Targeting NF-κB Signaling for Multiple Myeloma



Abstract
1. Background
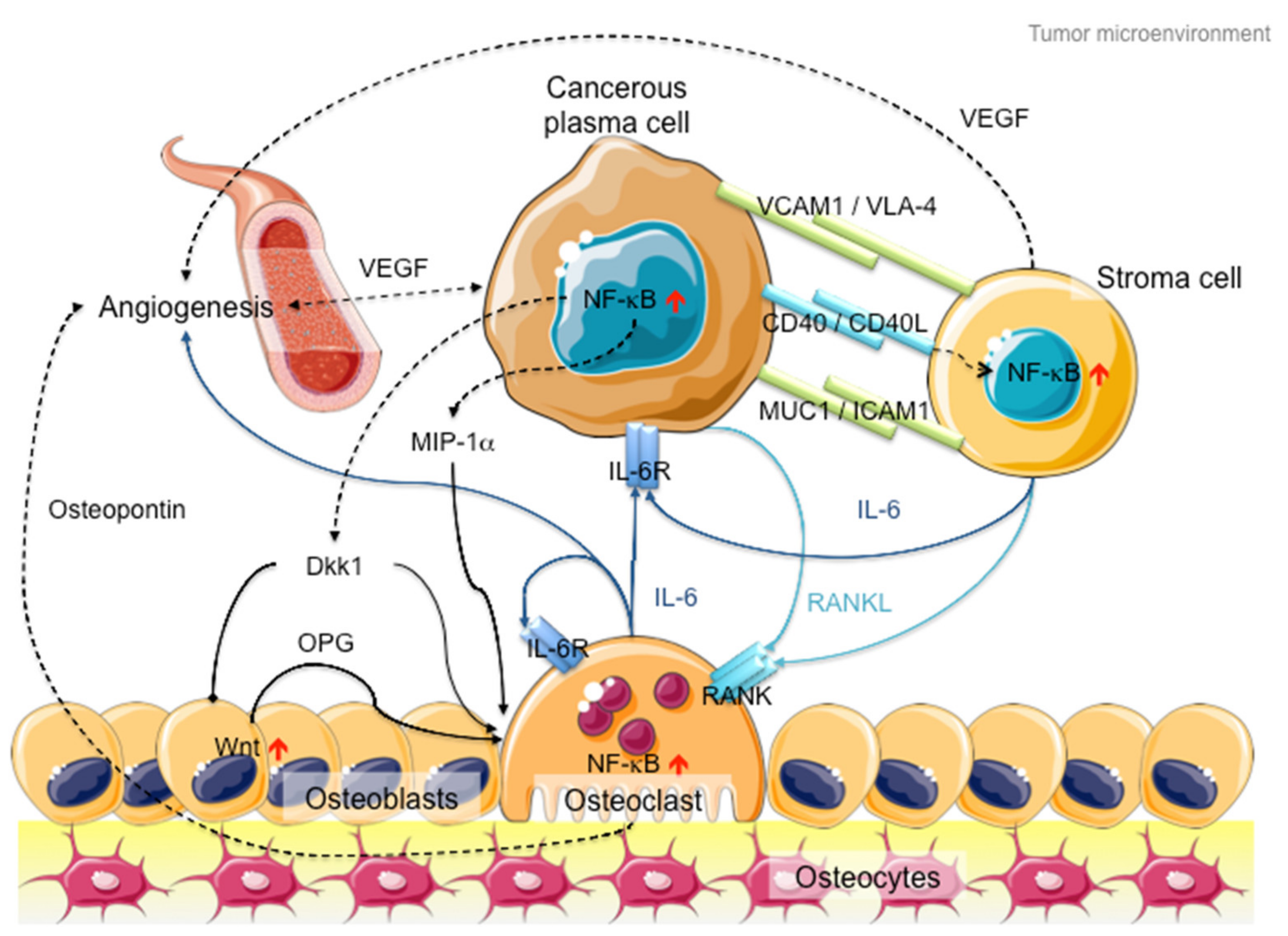
2. Multiple Myeloma and NF-κB Signaling
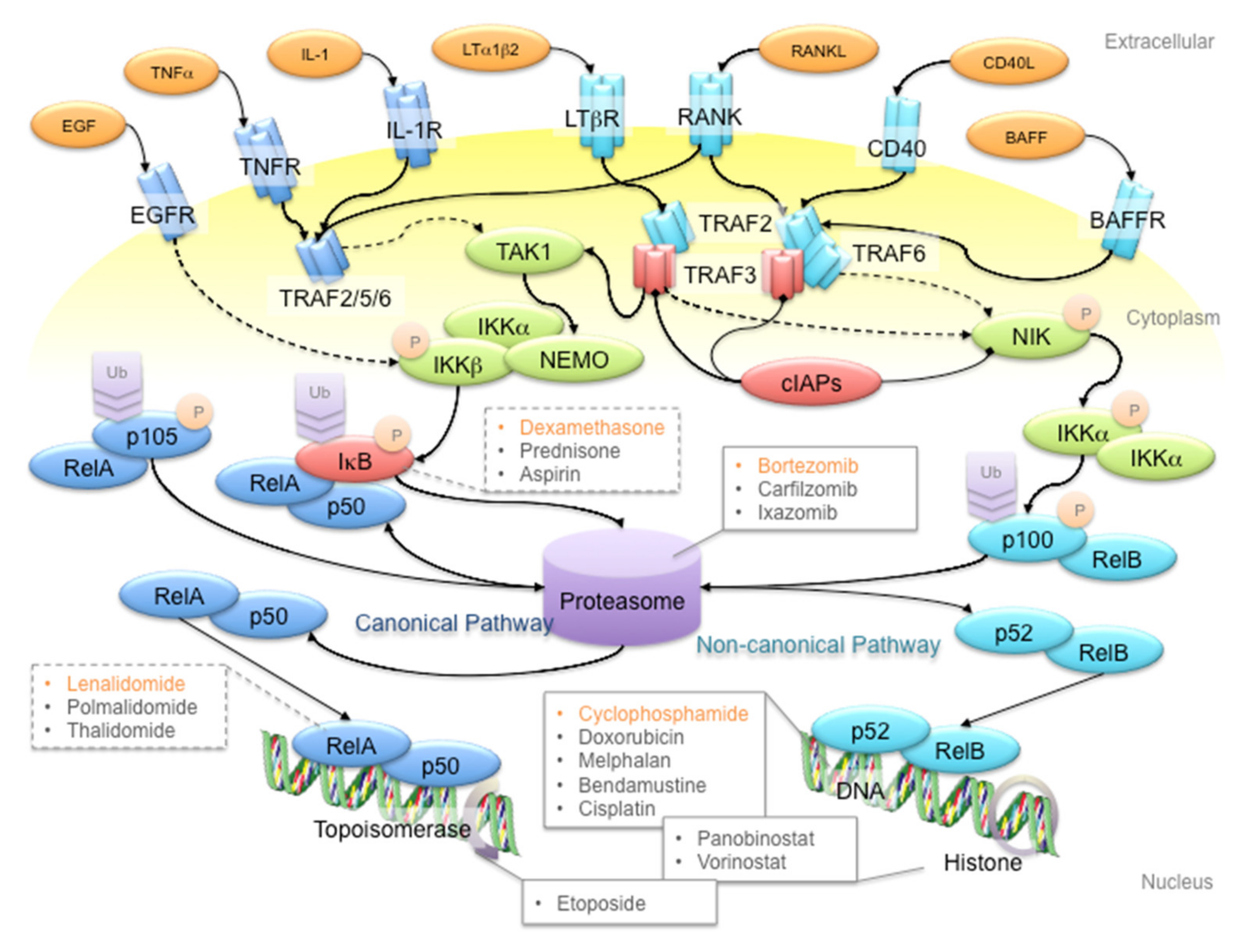
3. NF-κB Signaling: The Rose with Thorns in MM Treatment
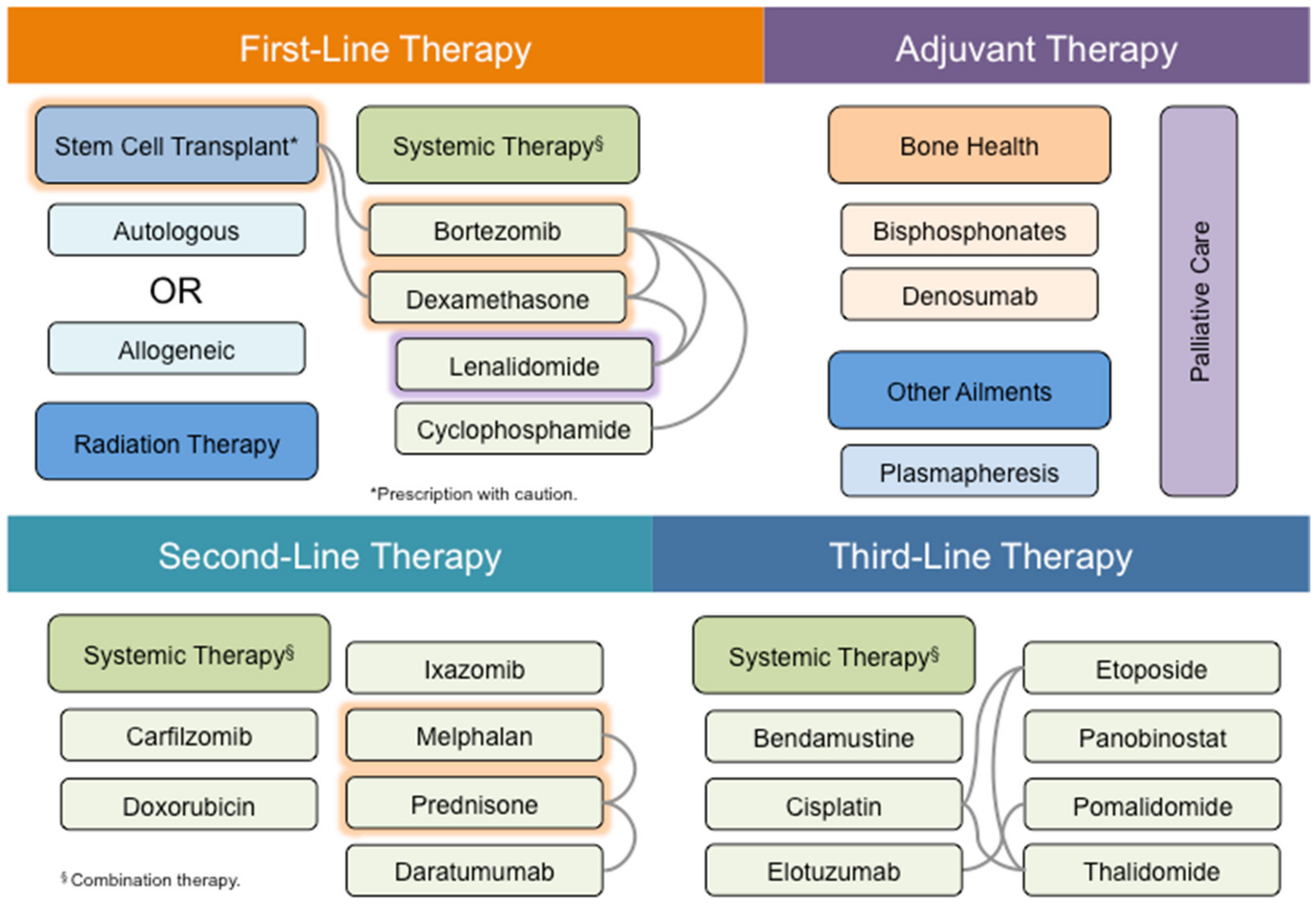
4. MM Therapy: A Steep Road to Success
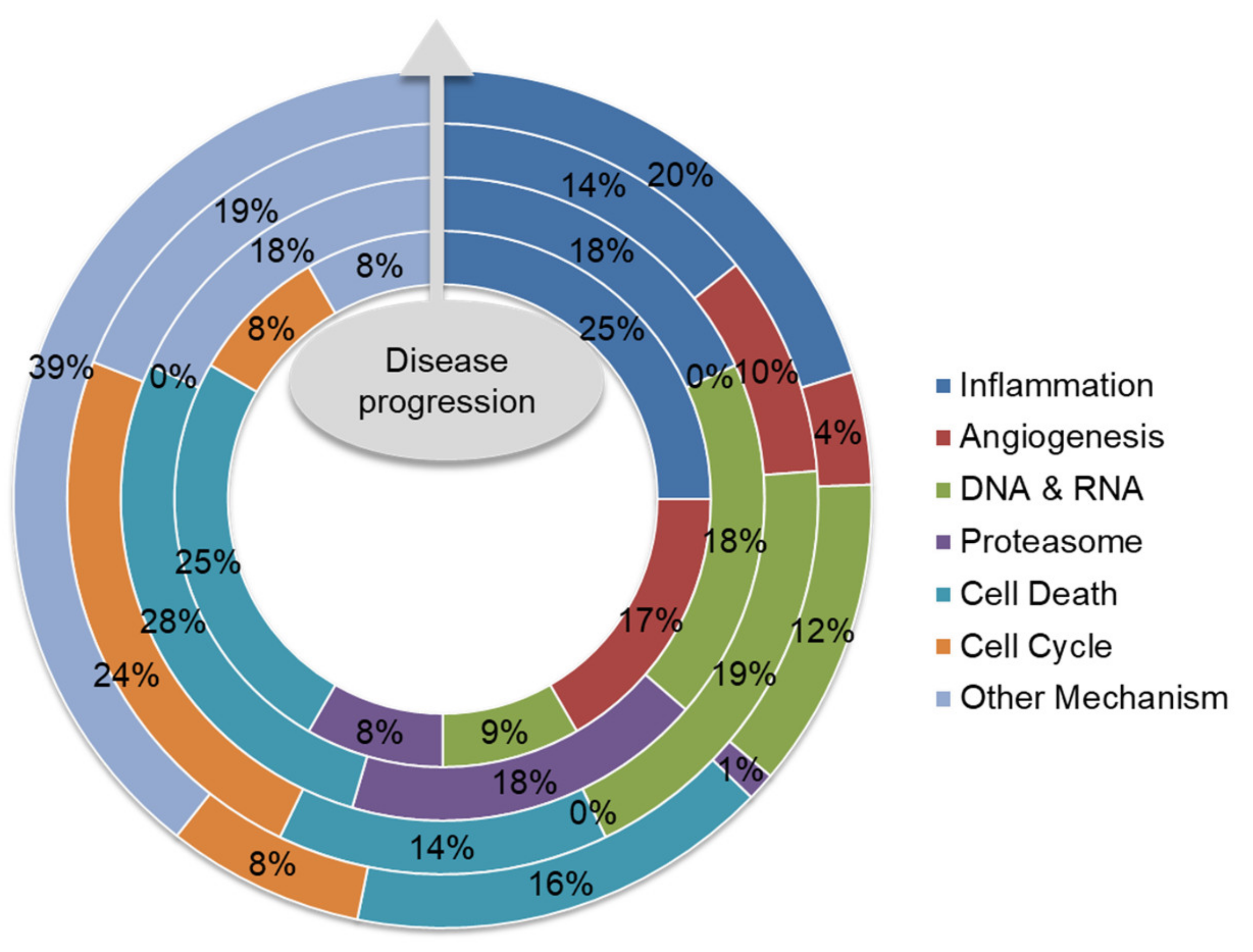
5. Future Direction: Biomarker-Guided Targeted Therapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kazandjian, D. Multiple myeloma epidemiology and survival: A unique malignancy. In Seminars in Oncology; WB Saunders: Philadelphia, PA, USA, 2016; pp. 676–681. [Google Scholar]
- Palumbo, A.; Bringhen, S.; Ludwig, H.; Dimopoulos, M.A.; Bladé, J.; Mateos, M.V.; Rosiñol, L.; Boccadoro, M.; Cavo, M.; Lokhorst, H.; et al. Personalized therapy in multiple myeloma according to patient age and vulnerability: A report of the European Myeloma Network (EMN). Blood 2011, 118, 4519–4529. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Thorsteinsdottir, S.; Dickman, P.W.; Landgren, O.; Blimark, C.; Hultcrantz, M.; Turesson, I.; Björkholm, M.; Kristinsson, S.Y. Dramatically improved survival in multiple myeloma patients in the recent decade: Results from a Swedish population-based study. Haematologica 2018, 103, e412–e415. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review (1975–2017). In Bethesda; 2020. Available online: https://seer.cancer.gov/csr/1975_2017/ (accessed on 3 May 2020).
