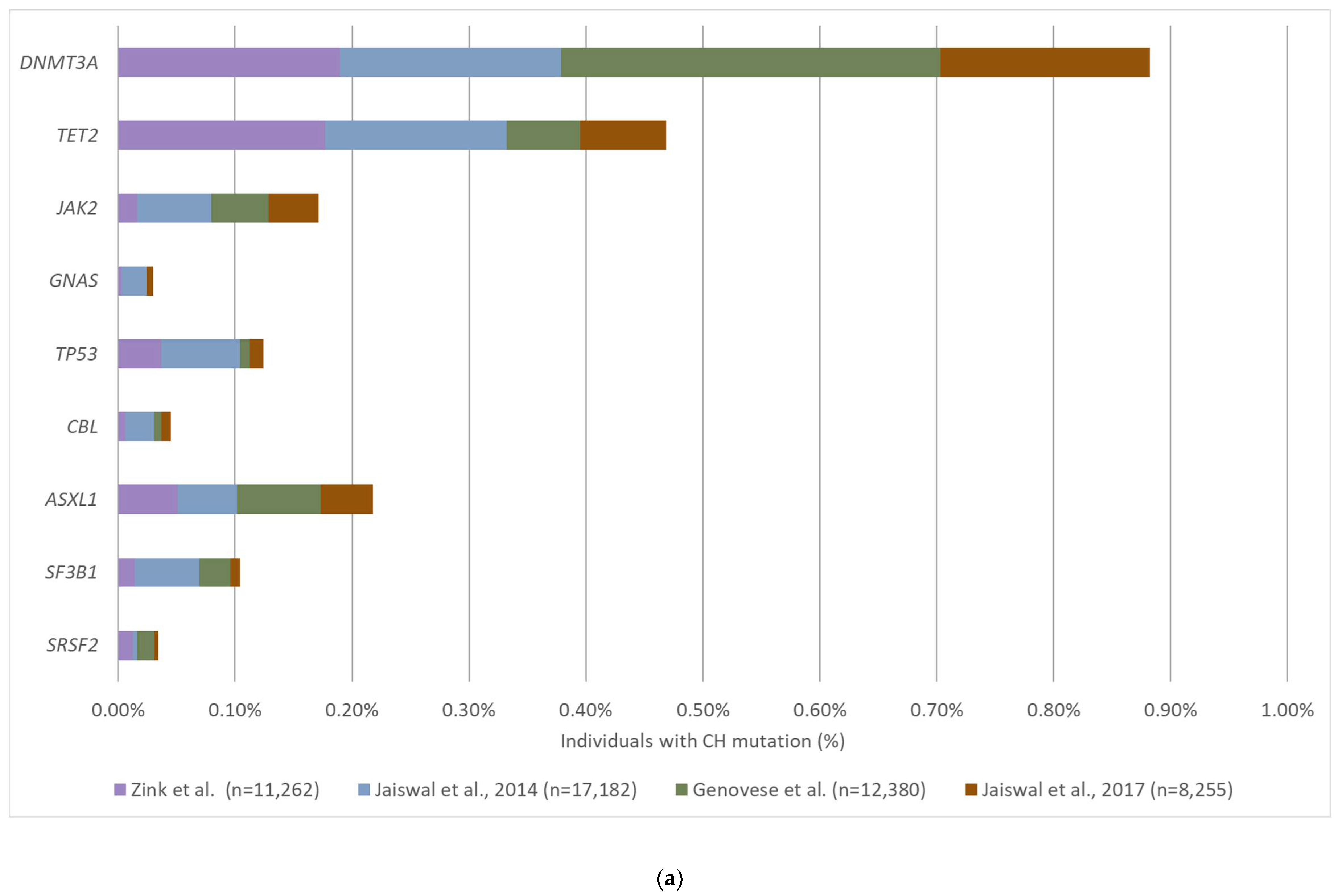
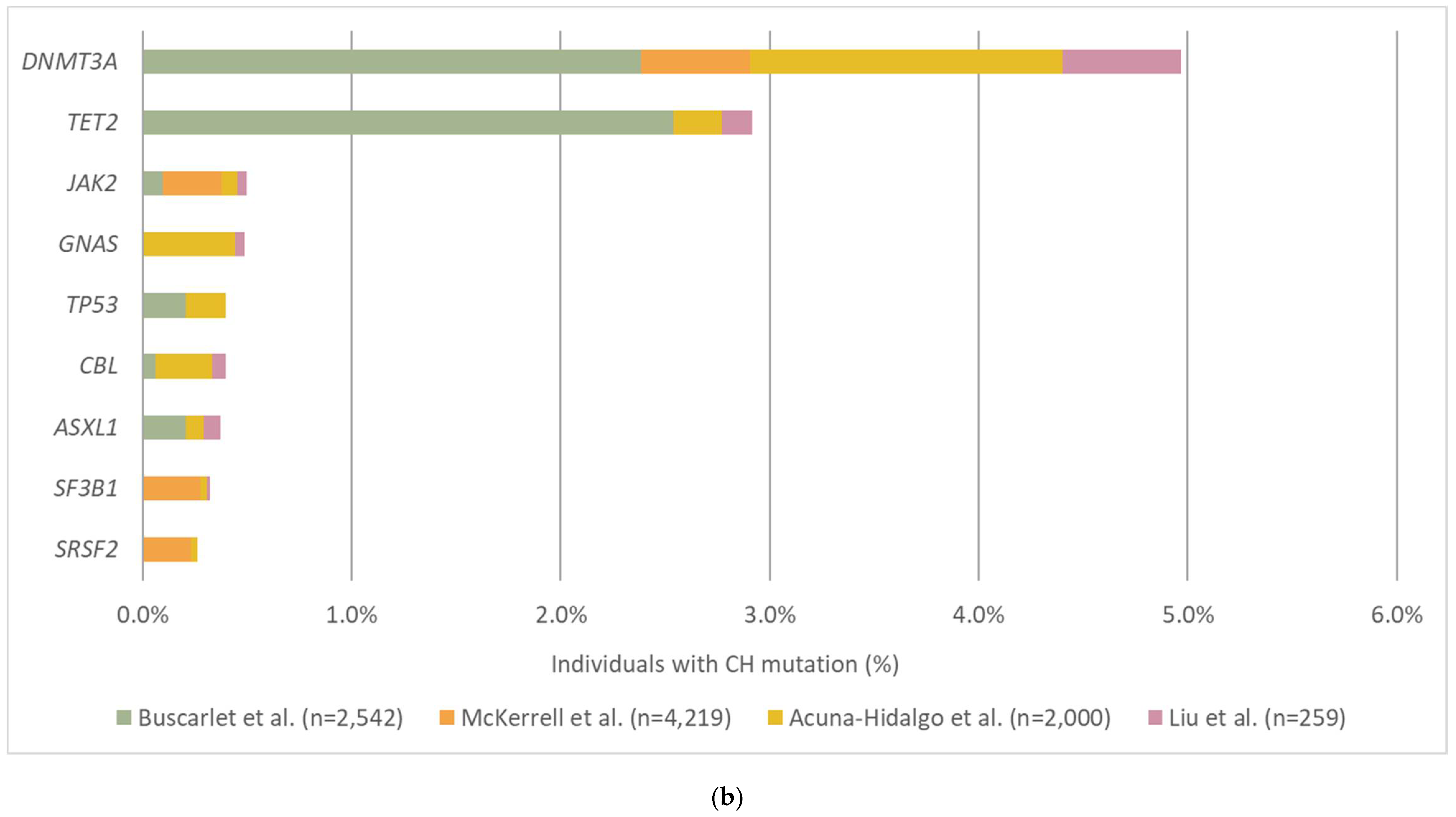
3.2. The Prevalence and Mutation Profile of CH
3.3. Clinical Implications of CH
3.3.1. Hematological Cancer
3.3.2. Therapy-Related Myeloid Neoplasms (t-MN)
3.3.3. Cardiovascular Disease (CVD)
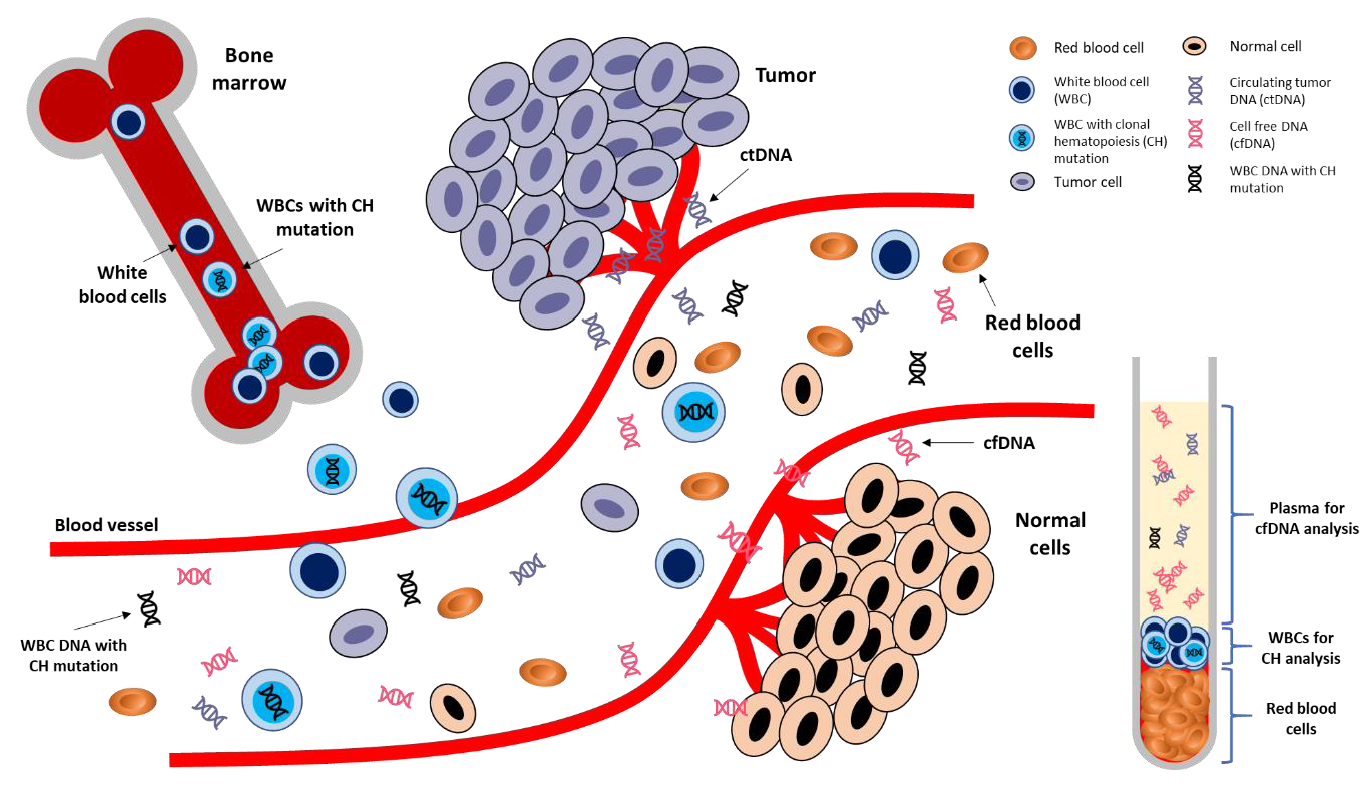
4. CH in Liquid Biopsy
4.1. Detection of CH from Plasma
4.2. Detection of CH from Tumor Tissues
4.3. Clinical Impact of CH Mutations on the Interpretation of Liquid Biopsy Results
4.4. Other Potential Sources of Non-Tumoral Somatic Mutations
5. Future Directions: Clinical Interpretation of CH
6. Conclusions
Funding
Conflicts of Interest
References
- Alix-Panabières, C. Perspective: The future of liquid biopsy. Nature 2020, 579, S9. [Google Scholar] [CrossRef]
- De Rubis, G.; Rajeev Krishnan, S.; Bebawy, M. Liquid Biopsies in Cancer Diagnosis, Monitoring, and Prognosis. Trends Pharmacol. Sci. 2019, 40, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Bellosillo, B.; Montagut, C. High-accuracy liquid biopsies. Nat. Med. 2019, 25, 1820–1821. [Google Scholar] [CrossRef] [PubMed]
- Lui, Y.Y.; Chik, K.W.; Chiu, R.W.; Ho, C.Y.; Lam, C.W.; Lo, Y.M. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin. Chem. 2002, 48, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Gillis, N.K.; Ball, M.; Zhang, Q.; Ma, Z.; Zhao, Y.; Yoder, S.J.; Balasis, M.E.; Mesa, T.E.; Sallman, D.A.; Lancet, J.E.; et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: A proof-of-concept, case-control study. Lancet Oncol. 2017, 18, 112–121. [Google Scholar] [CrossRef]
- Mandel, P.; Metais, P. Les acides nucleiques du plasma sanguin chez 1 homme. CR Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Kustanovich, A.; Schwartz, R.; Peretz, T.; Grinshpun, A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 2019, 20, 1057–1067. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Heitzer, E.; Auinger, L.; Speicher, M.R. Cell-Free DNA and Apoptosis: How Dead Cells Inform About the Living. Trends Mol. Med. 2020, 26, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Lui, Y.Y.; Dennis, Y.M. Circulating DNA in plasma and serum: Biology, preanalytical issues and diagnostic applications. Clin. Chem. Lab. Med. 2002, 40, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar] [PubMed]
- Anker, P.; Stroun, M.; Maurice, P.A. Spontaneous release of DNA by human blood lymphocytes as shown in an in vitro system. Cancer Res. 1975, 35, 2375–2382. [Google Scholar] [PubMed]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A., Jr.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.M.; Chan, K.C.; Sun, H.; Chen, E.Z.; Jiang, P.; Lun, F.M.; Zheng, Y.W.; Leung, T.Y.; Lau, T.K.; Cantor, C.R.; et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci. Transl. Med. 2010, 2, 61ra91. [Google Scholar] [CrossRef] [PubMed]
