MiR-302b as a Combinatorial Therapeutic Approach to Improve Cisplatin Chemotherapy Efficacy in Human Triple-Negative Breast Cancer

 ,
,  , , , ,
, , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
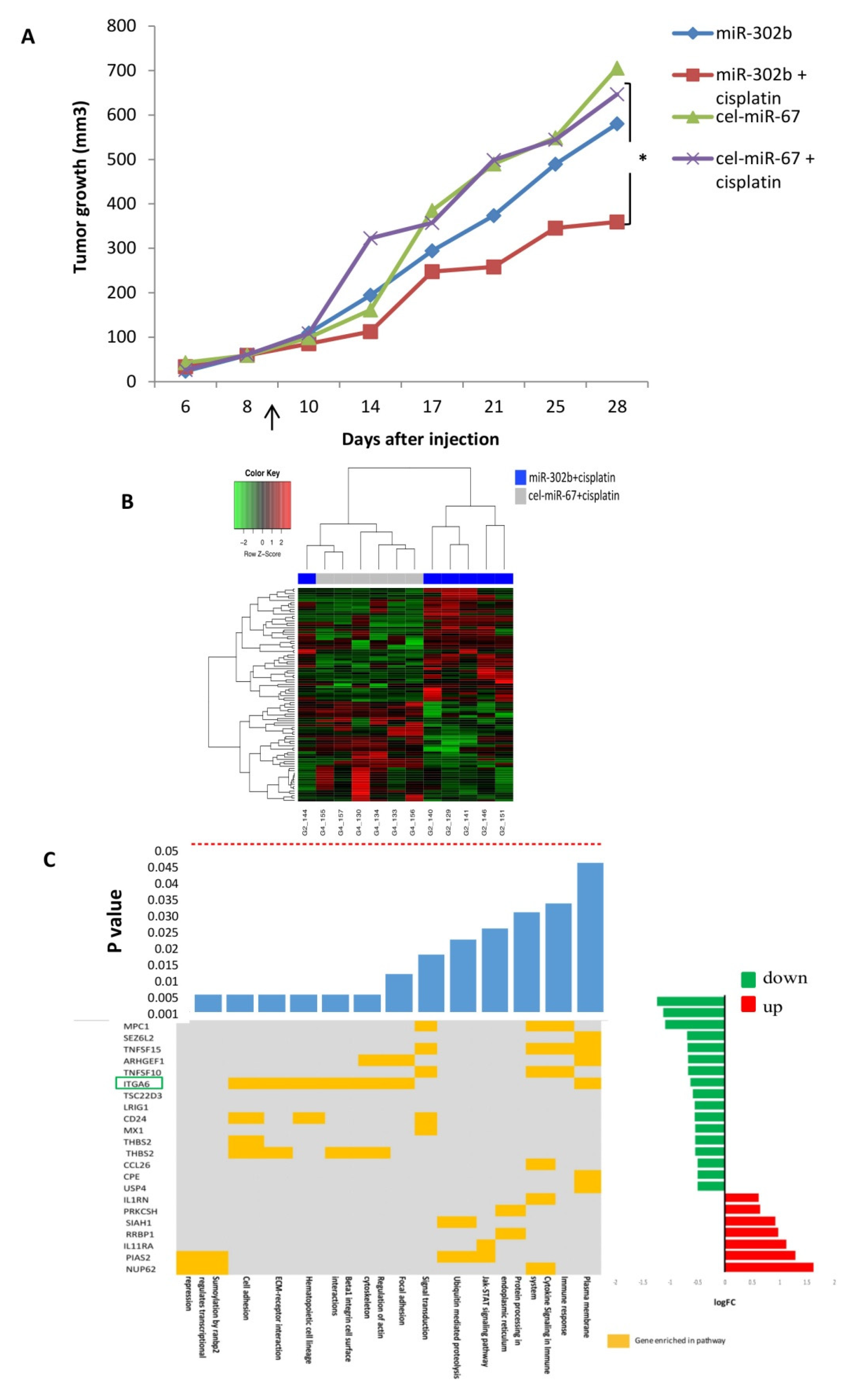
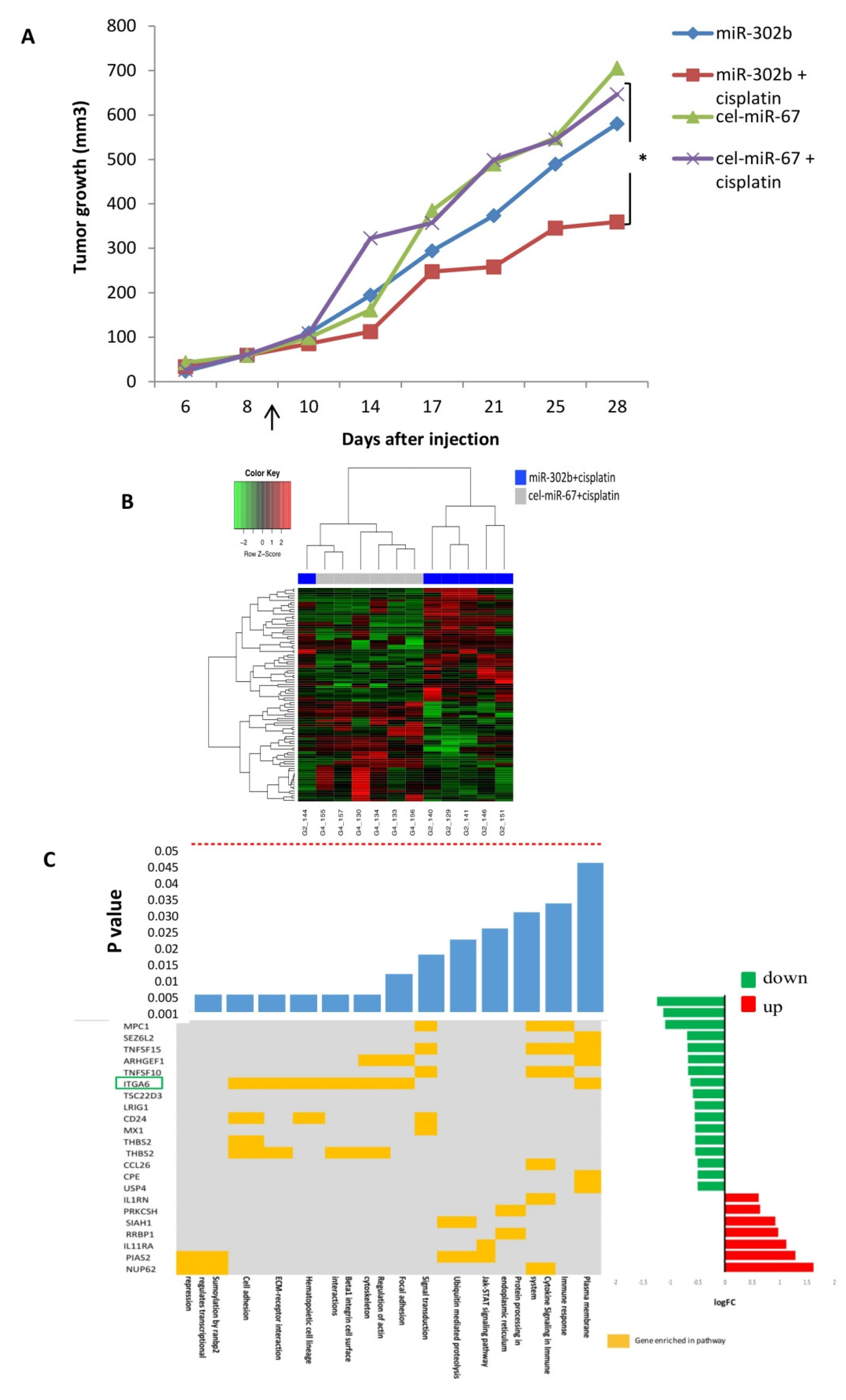
2.1. MiR-302b Enhances Sensitivity to Cisplatin In Vivo
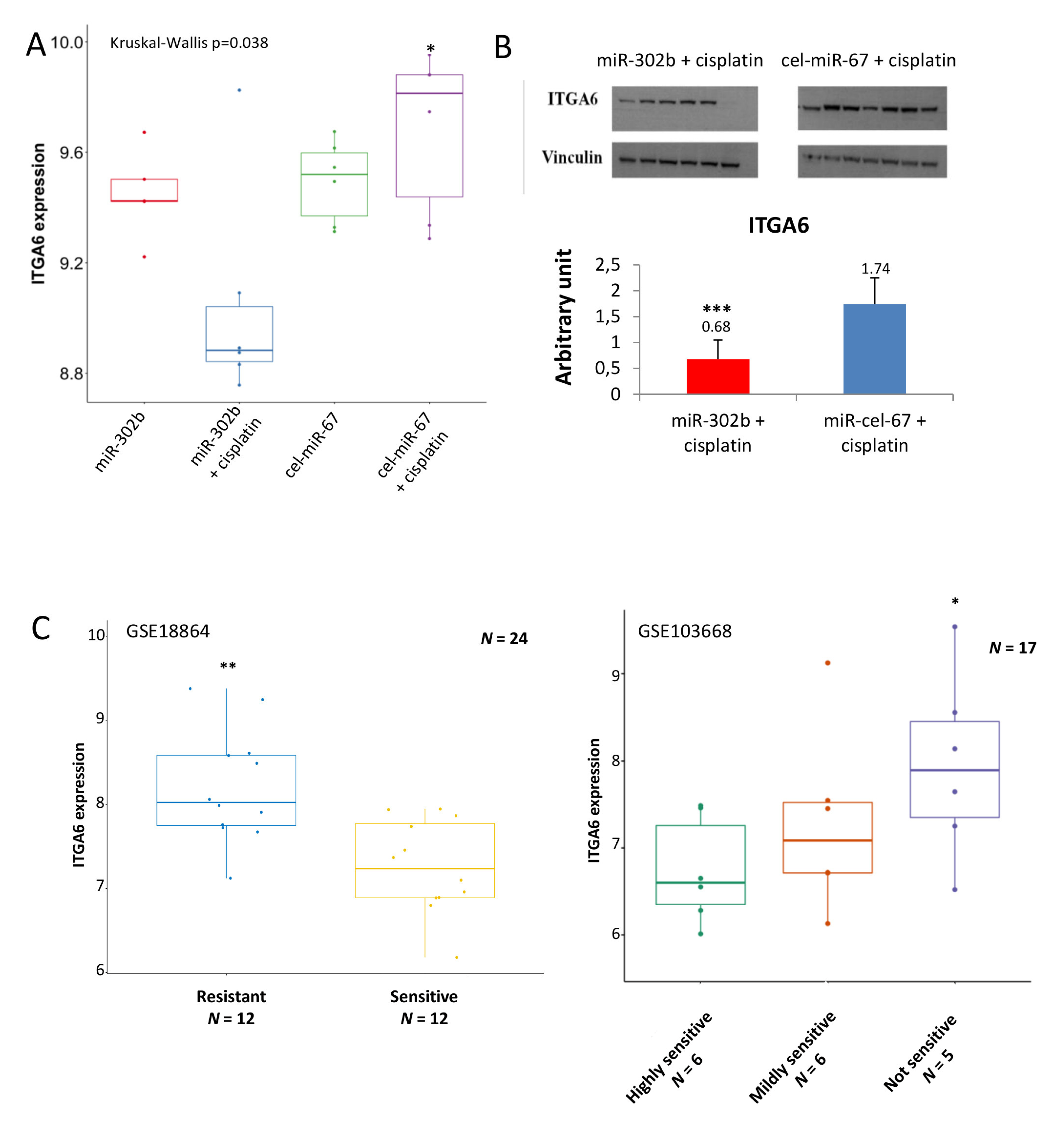
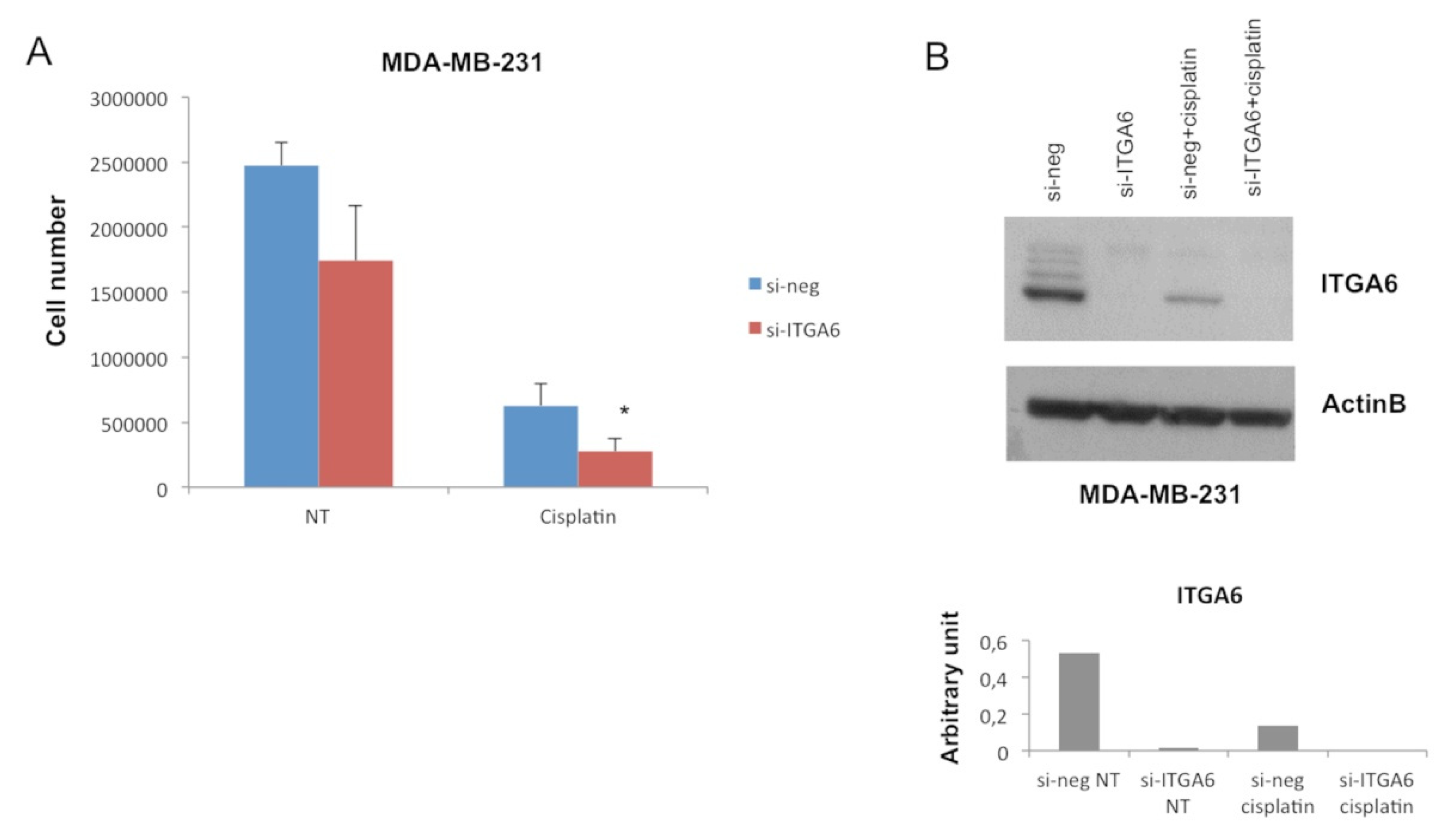
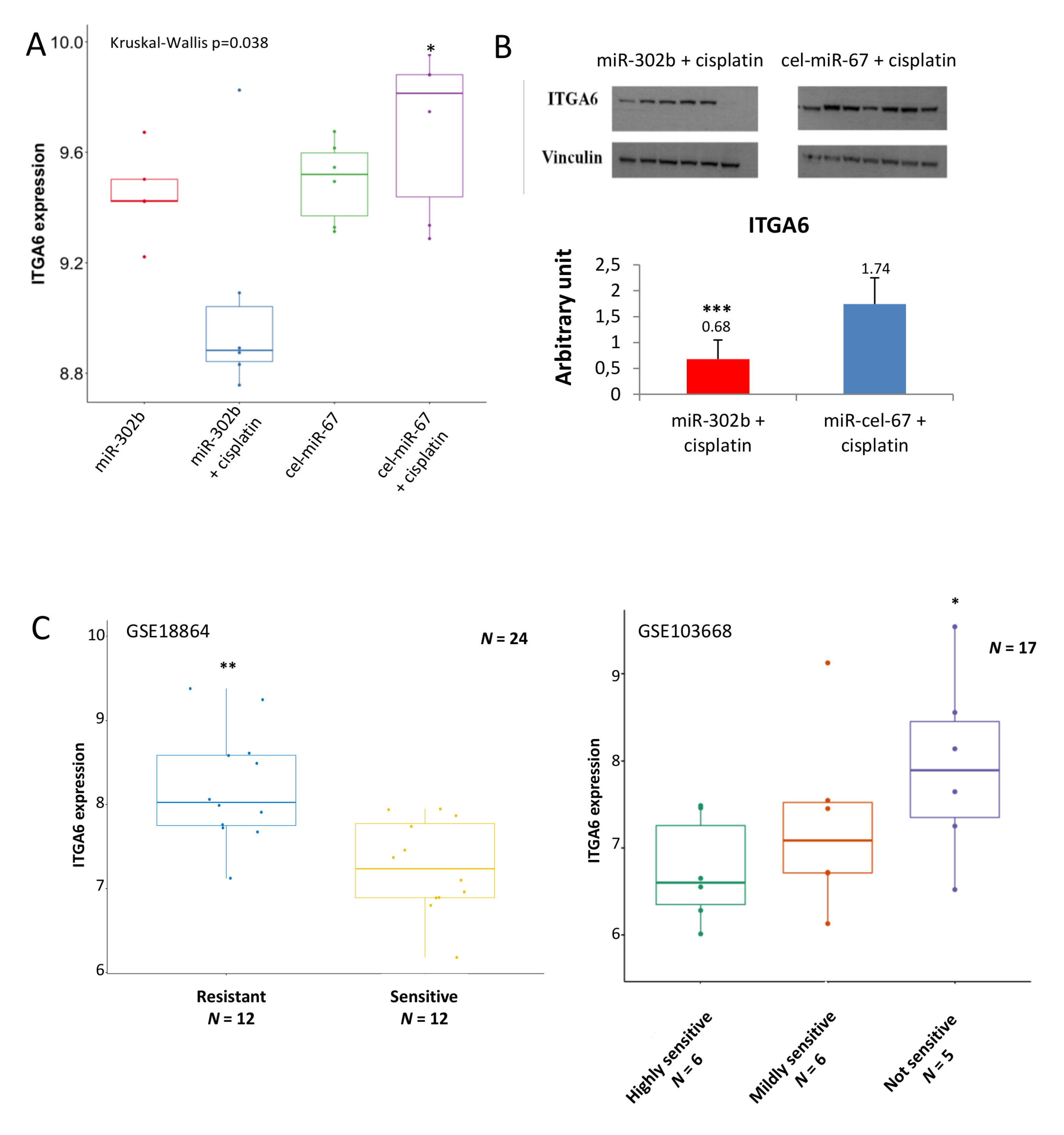
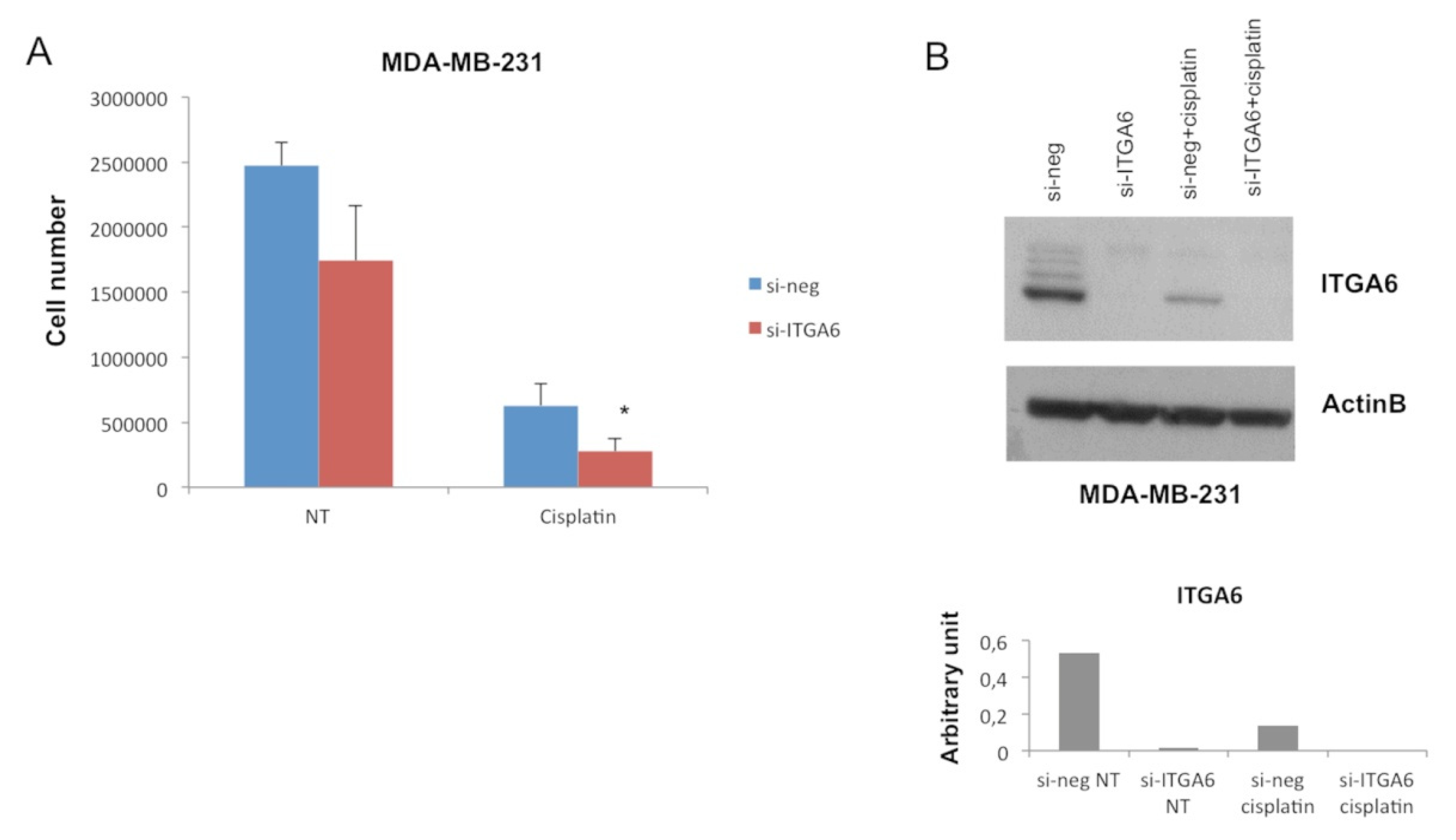
2.2. MiR-302b Enhances Cisplatin Response through the Down-Modulation of ITGA6 in Tumor Cells
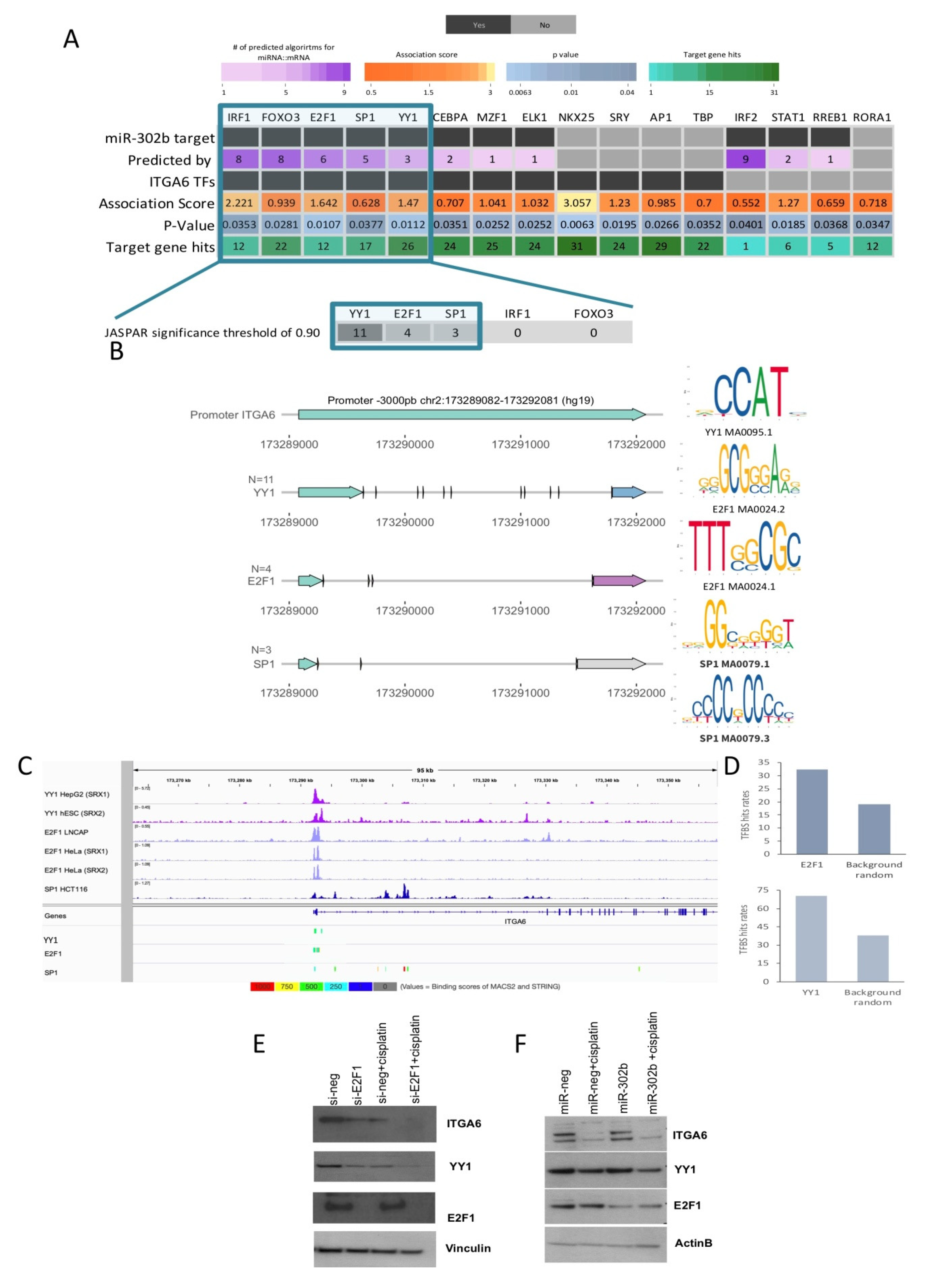
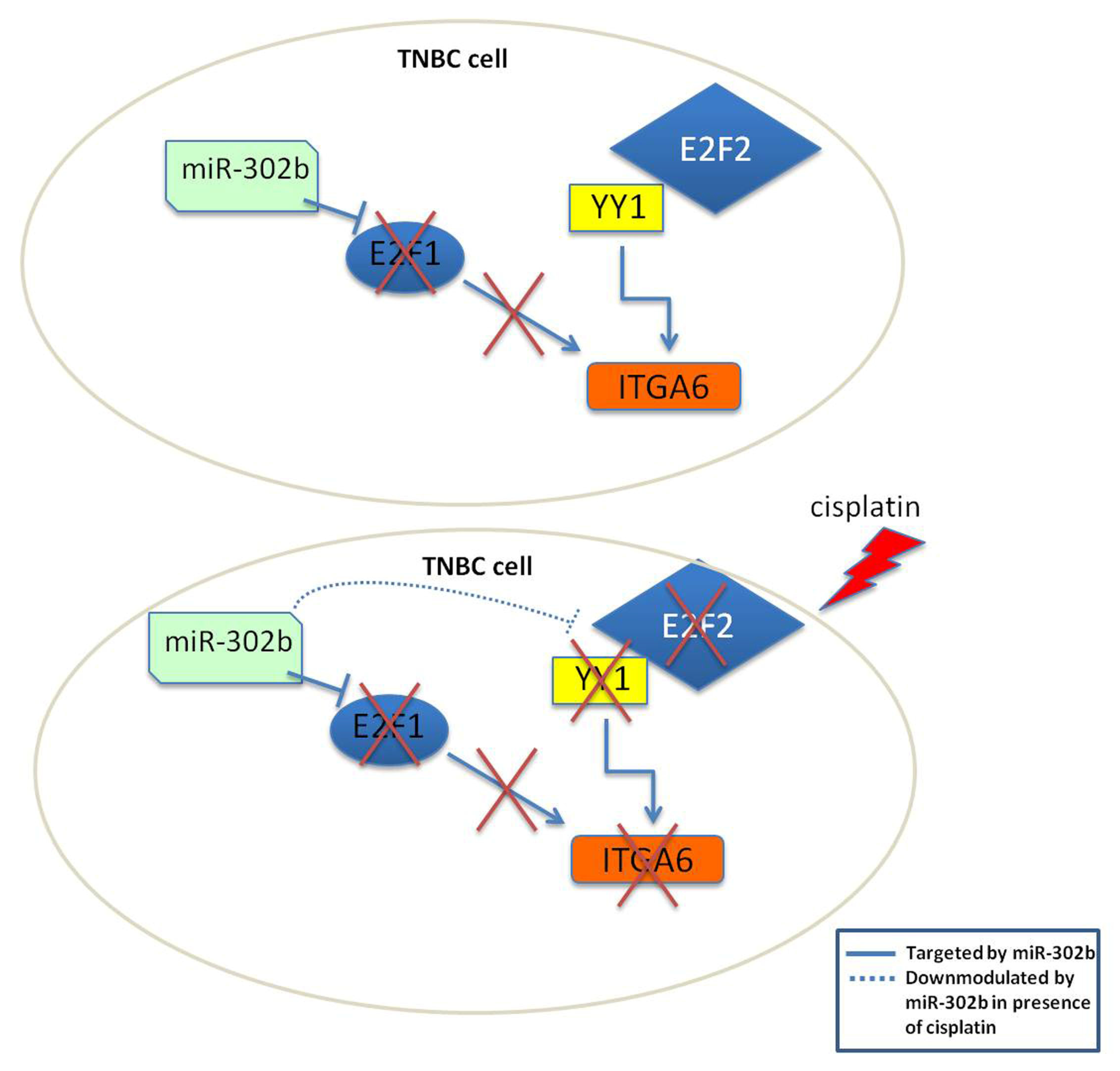
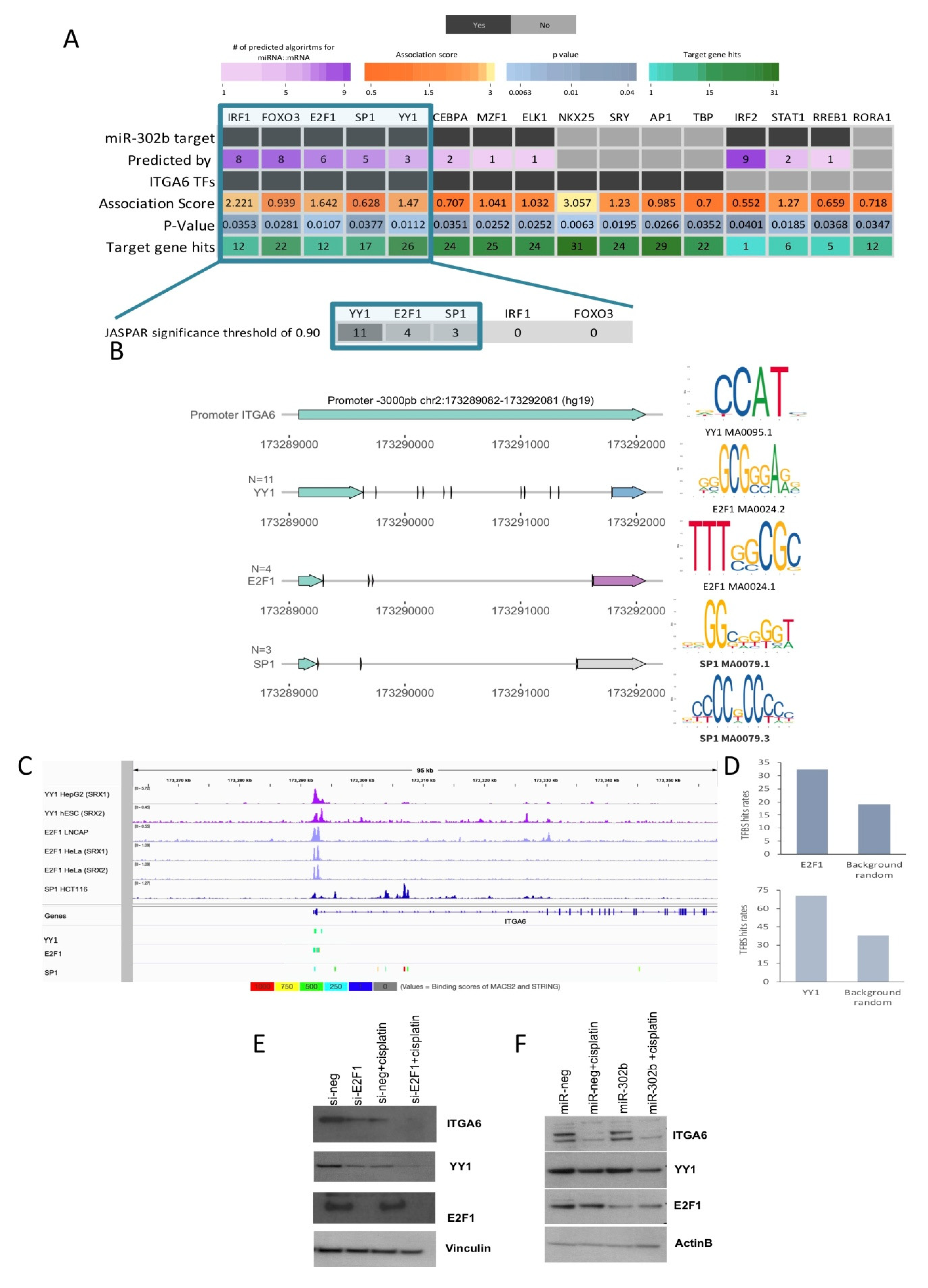
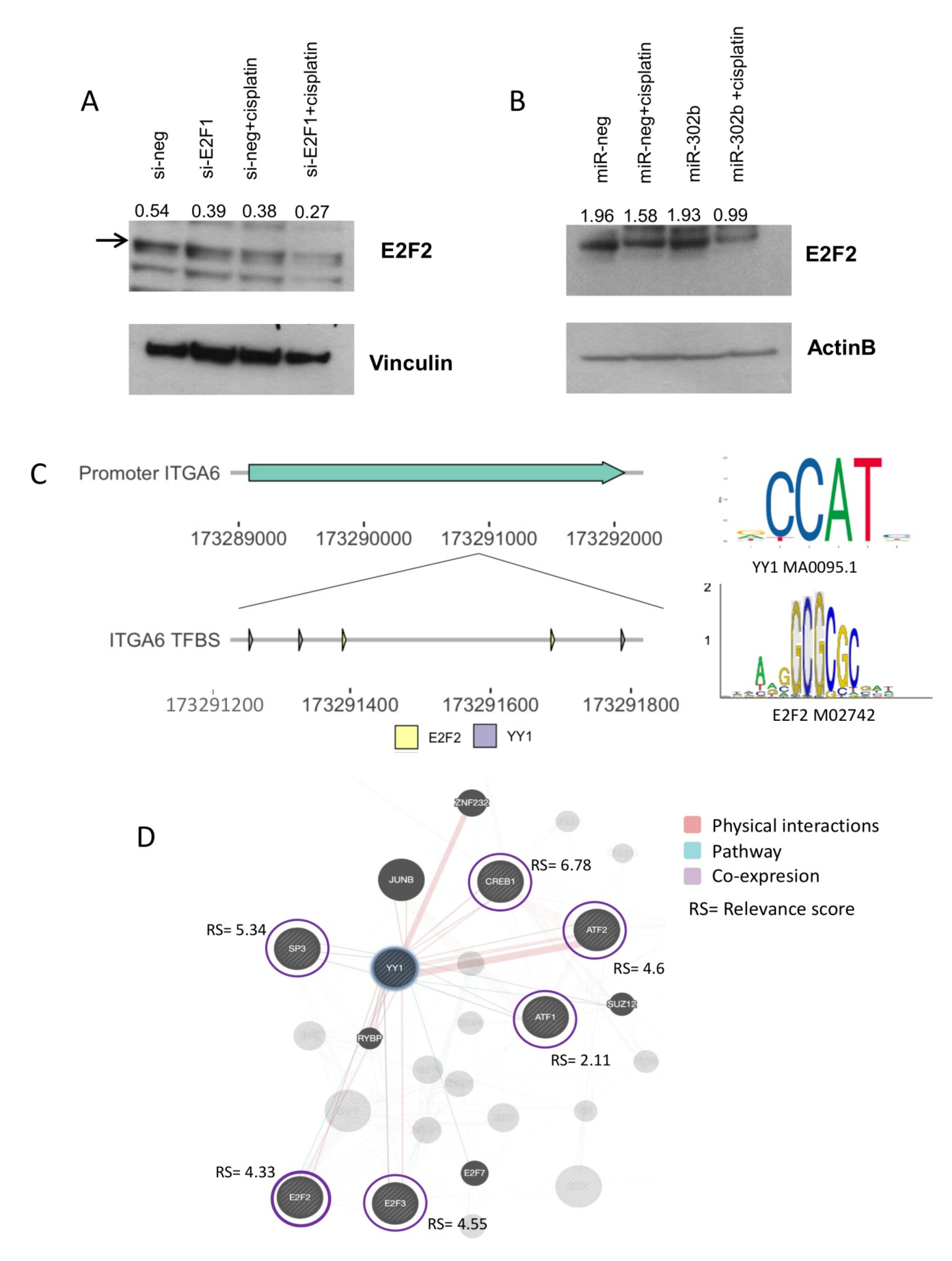
2.3. Combinatorial Treatment of miR-302b and Cisplatin Downregulates E2Fs Family and YY1, Which in Turn Downregulates ITGA6 Expression
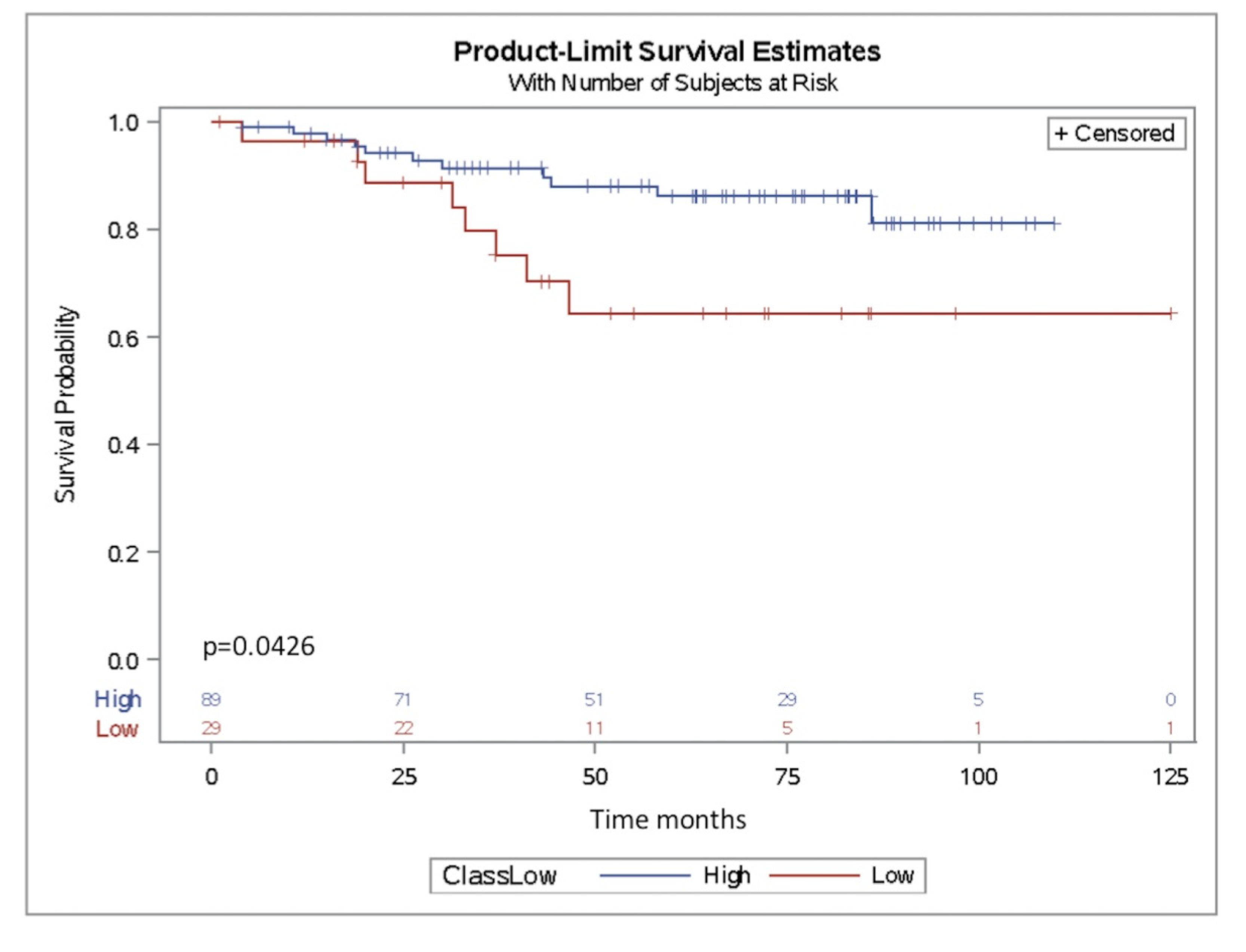
2.4. miR-302b as a Prognostic Factor in TNBC
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. In Vivo Experiment
4.3. mRNA Expression Profiling of In-Vivo Model Tumor Tissue
4.4. Gene Expression Analysis
4.5. Pathway Analysis
4.6. Independent In Silico Analysis of Cisplatin Mediated Genes and its Pathological Response Association
4.7. Transcription Factor Motif Analysis by Benchmark Databases
4.8. Data Mining of TNBC Patients from TCGA and ITAL–MEX Data Sets
4.9. miRNA and siRNA Transfection
4.10. Quantitative RT-PCR
4.11. Western Blot Analysis
4.12. miRNA Expression Profiling from TNBC Patients
