TERT Promoter Mutation as an Independent Prognostic Marker for Poor Prognosis MAPK Inhibitors-Treated Melanoma



Abstract
1. Introduction
2. Results
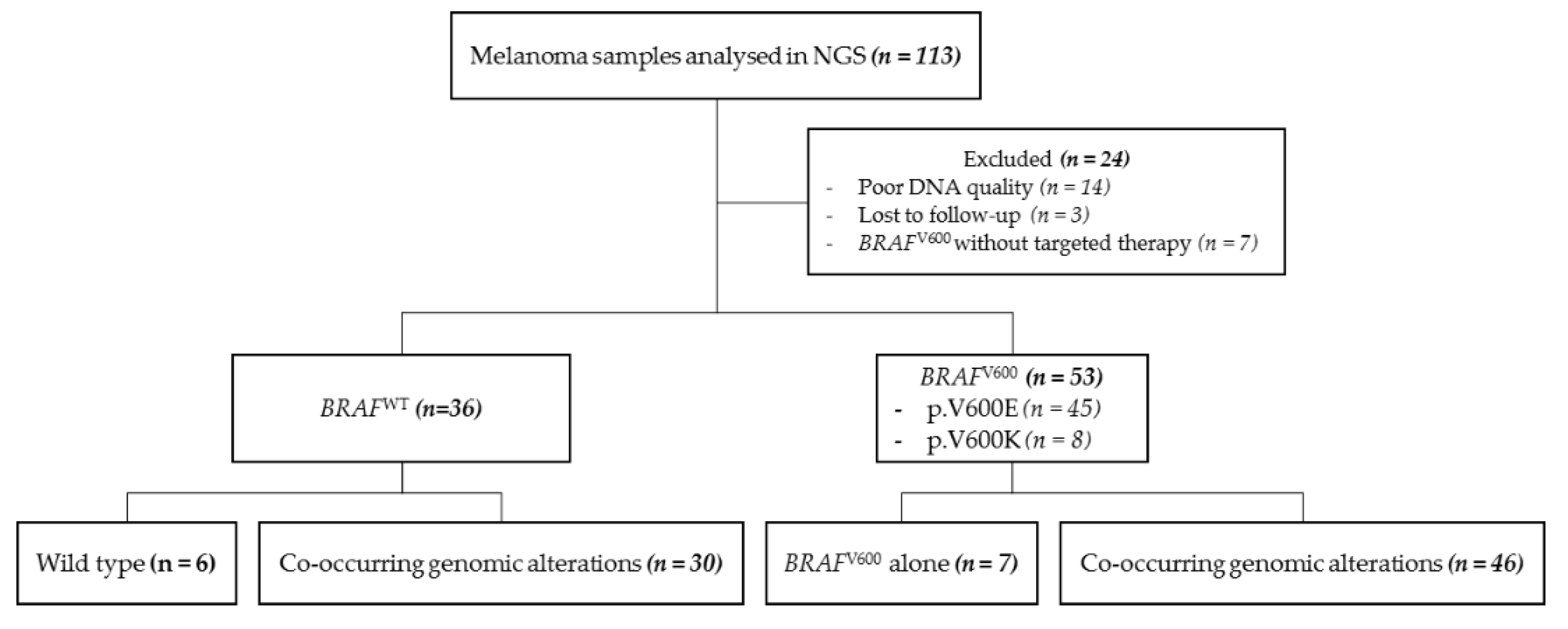
2.1. Patient Characteristics
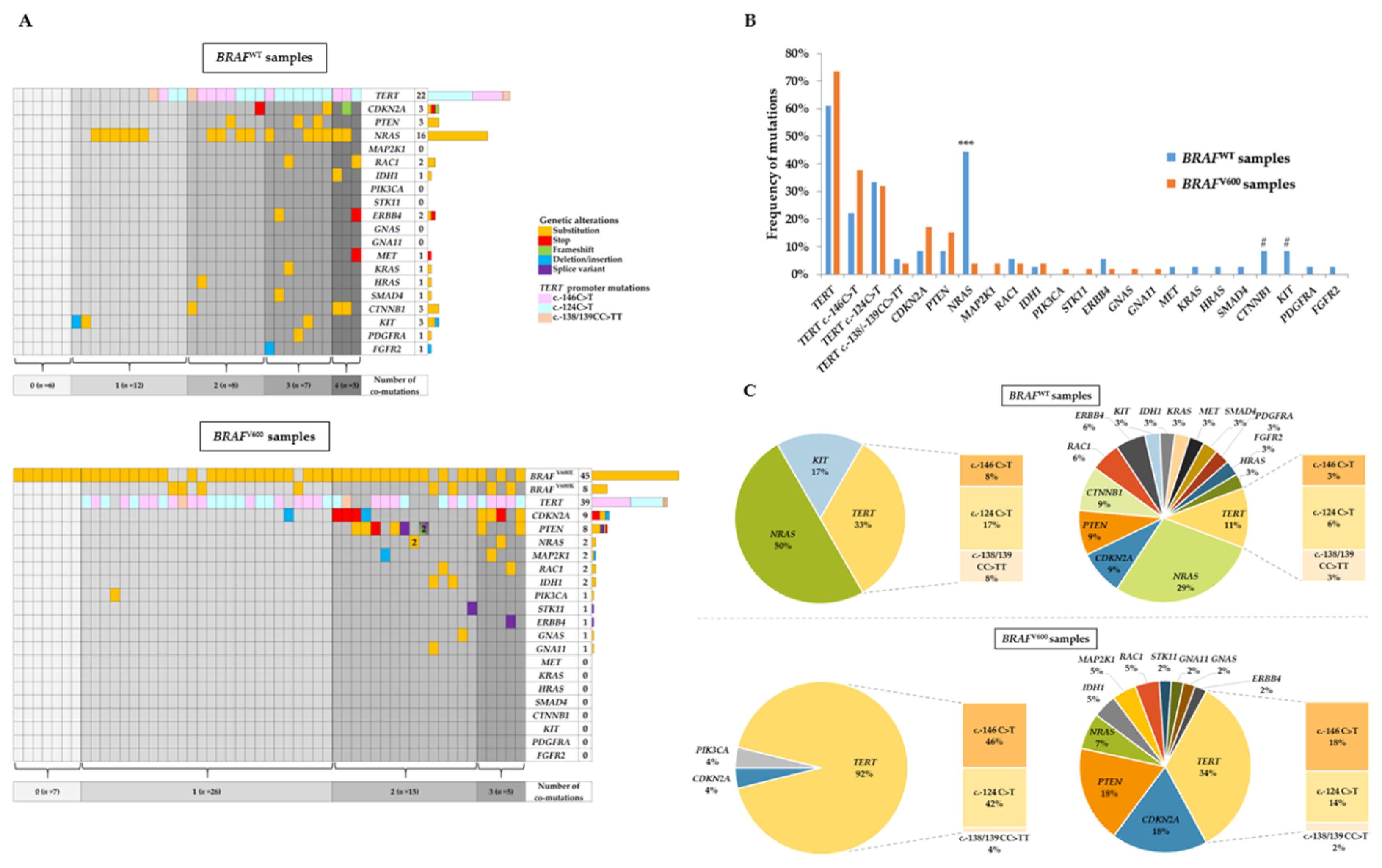