- Ulz, P.; Thallinger, G.G.; Auer, M.; Graf, R.; Kashofer, K.; Jahn, S.W.; Abete, L.; Pristauz, G.; Petru, E.; Geigl, J.B.; et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat. Genet. 2016, 48, 1273–1278. [Google Scholar] [CrossRef]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef]
- Sun, K.; Jiang, P.; Cheng, S.H.; Cheng, T.H.T.; Wong, J.; Wong, V.W.S.; Ng, S.S.M.; Ma, B.B.Y.; Leung, T.Y.; Chan, S.L.; et al. Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res. 2019, 29, 418–427. [Google Scholar] [CrossRef]
- Cristiano, S.; Leal, A.; Phallen, J.; Fiksel, J.; Adleff, V.; Bruhm, D.C.; Jensen, S.O.; Medina, J.E.; Hruban, C.; White, J.R.; et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019, 570, 385–389. [Google Scholar] [CrossRef]
- Khier, S.; Lohan, L. Kinetics of circulating cell-free DNA for biomedical applications: Critical appraisal of the literature. Future Sci. OA 2018, 4, FSO295. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef] [PubMed]
- Lam, W.K.J.; Gai, W.; Sun, K.; Wong, R.S.M.; Chan, R.W.Y.; Jiang, P.; Chan, N.P.H.; Hui, W.W.I.; Chan, A.W.H.; Szeto, C.C.; et al. DNA of Erythroid Origin Is Present in Human Plasma and Informs the Types of Anemia. Clin. Chem. 2017, 63, 1614–1623. [Google Scholar] [CrossRef] [PubMed]
- Stroun, M.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Beljanski, M. Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology 1989, 46, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Stroun, M.; Magnenat, J.L.; Nicod, L.P.; Kurt, A.M.; Lyautey, J.; Lederrey, C.; Anker, P. Microsatellite alterations in plasma DNA of small cell lung cancer patients. Nat. Med. 1996, 2, 1033–1035. [Google Scholar] [CrossRef] [PubMed]
- Anker, P.; Lefort, F.; Vasioukhin, V.; Lyautey, J.; Lederrey, C.; Chen, X.Q.; Stroun, M.; Mulcahy, H.E.; Farthing, M.J. K-ras mutations are found in DNA extracted from the plasma of patients with colorectal cancer. Gastroenterology 1997, 112, 1114–1120. [Google Scholar] [CrossRef]
- Theodor, L.; Melzer, E.; Sologov, M.; Idelman, G.; Friedman, E.; Bar-Meir, S. Detection of pancreatic carcinoma: Diagnostic value of K-ras mutations in circulating DNA from serum. Dig. Dis. Sci. 1999, 44, 2014–2019. [Google Scholar] [CrossRef]
- Namlos, H.M.; Boye, K.; Mishkin, S.J.; Baroy, T.; Lorenz, S.; Bjerkehagen, B.; Stratford, E.W.; Munthe, E.; Kudlow, B.A.; Myklebost, O.; et al. Noninvasive Detection of ctDNA Reveals Intratumor Heterogeneity and Is Associated with Tumor Burden in Gastrointestinal Stromal Tumor. Mol. Cancer Ther. 2018, 17, 2473–2480. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; Horswell, S.; Mitter, R.; Sakarya, O.; Constantin, T.; Salari, R.; Kirkizlar, E.; Sigurjonsson, S.; Pelham, R.; et al. Detection of ubiquitous and heterogeneous mutations in cell-free DNA from patients with early-stage non-small-cell lung cancer. Ann. Oncol. 2016, 27, 862–867. [Google Scholar] [CrossRef]