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Iorio, M.V.; Croce, C.M. MicroRNA dysregulation in cancer: Diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol. Med. 2012, 4, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, M.; Croce, C.M. MicroRNAs as therapeutic targets in chemoresistance. Drug Resist. Updat. 2013, 16, 47–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raguz, S.; Yague, E. Resistance to chemotherapy: New treatments and novel insights into an old problem. Br. J. Cancer. 2008, 99, 387–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Symmans, W.F.; Wei, C.; Gould, R.; Yu, X.; Zhang, Y.; Liu, M.; Walls, A.; Bousamra, A.; Ramineni, M.; Sinn, B.; et al. Long-Term Prognostic Risk After Neoadjuvant Chemotherapy Associated With Residual Cancer Burden and Breast Cancer Subtype. J. Clin. Oncol. 2017, 35, 1049–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebert, J.M.; Lester, R.; Powell, E.; Seal, M.; McCarthy, J. Advances in the systemic treatment of triple-negative breast cancer. Curr. Oncol. 2018, 25, S142–S150. [Google Scholar] [CrossRef] [Green Version]
- Peto, R.; Davies, C.; Godwin, J.; Gray, R.; Pan, H.C.; Clarke, M.; Cutter, D.; Darby, S.; McGale, P.; Taylor, C.; et al. Comparisons between different polychemotherapy regimens for early breast cancer: Meta-analyses of long-term outcome among 100,000 women in 123 randomised trials. Lancet 2012, 379, 432–444. [Google Scholar] [CrossRef] [Green Version]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Crown, J.; O’Shaughnessy, J.; Gullo, G. Emerging targeted therapies in triple-negative breast cancer. Ann. Oncol. 2012, 23, 56–65. [Google Scholar] [CrossRef]