2.2. NGS Analysis and Co-Occurring Genetic Alteration Detection
2.3. Correlation between Clinicopathological Features with BRAFV600 Mutation and TERT Promoter Mutation
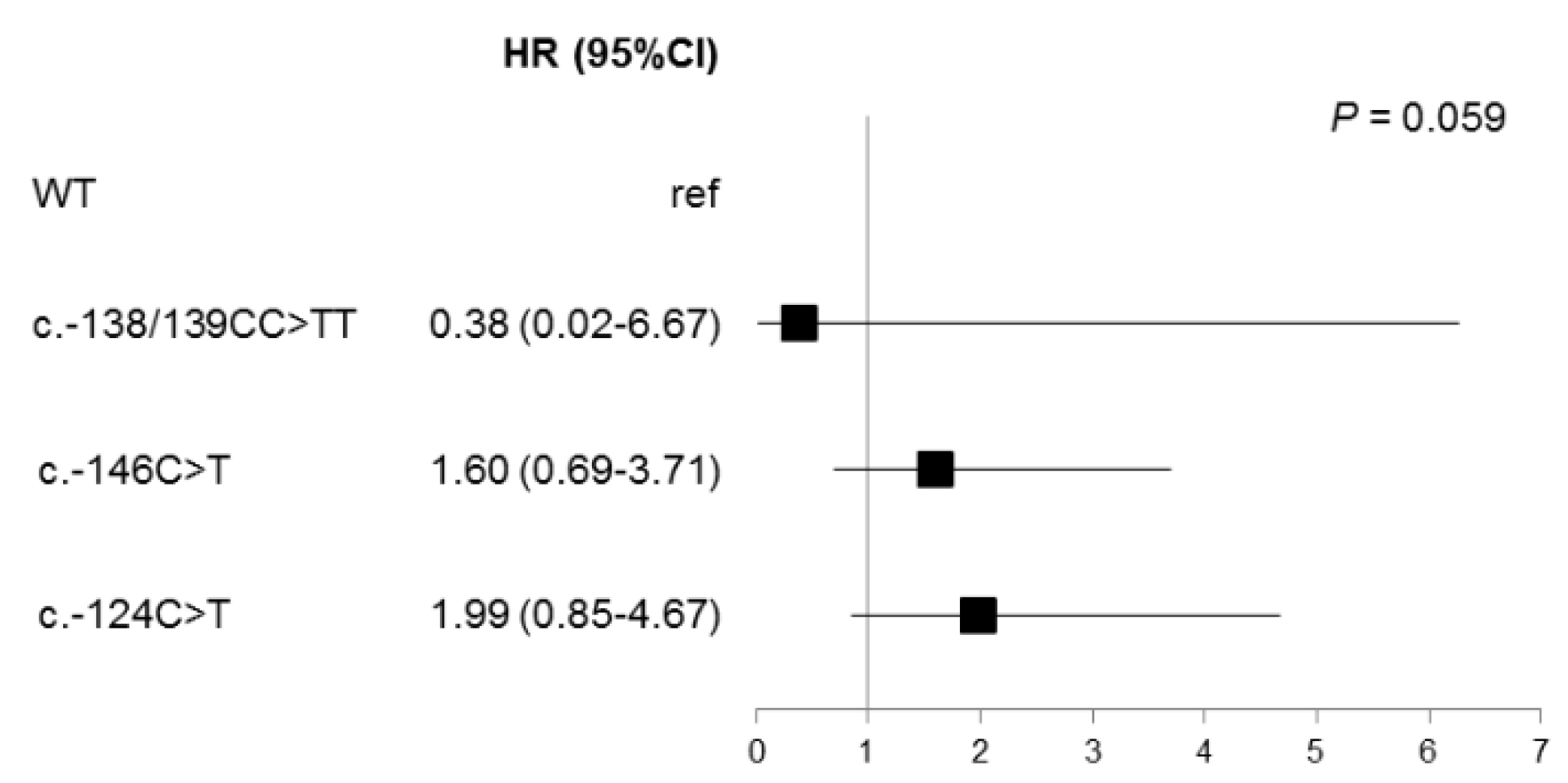
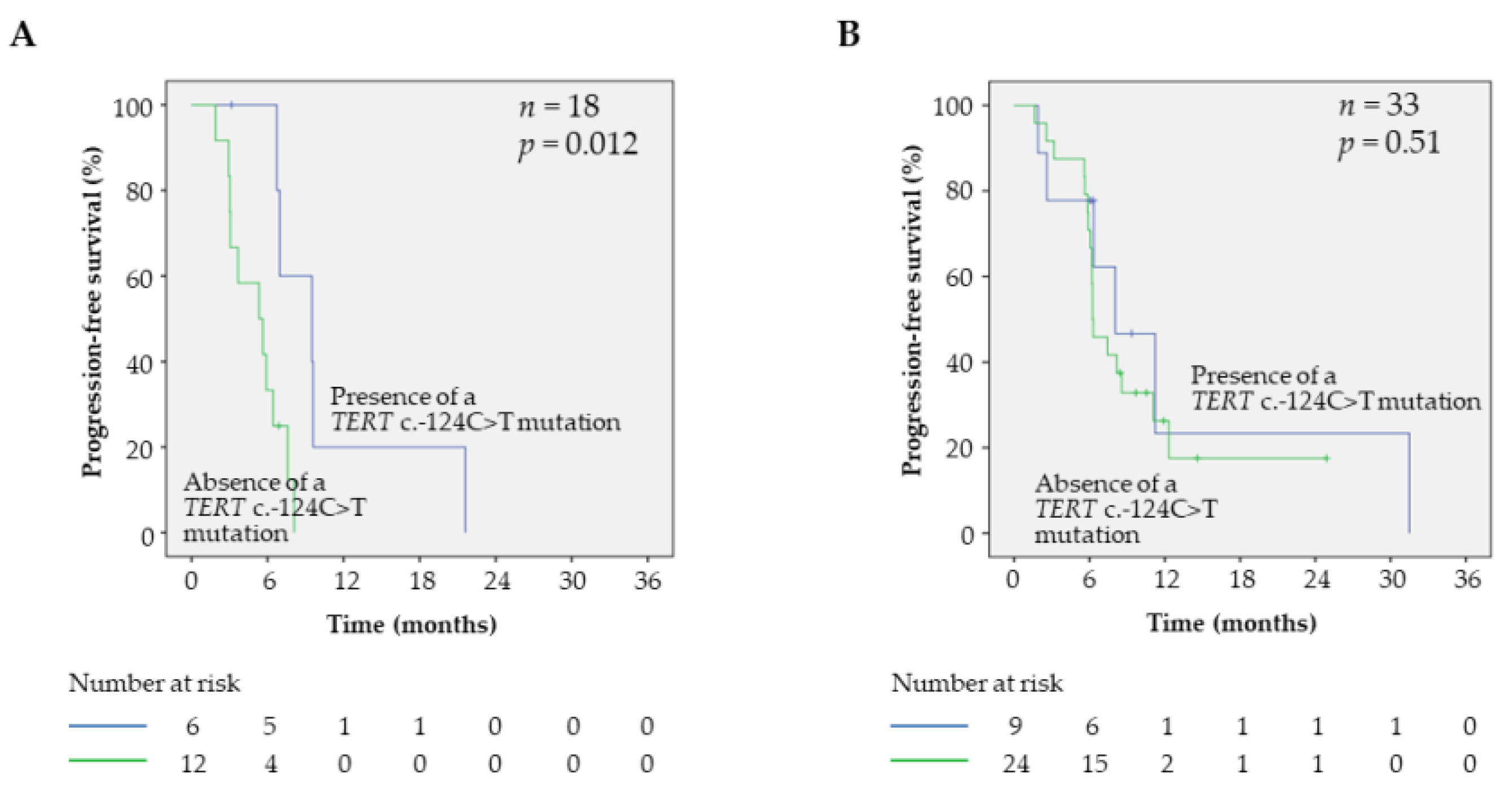
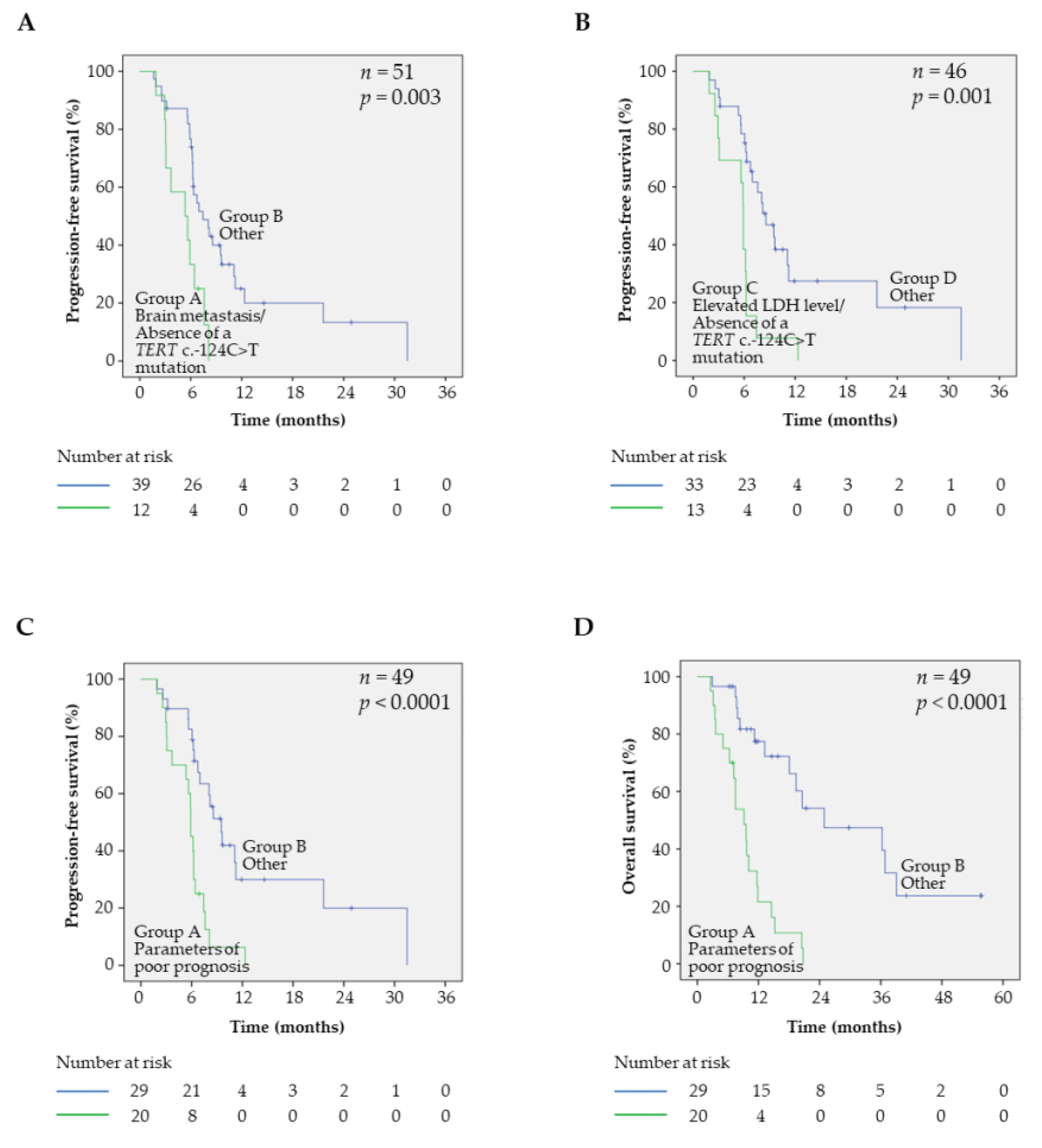
2.4. Prognostic Factors and PFS in BRAFV600 Samples
2.5. Prognostic Factors and OS in BRAFV600 Samples
3. Discussion
4. Materials and Methods
4.1. Patient and Sample Collection
4.2. NGS Analysis
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.-H.; Aiba, S.; Bröcker, E.-B.; LeBoit, P.E.; et al. Distinct Sets of Genetic Alterations in Melanoma. Available online: https://www.nejm.org/doi/10.1056/NEJMoa050092?url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org&rfr_dat=cr_pub++0www.ncbi.nlm.nih.gov (accessed on 29 May 2020).