- Li, G.; Pavlick, D.; Chung, J.H.; Bauer, T.; Tan, B.A.; Peguero, J.; Ward, P.; Kallab, A.; Bufill, J.; Hoffman, A.; et al. Genomic profiling of cell-free circulating tumor DNA in patients with colorectal cancer and its fidelity to the genomics of the tumor biopsy. J. Gastrointest. Oncol. 2019, 10, 831–840. [Google Scholar] [CrossRef]
- Devarakonda, S.; Sankararaman, S.; Herzog, B.H.; Gold, K.A.; Waqar, S.N.; Ward, J.P.; Raymond, V.M.; Lanman, R.B.; Chaudhuri, A.A.; Owonikoko, T.K.; et al. Circulating Tumor DNA Profiling in Small-Cell Lung Cancer Identifies Potentially Targetable Alterations. Clin. Cancer Res. 2019, 25, 6119–6126. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra224. [Google Scholar] [CrossRef] [PubMed]
- Cree, I.A.; Uttley, L.; Buckley Woods, H.; Kikuchi, H.; Reiman, A.; Harnan, S.; Whiteman, B.L.; Philips, S.T.; Messenger, M.; Cox, A.; et al. The evidence base for circulating tumour DNA blood-based biomarkers for the early detection of cancer: A systematic mapping review. BMC Cancer 2017, 17, 697. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.A.; Chabon, J.J.; Lovejoy, A.F.; Newman, A.M.; Stehr, H.; Azad, T.D.; Khodadoust, M.S.; Esfahani, M.S.; Liu, C.L.; Zhou, L.; et al. Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discov. 2017, 7, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra392. [Google Scholar] [CrossRef]
- Demuth, C.; Winther-Larsen, A.; Madsen, A.T.; Meldgaard, P.; Sorensen, B.S. A method for treatment monitoring using circulating tumour DNA in cancer patients without targetable mutations. Oncotarget 2018, 9, 31066–31076. [Google Scholar] [CrossRef]
- Long-Mira, E.; Ilie, M.; Chamorey, E.; Leduff-Blanc, F.; Montaudie, H.; Tanga, V.; Allegra, M.; Lespinet-Fabre, V.; Bordone, O.; Bonnetaud, C.; et al. Monitoring BRAF and NRAS mutations with cell-free circulating tumor DNA from metastatic melanoma patients. Oncotarget 2018, 9, 36238–36249. [Google Scholar] [CrossRef]
- Váraljai, R.; Wistuba-Hamprecht, K.; Seremet, T.; Diaz, J.M.S.; Nsengimana, J.; Sucker, A.; Griewank, K.; Placke, J.-M.; Horn, P.A.; Neuhoff, N.V.; et al. Application of Circulating Cell-Free Tumor DNA Profiles for Therapeutic Monitoring and Outcome Prediction in Genetically Heterogeneous Metastatic Melanoma. JCO Precis. Oncol. 2019, 3, 1–10. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef]
- Avanzini, S.; Kurtz, D.M.; Chabon, J.J.; Moding, E.J.; Hori, S.S.; Gambhir, S.S.; Alizadeh, A.A.; Diehn, M.; Reiter, J.G. A mathematical model of ctDNA shedding predicts tumor detection size. bioRxiv 2020. (Preprint). [Google Scholar] [CrossRef]
- Bauml, J.; Levy, B. Clonal Hematopoiesis: A New Layer in the Liquid Biopsy Story in Lung Cancer. Clin. Cancer Res. 2018, 24, 4352–4354. [Google Scholar] [CrossRef] [PubMed]
- Underhill, H.R.; Kitzman, J.O.; Hellwig, S.; Welker, N.C.; Daza, R.; Baker, D.N.; Gligorich, K.M.; Rostomily, R.C.; Bronner, M.P.; Shendure, J. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016, 12, e1006162. [Google Scholar] [CrossRef] [PubMed]