- Kelland, L.R. Preclinical perspectives on platinum resistance. Drugs 2000, 59, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.; Cheung, D.G.; Balsari, A.; Tagliabue, E.; Coppola, V.; Iorio, M.V.; Palmieri, D.; Croce, C.M. miR-302b enhances breast cancer cell sensitivity to cisplatin by regulating E2F1 and the cellular DNA damage response. Oncotarget 2016, 7, 786–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, M.R.; Lee, Y.; Kim, J.Y.; Kim, S.K.; Moon, S.H.; Lee, J.Y.; Cha, K.Y.; Chung, H.M.; Yoon, H.S.; Moon, S.Y.; et al. Human embryonic stem cells express a unique set of microRNAs. Dev. Biol. 2004, 270, 488–498. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Jin, P.; Wang, E.; Marincola, F.M.; Stroncek, D.F. MicroRNA and gene expression patterns in the differentiation of human embryonic stem cells. J. Transl. Med. 2009, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.H.; Shih, C.M.; Chang, W.C.; Cheng, C.H.; Lin, C.W.; Ho, K.H.; Su, P.C.; Chen, K.C. MicroRNA-302b-inhibited E2F3 transcription factor is related to all trans retinoic acid-induced glioma cell apoptosis. J. Neurochem. 2014, 131, 731–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cecco, L.; Berardi, M.; Sommariva, M.; Cataldo, A.; Canevari, S.; Mezzanzanica, D.; Iorio, M.V.; Tagliabue, E.; Balsari, A. Increased sensitivity to chemotherapy induced by CpG-ODN treatment is mediated by microRNA modulation. PLoS ONE 2013, 8, e58849. [Google Scholar] [CrossRef]