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Tarhini, A.; Rael, M.; Gupte-Singh, K.; O’Brien, E.; Ritchings, C.; Rao, S.; McDermott, D.F. Comparative efficacy of combination immunotherapy and targeted therapy in the treatment of BRAF-mutant advanced melanoma: A matching-adjusted indirect comparison. Immunotherapy 2019, 11, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Grob, J.-J.; Nathan, P.; Ribas, A.; Robert, C.; Schadendorf, D.; Lane, S.R.; Mak, C.; Legenne, P.; Flaherty, K.T.; et al. Factors predictive of response, disease progression, and overall survival after dabrafenib and trametinib combination treatment: A pooled analysis of individual patient data from randomised trials. Lancet Oncol. 2016, 17, 1743–1754. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Miraflor, A.P.; de Abreu, F.B.; Peterson, J.D.; Turner, S.A.; Amos, C.I.; Tsongalis, G.J.; Yan, S. Somatic mutation analysis in melanoma using targeted next generation sequencing. Exp. Mol. Pathol. 2017, 103, 172–177. [Google Scholar] [CrossRef]
- Ticha, I.; Hojny, J.; Michalkova, R.; Kodet, O.; Krkavcova, E.; Hajkova, N.; Nemejcova, K.; Bartu, M.; Jaksa, R.; Dura, M.; et al. A comprehensive evaluation of pathogenic mutations in primary cutaneous melanomas, including the identification of novel loss-of-function variants. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Zhao, X.; Little, P.; Hoyle, A.P.; Pegna, G.J.; Hayward, M.C.; Ivanova, A.; Parker, J.S.; Marron, D.L.; Soloway, M.G.; Jo, H.; et al. The Prognostic Significance of Low-Frequency Somatic Mutations in Metastatic Cutaneous Melanoma. Front. Oncol. 2019, 8. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational Landscape of Metastatic Cancer Revealed from Prospective Clinical Sequencing of 10,000 Patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Gopal, Y.N.V.; Deng, W.; Woodman, S.E.; Komurov, K.; Ram, P.; Smith, P.D.; Davies, M.A. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010, 70, 8736–8747. [Google Scholar] [CrossRef]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C.; et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; MacConaill, L.E.; Hahn, W.C.; et al. Dissecting Therapeutic Resistance to RAF Inhibition in Melanoma by Tumor Genomic Profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Q.; Liu, J.; Huang, L.; Qin, Y.; Hawley, T.; Seo, C.; Merlino, G.; Yu, Y. AXL/AKT axis mediated-resistance to BRAF inhibitor depends on PTEN status in melanoma. Oncogene 2018, 37, 3275. [Google Scholar] [CrossRef] [PubMed]
- Dutton-Regester, K.; Irwin, D.; Hunt, P.; Aoude, L.G.; Tembe, V.; Pupo, G.M.; Lanagan, C.; Carter, C.D.; O’Connor, L.; O’Rourke, M.; et al. A High-Throughput Panel for Identifying Clinically Relevant Mutation Profiles in Melanoma. Mol. Cancer Ther. 2012, 11, 888–897. [Google Scholar] [CrossRef]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef]
- Ellerhorst, J.A.; Greene, V.R.; Ekmekcioglu, S.; Warneke, C.L.; Johnson, M.M.; Cooke, C.P.; Wang, L.-E.; Prieto, V.G.; Gershenwald, J.E.; Wei, Q.; et al. Clinical Correlates of NRAS and BRAF Mutations in Primary Human Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 229–235. [Google Scholar] [CrossRef]
- Mann, G.J.; Pupo, G.M.; Campain, A.E.; Carter, C.D.; Schramm, S.-J.; Pianova, S.; Gerega, S.K.; De Silva, C.; Lai, K.; Wilmott, J.S.; et al. BRAF Mutation, NRAS Mutation, and the Absence of an Immune-Related Expressed Gene Profile Predict Poor Outcome in Patients with Stage III Melanoma. J. Invest. Dermatol. 2013, 133, 509–517. [Google Scholar] [CrossRef]
- Barbour, A.P.; Tang, Y.H.; Armour, N.; Dutton-Regester, K.; Krause, L.; Loffler, K.A.; Lambie, D.; Burmeister, B.; Thomas, J.; Smithers, B.M.; et al. BRAF mutation status is an independent prognostic factor for resected stage IIIB and IIIC melanoma: Implications for melanoma staging and adjuvant therapy. Eur. J. Cancer 2014, 50, 2668–2676. [Google Scholar] [CrossRef]