- Mouliere, F.; Robert, B.; Arnau Peyrotte, E.; Del Rio, M.; Ychou, M.; Molina, F.; Gongora, C.; Thierry, A.R. High fragmentation characterizes tumour-derived circulating DNA. PLoS ONE 2011, 6, e23418. [Google Scholar] [CrossRef]
- Jiang, P.; Chan, C.W.; Chan, K.C.; Cheng, S.H.; Wong, J.; Wong, V.W.; Wong, G.L.; Chan, S.L.; Mok, T.S.; Chan, H.L.; et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, E1317–E1325. [Google Scholar] [CrossRef]
- Champion, K.M.; Gilbert, J.G.; Asimakopoulos, F.A.; Hinshelwood, S.; Green, A.R. Clonal haemopoiesis in normal elderly women: Implications for the myeloproliferative disorders and myelodysplastic syndromes. Br. J. Haematol. 1997, 97, 920–926. [Google Scholar] [CrossRef]
- Fey, M.F.; Liechti-Gallati, S.; von Rohr, A.; Borisch, B.; Theilkas, L.; Schneider, V.; Oestreicher, M.; Nagel, S.; Ziemiecki, A.; Tobler, A. Clonality and X-inactivation patterns in hematopoietic cell populations detected by the highly informative M27 beta DNA probe. Blood 1994, 83, 931–938. [Google Scholar] [CrossRef]
- Lee-Six, H.; Obro, N.F.; Shepherd, M.S.; Grossmann, S.; Dawson, K.; Belmonte, M.; Osborne, R.J.; Huntly, B.J.P.; Martincorena, I.; Anderson, E.; et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 2018, 561, 473–478. [Google Scholar] [CrossRef]
- Steensma, D.P. Clinical consequences of clonal hematopoiesis of indeterminate potential. Blood Adv. 2018, 2, 3404–3410. [Google Scholar] [CrossRef]
- Genovese, G.; Kahler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Hidalgo, R.; Sengul, H.; Steehouwer, M.; van de Vorst, M.; Vermeulen, S.H.; Kiemeney, L.; Veltman, J.A.; Gilissen, C.; Hoischen, A. Ultra-sensitive Sequencing Identifies High Prevalence of Clonal Hematopoiesis-Associated Mutations throughout Adult Life. Am. J. Hum. Genet. 2017, 101, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Coombs, C.C.; Zehir, A.; Devlin, S.M.; Kishtagari, A.; Syed, A.; Jonsson, P.; Hyman, D.M.; Solit, D.B.; Robson, M.E.; Baselga, J.; et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017, 21, 374–382. [Google Scholar] [CrossRef] [PubMed]
- McKerrell, T.; Park, N.; Moreno, T.; Grove, C.S.; Ponstingl, H.; Stephens, J.; Crawley, C.; Craig, J.; Scott, M.A.; Hodkinson, C.; et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015, 10, 1239–1245. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Liu, J.; Chen, X.; Wang, J.; Zhou, S.; Wang, C.L.; Ye, M.Z.; Wang, X.Y.; Song, Y.; Wang, Y.Q.; Zhang, L.T.; et al. Biological background of the genomic variations of cf-DNA in healthy individuals. Ann. Oncol. 2019, 30, 464–470. [Google Scholar] [CrossRef]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Barhdadi, A.; Bourgoin, V.; Lepine, G.; Mollica, L.; Szuber, N.; Dube, M.P.; Busque, L. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 2017, 130, 753–762. [Google Scholar] [CrossRef]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Gao, J.; Adli, M.; Dey, A.; Trimarchi, T.; Chung, Y.R.; Kuscu, C.; Hricik, T.; Ndiaye-Lobry, D.; Lafave, L.M.; et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. 2013, 210, 2641–2659. [Google Scholar] [CrossRef]