- Wang, L.; Yao, J.; Zhang, X.; Guo, B.; Le, X.; Cubberly, M.; Li, Z.; Nan, K.; Song, T.; Huang, C. miRNA-302b suppresses human hepatocellular carcinoma by targeting AKT2. Mol. Cancer Res. 2014, 12, 190–202. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Yang, Q.; Zhang, L.; Zhou, S.; Ye, W.; Yao, Q.; Li, Z.; Huang, C.; Wen, Q.; Wang, J. miR-302b is a potential molecular marker of esophageal squamous cell carcinoma and functions as a tumor suppressor by targeting ErbB4. J. Exp. Clin. Cancer Res. 2014, 33, 10. [Google Scholar] [CrossRef] [Green Version]
- Zhu, R.; Yang, Y.; Tian, Y.; Bai, J.; Zhang, X.; Li, X.; Peng, Z.; He, Y.; Chen, L.; Pan, Q.; et al. Ascl2 knockdown results in tumor growth arrest by miRNA-302b-related inhibition of colon cancer progenitor cells. PLoS ONE 2012, 7, e32170. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Ahn, J.; Guo, D.; Votaw, J.R.; Shim, H. MicroRNA-302 replacement therapy sensitizes breast cancer cells to ionizing radiation. Pharm. Res. 2013, 30, 1008–1016. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhao, L.; Xiao, Q.; Jiang, L.; He, M.; Bai, X.; Ma, M.; Jiao, X.; Wei, M. miR-302a/b/c/d cooperatively inhibit BCRP expression to increase drug sensitivity in breast cancer cells. Gynecol. Oncol. 2016, 141, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, Y.; Jiang, L.; He, M.; Bai, X.; Yu, L.; Wei, M. MiR-302a/b/c/d cooperatively sensitizes breast cancer cells to adriamycin via suppressing P-glycoprotein(P-gp) by targeting MAP/ERK kinase