- Meckbach, D.; Keim, U.; Richter, S.; Leiter, U.; Eigentler, T.K.; Bauer, J.; Pflugfelder, A.; Büttner, P.; Garbe, C.; Weide, B. BRAF-V600 Mutations Have No Prognostic Impact in Stage IV Melanoma Patients Treated with Monochemotherapy. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Sperduto, P.W.; Jiang, W.; Brown, P.D.; Braunstein, S.; Sneed, P.; Wattson, D.A.; Shih, H.A.; Bangdiwala, A.; Shanley, R.; Lockney, N.A.; et al. The Prognostic Value of BRAF, C-KIT, and NRAS Mutations in Melanoma Patients with Brain Metastases. Int. J. Radiat. Oncol. Biol. Phys. 2017, 98, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, M.I.G.; Widmer, D.S.; Narechania, A.; Eichhoff, O.; Freiberger, S.N.; Wenzina, J.; Cheng, P.F.; Mihic-Probst, D.; Desalle, R.; Dummer, R.; et al. Co-existence of BRAF and NRAS driver mutations in the same melanoma cells results in heterogeneity of targeted therapy resistance. Oncotarget 2016, 7, 77163–77174. [Google Scholar] [CrossRef] [PubMed]
- Olbryt, M.; Pigłowski, W.; Rajczykowski, M.; Pfeifer, A.; Student, S.; Fiszer-Kierzkowska, A. Genetic Profiling of Advanced Melanoma: Candidate Mutations for Predicting Sensitivity and Resistance to Targeted Therapy. Target. Oncol. 2020, 15, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Barnet, M.B.; O’Toole, S.; Horvath, L.G.; Selinger, C.; Yu, B.; Ng, C.C.; Boyer, M.; Cooper, W.A.; Kao, S. EGFR–Co-Mutated Advanced NSCLC and Response to EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2017, 12, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.-L.; Gao, G.; Deng, H.-B.; Wang, F.; Wang, Z.-H.; Yang, Y. Co-occurring genetic alterations predict distant metastasis and poor efficacy of first-line EGFR-TKIs in EGFR-mutant NSCLC. J. Cancer Res. Clin. Oncol. 2019, 145, 2613–2624. [Google Scholar] [CrossRef]
- Griewank, K.G.; Murali, R.; Puig-Butille, J.A.; Schilling, B.; Livingstone, E.; Potrony, M.; Carrera, C.; Schimming, T.; Möller, I.; Schwamborn, M.; et al. TERT Promoter Mutation Status as an Independent Prognostic Factor in Cutaneous Melanoma. JNCI J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Vazquez, V.d.L.; Vicente, A.L.; Carloni, A.; Berardinelli, G.; Soares, P.; Scapulatempo, C.; Martinho, O.; Reis, R.M. Molecular profiling, including TERT promoter mutations, of acral lentiginous melanomas. Melanoma Res. 2016, 26, 93–99. [Google Scholar] [CrossRef]
- Bai, X.; Kong, Y.; Chi, Z.; Sheng, X.; Cui, C.; Wang, X.; Mao, L.; Tang, B.; Li, S.; Lian, B.; et al. MAPK Pathway and TERT Promoter Gene Mutation Pattern and Its Prognostic Value in Melanoma Patients: A Retrospective Study of 2,793 Cases. Clin. Cancer Res. 2017, 23, 6120–6127. [Google Scholar] [CrossRef]
- Heidenreich, B.; Nagore, E.; Rachakonda, P.S.; Garcia-Casado, Z.; Requena, C.; Traves, V.; Becker, J.; Soufir, N.; Hemminki, K.; Kumar, R. Telomerase reverse transcriptase promoter mutations in primary cutaneous melanoma. Nat. Commun. 2014, 5, 1–7. [Google Scholar] [CrossRef]
- Macerola, E.; Loggini, B.; Giannini, R.; Garavello, G.; Giordano, M.; Proietti, A.; Niccoli, C.; Basolo, F.; Fontanini, G. Coexistence of TERT promoter and BRAF mutations in cutaneous melanoma is associated with more clinicopathological features of aggressiveness. Virchows Arch. 2015, 467, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Nagore, E.; Heidenreich, B.; Rachakonda, S.; Garcia-Casado, Z.; Requena, C.; Soriano, V.; Frank, C.; Traves, V.; Quecedo, E.; Sanjuan-Gimenez, J.; et al. TERT promoter mutations in melanoma survival. Int. J. Cancer 2016, 139, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Del Bianco, P.; Stagni, C.; Giunco, S.; Fabozzi, A.; Elefanti, L.; Pellegrini, S.; Vecchiato, A.; Pigozzo, J.; Zamuner, C.; De Rossi, A.; et al. TERT Promoter Mutations Differently Correlate with the Clinical Outcome of MAPK Inhibitor-Treated Melanoma Patients. Cancers 2020, 12, 946. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhang, T.; Zhu, G.; Xing, M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Vinagre, J.; Almeida, A.; Pópulo, H.; Batista, R.; Lyra, J.; Pinto, V.; Coelho, R.; Celestino, R.; Prazeres, H.; Lima, L.; et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 2013, 4, 2185. [Google Scholar] [CrossRef]
- Pópulo, H.; Boaventura, P.; Vinagre, J.; Batista, R.; Mendes, A.; Caldas, R.; Pardal, J.; Azevedo, F.; Honavar, M.; Guimarães, I.; et al. TERT Promoter Mutations in Skin Cancer: The Effects of Sun Exposure and X-Irradiation. J. Invest. Dermatol. 2014, 134, 2251–2257. [Google Scholar] [CrossRef]