- Shepherd, M.S.; Li, J.; Wilson, N.K.; Oedekoven, C.A.; Li, J.; Belmonte, M.; Fink, J.; Prick, J.C.M.; Pask, D.C.; Hamilton, T.L.; et al. Single-cell approaches identify the molecular network driving malignant hematopoietic stem cell self-renewal. Blood 2018, 132, 791–803. [Google Scholar] [CrossRef]
- Nagase, R.; Inoue, D.; Pastore, A.; Fujino, T.; Hou, H.A.; Yamasaki, N.; Goyama, S.; Saika, M.; Kanai, A.; Sera, Y.; et al. Expression of mutant Asxl1 perturbs hematopoiesis and promotes susceptibility to leukemic transformation. J. Exp. Med. 2018, 215, 1729–1747. [Google Scholar] [CrossRef] [PubMed]
- Kent, D.G.; Li, J.; Tanna, H.; Fink, J.; Kirschner, K.; Pask, D.C.; Silber, Y.; Hamilton, T.L.; Sneade, R.; Simons, B.D.; et al. Self-renewal of single mouse hematopoietic stem cells is reduced by JAK2V617F without compromising progenitor cell expansion. PLoS Biol. 2013, 11, e1001576. [Google Scholar] [CrossRef] [PubMed]
- Ptashkin, R.N.; Mandelker, D.L.; Coombs, C.C.; Bolton, K.; Yelskaya, Z.; Hyman, D.M.; Solit, D.B.; Baselga, J.; Arcila, M.E.; Ladanyi, M.; et al. Prevalence of Clonal Hematopoiesis Mutations in Tumor-Only Clinical Genomic Profiling of Solid Tumors. JAMA Oncol. 2018, 4, 1589–1593. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ulrich, B.C.; Supplee, J.; Kuang, Y.; Lizotte, P.H.; Feeney, N.B.; Guibert, N.M.; Awad, M.M.; Wong, K.K.; Janne, P.A.; et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin. Cancer Res. 2018, 24, 4437–4443. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.; Mencia-Trinchant, N.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Ballman, K.V.; et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 2018, 24, 1015–1023. [Google Scholar] [CrossRef]
- Abelson, S.; Collord, G.; Ng, S.W.K.; Weissbrod, O.; Mendelson Cohen, N.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.; Sundaravadanam, Y.; et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559, 400–404. [Google Scholar] [CrossRef]
- Gibson, C.J.; Steensma, D.P. New Insights from Studies of Clonal Hematopoiesis. Clin. Cancer Res. 2018, 24, 4633–4642. [Google Scholar] [CrossRef]
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S.; et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015, 518, 552–555. [Google Scholar] [CrossRef]
- Bick, A.G.; Pirruccello, J.P.; Griffin, G.K.; Gupta, N.; Gabriel, S.; Saleheen, D.; Libby, P.; Kathiresan, S.; Natarajan, P. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation 2020, 141, 124–131. [Google Scholar] [CrossRef]
- Dorsheimer, L.; Assmus, B.; Rasper, T.; Ortmann, C.A.; Ecke, A.; Abou-El-Ardat, K.; Schmid, T.; Brune, B.; Wagner, S.; Serve, H.; et al. Association of Mutations Contributing to Clonal Hematopoiesis With Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 2019, 4, 25–33. [Google Scholar] [CrossRef]