kinase 1 (MEKK1). J. Exp. Clin. Cancer Res. 2016, 35, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Zhou, Z. Downregulation of miR-302b is associated with poor prognosis and tumor progression of breast cancer. Breast Cancer 2020, 27, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Miragaya, J.; Gonzalez-Suarez, E. Tumor-initiating CD49f cells are a hallmark of chemoresistant triple negative breast cancer. Mol. Cell Oncol. 2017, 4, e1338208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, A.T.; Arenillas, D.J.; Worsley Hunt, R.; Wasserman, W.W. oPOSSUM-3: Advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3 (Bethesda) 2012, 2, 987–1002. [Google Scholar] [CrossRef]
- Roider, H.G.; Kanhere, A.; Manke, T.; Vingron, M. Predicting transcription factor affinities to DNA from a biophysical model. Bioinformatics 2007, 23, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Roider, H.G.; Manke, T.; O’Keeffe, S.; Vingron, M.; Haas, S.A. PASTAA: Identifying transcription factors associated with sets of co-regulated genes. Bioinformatics 2009, 25, 435–442. [Google Scholar] [CrossRef] [Green Version]
- Stormo, G.D. Modeling the specificity of protein-DNA interactions. Quant. Biol. 2013, 1, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Wasserman, W.W.; Sandelin, A. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet. 2004, 5, 276–287. [Google Scholar] [CrossRef]