- Heidenreich, B.; Kumar, R. TERT promoter mutations in telomere biology. Mutat. Res. 2017, 771, 15–31. [Google Scholar] [CrossRef]
- Hsu, C.P.; Hsu, N.Y.; Lee, L.W.; Ko, J.L. Ets2 binding site single nucleotide polymorphism at the hTERT gene promoter—effect on telomerase expression and telomere length maintenance in non-small cell lung cancer. Eur J Cancer 2006, 42, 1466–1474. [Google Scholar] [CrossRef]
- Nagore, E.; Rachakonda, S.; Kumar, R. TERT promoter mutations in melanoma survival. Oncotarget 2019, 10, 1546–1548. [Google Scholar] [CrossRef]
- Andrés-Lencina, J.J.; Rachakonda, S.; García-Casado, Z.; Srinivas, N.; Skorokhod, A.; Requena, C.; Soriano, V.; Kumar, R.; Nagore, E. TERT promoter mutation subtypes and survival in stage I and II melanoma patients. Int. J. Cancer 2019, 144, 1027–1036. [Google Scholar] [CrossRef]
- Batista, R.; Cruvinel-Carloni, A.; Vinagre, J.; Peixoto, J.; Catarino, T.A.; Campanella, N.C.; Menezes, W.; Becker, A.P.; de Almeida, G.C.; Matsushita, M.M.; et al. The prognostic impact of TERT promoter mutations in glioblastomas is modified by the rs2853669 single nucleotide polymorphism. Int. J. Cancer 2016, 139, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Hosen, I.; Gousias, K.; Rachakonda, S.; Heidenreich, B.; Gessi, M.; Schramm, J.; Hemminki, K.; Waha, A.; Kumar, R. TERT promoter mutations: A novel independent prognostic factor in primary glioblastomas. Neuro Oncol. 2015, 17, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Spiegl-Kreinecker, S.; Lotsch, D.; Ghanim, B.; Pirker, C.; Mohr, T.; Laaber, M.; Weis, S.; Olschowski, A.; Webersinke, G.; Pichler, J.; et al. Prognostic quality of activating TERT promoter mutations in glioblastoma: Interaction with the rs2853669 polymorphism and patient age at diagnosis. Neuro Oncol. 2015, 17, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Nencha, U.; Rahimian, A.; Giry, M.; Sechi, A.; Mokhtari, K.; Polivka, M.; Schmitt, Y.; Di Stefano, A.L.; Alentorn, A.; Labussiere, M.; et al. TERT promoter mutations and rs2853669 polymorphism: Prognostic impact and interactions with common alterations in glioblastomas. J. Neurooncol. 2016, 126, 441–446. [Google Scholar] [CrossRef]
- Vendrell, J.A.; Grand, D.; Rouquette, I.; Costes, V.; Icher, S.; Selves, J.; Larrieux, M.; Barbe, A.; Brousset, P.; Solassol, J. High-throughput detection of clinically targetable alterations using next-generation sequencing. Oncotarget 2017, 8, 40345–40358. [Google Scholar] [CrossRef]
- Mariette, J.; Escudié, F.; Bardou, P.; Nabihoudine, I.; Noirot, C.; Trotard, M.-S.; Gaspin, C.; Klopp, C. Jflow: A workflow management system for web applications. Bioinformatics 2016, 32, 456. [Google Scholar] [CrossRef][Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicopathological Features | BRAF Status | TERT Promoter Status | |||||
|---|---|---|---|---|---|---|---|
| BRAFWT (n = 36) | BRAFV600 (n = 53) | p-Value | WT (n = 28) | Mutated (n = 61) | p-Value | ||
| Age | ≤60 | 4 (11.1) | 28 (52.8) | <0.001 | 9 (32.1) | 23 (37.7) | 0.61 |
| >60 | 32 (88.9) | 25 (47.2) | 19 (67.9) | 38 (62.3) | |||
| Sex | Male | 22 (61.1) | 28 (52.8) | 0.44 | 15 (53.6) | 35 (57.4) | 0.74 |
| Female | 14 (38.9) | 25 (47.2) | 13 (46.4) | 26 (42.6) | |||
| Histological type | NM | 8 (25.8) | 10 (20.8) | 0.18 | 5 (20) | 13 (24.1) | 0.03 |
| SSM | 15 (48.4) | 24 (50) | 10 (40) | 29 (53.7) | |||
| MUP | 3 (9.7) | 9 (18.8) | 3 (12) | 9 (16.7) | |||
| Unclassified | 0 (0) | 3 (6.3) | 1 (4) | 2 (3.7) | |||
| ALM | 5 (16.1) | 2 (4.2) | 6 (24) | 1 (1.9) | |||
| Missing data | 5 | 5 | 3 | 7 | |||
| Primary tumor site | Head/neck | 7 (20.6) | 11 (20.8) | 0.06 | 1 (3.7) | 17 (28.3) | 0.003 |
| Upper limbs | 4 (11.8) | 2 (3.8) | 1 (3.7) | 5 (8.3) | |||
| Trunk | 6 (17.6) | 21 (39.6) | 8 (29.6) | 19 (31.7) | |||
| Lower limbs | 9 (26.5) | 8 (15.1) | 8 (29.6) | 9 (15.0) | |||
| MUP | 3 (8.8) | 9 (17) | 3 (11.1) | 9 (15.0) | |||
| Acral | 5 (14.7) | 2 (3.8) | 6 (22.2) | 1 (1.7) | |||
| Missing data | 2 | 0 | 1 | 1 | |||
| Breslow thickness * | <1 mm | 1 (3.3) | 4 (10.2) | 0.25 | 2 (8.7) | 3 (6.5) | 0.66 |