- Mas-Peiro, S.; Hoffmann, J.; Fichtlscherer, S.; Dorsheimer, L.; Rieger, M.A.; Dimmeler, S.; Vasa-Nicotera, M.; Zeiher, A.M. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur. Heart J. 2020, 41, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cuesta, L.; Perdomo, S.; Avogbe, P.H.; Leblay, N.; Delhomme, T.M.; Gaborieau, V.; Abedi-Ardekani, B.; Chanudet, E.; Olivier, M.; Zaridze, D.; et al. Identification of Circulating Tumor DNA for the Early Detection of Small-cell Lung Cancer. EBioMedicine 2016, 10, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Razavi, P.; Li, B.T.; Brown, D.N.; Jung, B.; Hubbell, E.; Shen, R.; Abida, W.; Juluru, K.; De Bruijn, I.; Hou, C.; et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 2019, 25, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Chabon, J.J.; Hamilton, E.G.; Kurtz, D.M.; Esfahani, M.S.; Moding, E.J.; Stehr, H.; Schroers-Martin, J.; Nabet, B.Y.; Chen, B.; Chaudhuri, A.A.; et al. Integrating genomic features for non-invasive early lung cancer detection. Nature 2020, 580, 245–251. [Google Scholar] [CrossRef]
- Xia, L.; Li, Z.; Zhou, B.; Tian, G.; Zeng, L.; Dai, H.; Li, X.; Liu, C.; Lu, S.; Xu, F.; et al. Statistical analysis of mutant allele frequency level of circulating cell-free DNA and blood cells in healthy individuals. Sci. Rep. 2017, 7, 7526. [Google Scholar] [CrossRef]
- Chan, H.T.; Nagayama, S.; Chin, Y.M.; Otaki, M.; Hayashi, R.; Kiyotni, K.; Fukunaa, Y.; Ueno, M.; Nakamura, Y.; Low, S.-K. Clinical significance of clonal hematopoiesis in the interpretation of blood liquid biopsy. Mol. Oncol. 2020, 14, 1719–1730. [Google Scholar]
- Leal, A.; van Grieken, N.C.T.; Palsgrove, D.N.; Phallen, J.; Medina, J.E.; Hruban, C.; Broeckaert, M.A.M.; Anagnostou, V.; Adleff, V.; Bruhm, D.C.; et al. White blood cell and cell-free DNA analyses for detection of residual disease in gastric cancer. Nat. Commun. 2020, 11, 525. [Google Scholar] [CrossRef]
- Mayrhofer, M.; De Laere, B.; Whitington, T.; Van Oyen, P.; Ghysel, C.; Ampe, J.; Ost, P.; Demey, W.; Hoekx, L.; Schrijvers, D.; et al. Cell-free DNA profiling of metastatic prostate cancer reveals microsatellite instability, structural rearrangements and clonal hematopoiesis. Genome Med. 2018, 10, 85. [Google Scholar] [CrossRef]
- Marass, F.; Stephens, D.; Ptashkin, R.; Zehir, A.; Berger, M.F.; Solit, D.B.; Diaz, L.A.; Tsui, D.W.Y. Fragment Size Analysis May Distinguish Clonal Hematopoiesis from Tumor-Derived Mutations in Cell-Free DNA. Clin. Chem. 2020, 66, 616–618. [Google Scholar] [CrossRef]
- Osorio, F.G.; Rosendahl Huber, A.; Oka, R.; Verheul, M.; Patel, S.H.; Hasaart, K.; de la Fonteijne, L.; Varela, I.; Camargo, F.D.; van Boxtel, R. Somatic Mutations Reveal Lineage Relationships and Age-Related Mutagenesis in Human Hematopoiesis. Cell Rep. 2018, 25, 2308–2316. [Google Scholar] [CrossRef]
- Gibson, C.J.; Lindsley, R.C.; Tchekmedyian, V.; Mar, B.G.; Shi, J.; Jaiswal, S.; Bosworth, A.; Francisco, L.; He, J.; Bansal, A.; et al. Clonal Hematopoiesis Associated With Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma. J. Clin. Oncol. 2017, 35, 1598–1605. [Google Scholar] [CrossRef] [PubMed]