- Oki, S.; Ohta, T.; Shioi, G.; Hatanaka, H.; Ogasawara, O.; Okuda, Y.; Kawaji, H.; Nakaki, R.; Sese, J.; Meno, C. ChIP-Atlas: A data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Schlisio, S.; Halperin, T.; Vidal, M.; Nevins, J.R. Interaction of YY1 with E2Fs, mediated by RYBP, provides a mechanism for specificity of E2F function. EMBO J. 2002, 21, 5775–5786. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinformatics 2016, 54, 1.30.31–31.30.33. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Zhou, R.; Zhao, Y.; Wu, G. Integrin alpha6/Akt/Erk signaling is essential for human breast cancer resistance to radiotherapy. Sci. Rep. 2016, 6, 33376. [Google Scholar] [CrossRef] [PubMed]
- Bigoni-Ordonez, G.D.; Czarnowski, D.; Parsons, T.; Madlambayan, G.J.; Villa-Diaz, L.G. Integrin α6 (CD49f), The Microenvironment and Cancer Stem Cells. Curr. Stem Cell Res. Ther. 2019, 14, 428–436. [Google Scholar] [CrossRef]
- Sarvagalla, S.; Kolapalli, S.P.; Vallabhapurapu, S. The Two Sides of YY1 in Cancer: A Friend and a Foe. Front. Oncol. 2019, 9, 428–436. [Google Scholar] [CrossRef] [Green Version]
- Thomassen, M.; Tan, Q.; A Kruse, T. Gene expression meta-analysis identifies metastatic pathways and transcription factors in breast cancer. BMC Cancer 2008, 8, 394. [Google Scholar] [CrossRef] [Green Version]
- Qiao, K.; Ning, S.; Wan, L.; Wu, H.; Wang, Q.; Zhang, X.; Xu, S.; Pang, D. LINC00673 is activated by YY1 and promotes the proliferation of breast cancer cells via the miR-515-5p/MARK4/Hippo signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 418. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Li, R.; Gan, Y. Knockdown of Yin Yang 1 enhances anticancer effects of cisplatin through protein phosphatase 2A-mediated T308 dephosphorylation of AKT. Cell Death Dis. 2018, 9, 747. [Google Scholar] [CrossRef] [Green Version]