| 1–1.99 mm | 4 (13.3) | 11 (28.2) | 3 (13.0) | 12 (26.1) | |||
| 2–3.99 mm | 8 (26.6) | 9 (23.1) | 6 (26.1) | 11 (23.9) | |||
| ≥4 mm | 17 (56.6) | 15 (38.4) | 12 (52.2) | 20 (43.5) | |||
| Missing data | 3 | 5 | 2 | 6 | |||
| Clark level * | II | 0 (0) | 2 (5.4) | 0.09 | 0 (0) | 2 (4.6) | 0.66 |
| III | 2 (6.9) | 10 (27.0) | 3 (13.6) | 9 (20.4) | |||
| IV | 21 (72.4) | 20 (54.1) | 15 (68.2) | 26 (59.1) | |||
| V | 6 (20.7) | 5 (13.5) | 4 (18.2) | 7 (15.9) | |||
| Missing data | 4 | 7 | 3 | 8 | |||
| AJCC | I | 3 (8.8) | 7 (14.0) | 0.56 | 3 (11.1) | 7 (12.3) | 0.97 |
| II | 16 (47.1) | 17 (34.0) | 10 (37.0) | 23 (40.4) | |||
| III | 8 (23.5) | 11 (22.0) | 6 (22.2) | 13 (22.8) | |||
| IV | 7 (20.6) | 15 (30.0) | 8 (29.6) | 14 (24.6) | |||
| Missing data | 2 | 3 | 1 | 4 | |||
| Clinical Parameters | Samples (n) | HR | 95% CI | p-Value |
|---|---|---|---|---|
| Sex (Male; Female) | 51 | 1.16 | 0.61–2.20 | NS (0.65) |
| Age (≤60; >60-year-old) | 51 | 1.66 | 0.88–3.14 | NS (0.12) |
| LDH level (normal; high) | 46 | 2.38 | 1.19–4.75 | 0.01 |
| Brain metastases (absence; presence) | 51 | 1.72 | 0.90–3.29 | NS (0.09) |
| Histological type (NM; SSM; MUP; ALM; unclassified) | 45 | 1.35 | 0.93–1.95 | NS (0.11) |
| Breslow thickness (<1 mm; 1–2 mm; 2–4 mm; >4) | 37 | 1.10 | 0.75–1.62 | NS (0.61) |
| Treatment modalities (monotherapy; bitherapy) | 50 | 1.03 | 0.40–2.65 | NS (0.95) |
| Co-occurring mutation (absence; presence) | 51 | 0.49 | 0.20–1.20 | NS (0.12) |
| TERT promoter mutation (absence; presence) | 51 | 0.58 | 0.29–1.17 | NS (0.13) |
| CDKN2A mutation (absence; presence) | 51 | 1.50 | 0.65–3.44 | NS (0.34) |
| PTEN mutation (absence; presence) | 51 | 1.78 | 0.65–3.40 | NS (0.35) |
| Clinical Parameters | HR | 95% CI | p-Value |
|---|---|---|---|
| LDH level (normal; high) | 2.86 | 1.36–6.02 | 0.006 |
| Brain metastasis (absence; presence) | 2.34 | 1.03–5.30 | 0.04 |
| TERT c.-124C>T mutation (absence; presence) | 1.37 | 1.08–1.73 | 0.009 |
| Clinical Parameters | Samples (n) | HR | 95% CI | p-Value |
|---|---|---|---|---|
| Sex (Male; Female) | 51 | 2.20 | 1.10–4.42 | 0.03 |
| Age (≤60; >60-year-old) | 51 | 1.63 | 0.83–3.20 | NS (0.15) |
| LDH level (normal; high) | 46 | 1.71 | 0.83–3.52 | NS (0.15) |
| Brain metastases (absence; presence) | 51 | 2.21 | 1.08–4.50 | 0.03 |
| Histological type (NM; SSM; MUP; ALM; unclassified) | 45 | 1.27 | 0.89–1.82 | NS (0.19) |
| Breslow thickness (<1 mm; 1–2 mm; 2–4 mm; >4) | 37 | 1.02 | 0.67–1.55 | NS (0.93) |
| Treatment modalities (monotherapy; bitherapy) | 50 | 0.61 | 0.25–1.49 | NS (0.27) |
| Co-occurring mutation (absence; presence) | 51 | 0.68 | 0.28–1.65 | NS (0.39) |
| TERT promoter mutation (absence; presence) | 51 | 0.58 | 0.28–1.21 | NS (0.15) |
| CDKN2A mutation (absence; presence) | 51 | 1.41 | 0.58–3.45 | NS (0.45) |
| PTEN mutation (absence; presence) | 51 | 1.44 | 0.63–3.31 | NS (0.39) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blateau, P.; Coyaud, E.; Laurent, E.; Béganton, B.; Ducros, V.; Chauchard, G.; Vendrell, J.A.; Solassol, J. TERT Promoter Mutation as an Independent Prognostic Marker for Poor Prognosis MAPK Inhibitors-Treated Melanoma. Cancers 2020, 12, 2224. https://doi.org/10.3390/cancers12082224
Blateau P, Coyaud E, Laurent E, Béganton B, Ducros V, Chauchard G, Vendrell JA, Solassol J. TERT Promoter Mutation as an Independent Prognostic Marker for Poor Prognosis MAPK Inhibitors-Treated Melanoma. Cancers. 2020; 12(8):2224. https://doi.org/10.3390/cancers12082224
Chicago/Turabian StyleBlateau, Pauline, Etienne Coyaud, Estelle Laurent, Benoit Béganton, Vincent Ducros, Géraldine Chauchard, Julie A. Vendrell, and Jérôme Solassol. 2020. "TERT Promoter Mutation as an Independent Prognostic Marker for Poor Prognosis MAPK Inhibitors-Treated Melanoma" Cancers 12, no. 8: 2224. https://doi.org/10.3390/cancers12082224
APA StyleBlateau, P., Coyaud, E., Laurent, E., Béganton, B., Ducros, V., Chauchard, G., Vendrell, J. A., & Solassol, J. (2020). TERT Promoter Mutation as an Independent Prognostic Marker for Poor Prognosis MAPK Inhibitors-Treated Melanoma. Cancers, 12(8), 2224. https://doi.org/10.3390/cancers12082224