- Bacon, J.V.W.; Annala, M.; Soleimani, M.; Lavoie, J.M.; So, A.; Gleave, M.E.; Fazli, L.; Wang, G.; Chi, K.N.; Kollmannsberger, C.K.; et al. Plasma Circulating Tumor DNA and Clonal Hematopoiesis in Metastatic Renal Cell Carcinoma. Clin. Genitourin. Cancer 2020, 18, 331–332. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Cohen, J.D.; Wang, Y.; Christie, M.; Simons, K.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; McKendrick, J.; et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol. 2019, 5, 1710–1717. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Cohen, J.D.; Kinde, I.; Ptak, J.; Popoli, M.; Schaefer, J.; Silliman, N.; Dobbyn, L.; Tie, J.; et al. Prognostic Potential of Circulating Tumor DNA Measurement in Postoperative Surveillance of Nonmetastatic Colorectal Cancer. JAMA Oncol. 2019, 5, 1118–1123. [Google Scholar] [CrossRef]
- Chen, Y.H.; Hancock, B.A.; Solzak, J.P.; Brinza, D.; Scafe, C.; Miller, K.D.; Radovich, M. Next-generation sequencing of circulating tumor DNA to predict recurrence in triple-negative breast cancer patients with residual disease after neoadjuvant chemotherapy. NPJ Breast Cancer 2017, 3, 24. [Google Scholar] [CrossRef]
- Ng, S.B.; Chua, C.; Ng, M.; Gan, A.; Poon, P.S.; Teo, M.; Fu, C.; Leow, W.Q.; Lim, K.H.; Chung, A.; et al. Individualised multiplexed circulating tumour DNA assays for monitoring of tumour presence in patients after colorectal cancer surgery. Sci. Rep. 2017, 7, 40737. [Google Scholar] [CrossRef]
- Scholer, L.V.; Reinert, T.; Orntoft, M.W.; Kassentoft, C.G.; Arnadottir, S.S.; Vang, S.; Nordentoft, I.; Knudsen, M.; Lamy, P.; Andreasen, D.; et al. Clinical Implications of Monitoring Circulating Tumor DNA in Patients with Colorectal Cancer. Clin. Cancer Res. 2017, 23, 5437–5445. [Google Scholar] [CrossRef]
- Sun, K. Clonal hematopoiesis: Background player in plasma cell-free DNA variants. Ann. Transl. Med. 2019, 7, S384. [Google Scholar] [CrossRef] [PubMed]
- Cescon, D.W.; Bratman, S.V.; Chan, S.M.; Siu, L.L. Circulating tumor DNA and liquid biopsy in oncology. Nat. Cancer 2020, 1, 276–290. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequencing Method | Study Size | Participants | Age Range | Depth * | Reported LOD (%) | CH Prevalence | Study |
|---|---|---|---|---|---|---|---|
| WGS | 11,262 | Icelanders with various diseases | 10–100 | 36x | 10% | 12% | Zink et al., 2017 [51] |
| WES | 17,182 | Healthy controls | 19–108 | 84x | 3.5% | Age 70–79: 9.5% Age 80–89: 11.7% Age 90–108: 18.4% | Jaiswal et al., 2014 [5] |
| WES | 12,380 | Healthy controls | 19–93 | NR | 5% | Age < 50: 1% Age > 65: 10% | Genovese et al., 2014 [50] |
| WES | 2728 | Patients with solid tumors | 10–90 | 108x | 3% | Age > 70: 5% | Xie et al., 2014 [58] |