- Attwooll, C.; Denchi, E.L.; Helin, K. The E2F family: Specific functions and overlapping interests. EMBO J. 2004, 23, 4709–4716. [Google Scholar] [CrossRef]
- Johnson, J.; Thijssen, B.; McDermott, U.; Garnett, M.; Wessels, L.F.; Bernards, R. Targeting the RB-E2F pathway in breast cancer. Oncogene 2016, 35, 4829–4835. [Google Scholar] [CrossRef] [Green Version]
- Meng, P.; Ghosh, R. Transcription addiction: Can we garner the Yin and Yang functions of E2F1 for cancer therapy? Cell Death Dis. 2014, 5, e1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, G.K. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat. Appl. Genet. Mol. Boil. 2004, 3. [Google Scholar] [CrossRef] [PubMed]
- Dunning, M.; Lynch, A.; Eldridge, M. Illuminahumanv4.db: Illumina HumanHT12v4 Annotation Data (Chip IlluminaHumanv4), R Package Version 1.26.0; Bioconductor, Roswell Park Comprehensive Cancer Center, 2015. [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.innatedb.com/ (accessed on 17 January 2017).
- Jiao, X.; Sherman, B.T.; Huang, D.W.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef] [Green Version]
- Dweep, H.; Gretz, N. miRWalk2.0: A comprehensive atlas of microRNA-target interactions. Nat. Methods 2015, 12, 697. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Briefings Bioinform. 2012, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Available online: http://xena.ucsc.edu (accessed on 12 May 2020).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 002832. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://tools.sschmeier.com/tcof/home/ (accessed on 15 May 2020).


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cataldo, A.; Romero-Cordoba, S.; Plantamura, I.; Cosentino, G.; Hidalgo-Miranda, A.; Tagliabue, E.; Iorio, M.V. MiR-302b as a Combinatorial Therapeutic Approach to Improve Cisplatin Chemotherapy Efficacy in Human Triple-Negative Breast Cancer. Cancers 2020, 12, 2261. https://doi.org/10.3390/cancers12082261
Cataldo A, Romero-Cordoba S, Plantamura I, Cosentino G, Hidalgo-Miranda A, Tagliabue E, Iorio MV. MiR-302b as a Combinatorial Therapeutic Approach to Improve Cisplatin Chemotherapy Efficacy in Human Triple-Negative Breast Cancer. Cancers. 2020; 12(8):2261. https://doi.org/10.3390/cancers12082261
Chicago/Turabian StyleCataldo, Alessandra, Sandra Romero-Cordoba, Ilaria Plantamura, Giulia Cosentino, Alfredo Hidalgo-Miranda, Elda Tagliabue, and Marilena V. Iorio. 2020. "MiR-302b as a Combinatorial Therapeutic Approach to Improve Cisplatin Chemotherapy Efficacy in Human Triple-Negative Breast Cancer" Cancers 12, no. 8: 2261. https://doi.org/10.3390/cancers12082261
APA StyleCataldo, A., Romero-Cordoba, S., Plantamura, I., Cosentino, G., Hidalgo-Miranda, A., Tagliabue, E., & Iorio, M. V. (2020). MiR-302b as a Combinatorial Therapeutic Approach to Improve Cisplatin Chemotherapy Efficacy in Human Triple-Negative Breast Cancer. Cancers, 12(8), 2261. https://doi.org/10.3390/cancers12082261