| WES | 8255 | Patients with cardiovascular disease and healthy controls | Median: 60 | NR | 3% | CVD: 17% Controls: 10% | Jaiswal et al., 2017 [55] |
| Targeted NGS (15 hotspot mutations) | 4219 | Healthy controls | 17–98 | 1000x | 0.8% | Age < 60: 0.8% Age ≥ 90: 19.5% | McKerrell et al., 2015 [54] |
| smMIPs | 2000 | Healthy controls | 20–69 | 845x | 0.1% | Age 60–69: >20% | Acuna-Hidalgo et al., 2017 [52] |
| Targeted NGS | 8810 | Patients with solid tumors | 1–98 | 419x | 1% | 25% | Coombs et al., 2017 [53] |
| Targeted NGS (19 genes) | 2530 | Women without a known hematological disorder | 55–101 | 4000x | 0.1% | 13.7% | Buscarlet et al., 2017 [57] |
| Targeted NGS (559 genes) | 259 | Healthy controls | Median: 47 | 6200x (Collapsed: 680x) | 0.1% | Age > 50: 76% | Liu et al., 2019 [56] |
| Cancer Type | Stage | Study Size | Gene Panel | Depth * | Reported LOD (%) | Prevalence of CH Detection from Plasma cfDNA Analysis | Study |
|---|---|---|---|---|---|---|---|
| SCLC | I–IV | SCLC: 51 Healthy controls: 123 | TP53 | NR | NR | SCLC: 5.3% Controls: 15% | Fernandez-Cuesta et al., 2016 [72] |
| Cancer-free | - | 821 | 50 genes | 40,000x | 0.10% | 89% | Xia et al., 2017 [75] |
| NSCLC | III–IV | 122 | Focused on TP53 analysis | NR | NR | 15% | Hu et al., 2018 [64] |
| Prostate | IV | 217 | 305 genes | 814x | 1% | 15% | Mayrhofer et al., 2018 [78] |
| Cancer-free | - | 259 | 599 genes | cfDNA: 6200x white blood cells: 406x | 0.25% | 66% | Liu et al., 2019 [56] |
| Various solid tumors | IV | Cancer:124 Healthy controls: 47 | 508 genes | 60,000x (collapsed: 4500x) | 0.1% | Cancer: 53% Controls: 82% | Razavi et al., 2019 [73] |
| Gastric | I–IV | 788 | 58 genes | 30,000x | 0.1% | 44% | Leal et al., 2020 [77] |
| Renal cell carcinoma | IV | 55 | 981 genes | Collapsed: 938x | 1% | 20% | Bacon et al., 2020 [82] |
| NSCLC | I–III | NSCLC: 104 Healthy controls: 98 | 255 genes | Collapsed: 4000–5000x | 0.01% | NSCLC: 58% Controls: 90% | Chabon et al., 2020 [74] |
| CRC | I–IV | 38 | 52 genes | 48,000x (collapsed: 4000x) | 0.1% | 17% | Chan et al., 2020 [76] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, H.T.; Chin, Y.M.; Nakamura, Y.; Low, S.-K. Clonal Hematopoiesis in Liquid Biopsy: From Biological Noise to Valuable Clinical Implications. Cancers 2020, 12, 2277. https://doi.org/10.3390/cancers12082277
Chan HT, Chin YM, Nakamura Y, Low S-K. Clonal Hematopoiesis in Liquid Biopsy: From Biological Noise to Valuable Clinical Implications. Cancers. 2020; 12(8):2277. https://doi.org/10.3390/cancers12082277
Chicago/Turabian StyleChan, Hiu Ting, Yoon Ming Chin, Yusuke Nakamura, and Siew-Kee Low. 2020. "Clonal Hematopoiesis in Liquid Biopsy: From Biological Noise to Valuable Clinical Implications" Cancers 12, no. 8: 2277. https://doi.org/10.3390/cancers12082277
APA StyleChan, H. T., Chin, Y. M., Nakamura, Y., & Low, S.-K. (2020). Clonal Hematopoiesis in Liquid Biopsy: From Biological Noise to Valuable Clinical Implications. Cancers, 12(8), 2277. https://doi.org/10.3390/cancers12082277