- Fonseca, R.; Abouzaid, S.; Bonafede, M.; Cai, Q.; Parikh, K.; Cosler, L.; Richardson, P. Trends in overall survival and costs of multiple myeloma, 2000–2014. Leukemia 2017, 31, 1915–1921. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Jakubowiak, A.J.; McCarthy, P.L.; Orlowski, R.Z.; Attal, M.; Bladé, J.; Goldschmidt, H.; Weisel, K.C.; Ramasamy, K.; Zweegman, S.; et al. Developments in continuous therapy and maintenance treatment approaches for patients with newly diagnosed multiple myeloma. Blood Cancer J. 2020, 10, 17. [Google Scholar] [CrossRef]
- Cildir, G.; Low, K.C.; Tergaonkar, V. Noncanonical NF-κB Signaling in Health and Disease. Trends Mol. Med. 2016, 22, 414–429. [Google Scholar] [CrossRef]
- Puar, Y.R.; Shanmugam, M.K.; Fan, L.; Arfuso, F.; Sethi, G.; Tergaonkar, V. Evidence for the involvement of the master transcription factor NF-κB in cancer initiation and progression. Biomedicines 2018, 6, 82. [Google Scholar] [CrossRef]
- Chew, C.L.; Conos, S.A.; Unal, B.; Tergaonkar, V. Noncoding RNAs: Master Regulators of Inflammatory Signaling. Trends Mol. Med. 2018, 24, 66–84. [Google Scholar] [CrossRef]
- Chesi, M.; Bergsagel, P.L. Molecular pathogenesis of multiple myeloma: Basic and clinical updates. Int. J. Hematol. 2013, 97, 313–323. [Google Scholar] [CrossRef]
- Bergsagel, P.L.; Nardini, E.; Brents, L.; Chesi, M.; Kuehl, W.M. IgH translocations in multiple myeloma: A nearly universal event that rarely involves c-myc. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 1997; pp. 283–287. [Google Scholar]
- Fonseca, R.; Debes-Marun, C.S.; Picken, E.B.; Dewald, G.W.; Bryant, S.C.; Winkler, J.M.; Blood, E.; Oken, M.; Santana-Dávila, R.; González-Paz, N.; et al. The recurrent IgH translocations are highly associated with nonhyperdiploid variant multiple myeloma. Blood 2003, 102, 2562–2567. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, Y.N.; Michael Kuehl, W. A critical role for the NFκB pathway in multiple myeloma. Oncotarget 2010, 1, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Flynt, E.; Bisht, K.; Sridharan, V.; Ortiz, M.; Towfic, F.; Thakurta, A. Prognosis, Biology, and Targeting of TP53 Dysregulation in Multiple Myeloma. Cells 2020, 9, 287. [Google Scholar] [CrossRef]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.-J.; Van Wier, S.; Tiedemann, R.; Shi, C.-X.; Sebag, M.; et al. Promiscuous Mutations Activate the Noncanonical NF-κB Pathway in Multiple Myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef]
- Walker, B.A.; Wardell, C.P.; Chiecchio, L.; Smith, E.M.; Boyd, K.; Neri, A.; Davies, F.E.; Ross, F.M.; Morgan, G.J. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood 2011, 117, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Hodge, D.R.; Peng, B.; Cherry, J.C.; Hurt, E.M.; Fox, S.D.; Kelley, J.A.; Munroe, D.J.; Farrar, W.L. Interleukin 6 supports the maintenance of p53 tumor suppressor gene promoter methylation. Cancer Res. 2005, 65, 4673–4682. [Google Scholar] [CrossRef]
- De Smedt, E.; Lui, H.; Maes, K.; De Veirman, K.; Menu, E.; Vanderkerken, K.; De Bruyne, E. The epigenome in multiple myeloma: Impact on tumor cell plasticity and drug response. Front. Oncol. 2018, 8, 566. [Google Scholar] [CrossRef]
- Iannetti, A.; Ledoux, A.C.; Tudhope, S.J.; Sellier, H.; Zhao, B.; Mowla, S.; Moore, A.; Hummerich, H.; Gewurz, B.E.; Cockell, S.J.; et al. Regulation of p53 and Rb Links the Alternative NF-κB Pathway to EZH2 Expression and Cell Senescence. PLoS Genet. 2014, 10, e1004642. [Google Scholar] [CrossRef]
- Rizq, O.; Mimura, N.; Oshima, M.; Saraya, A.; Koide, S.; Kato, Y.; Aoyama, K.; Nakajima-Takagi, Y.; Wang, C.; Chiba, T.; et al. Dual Inhibition of EZH2 and EZH1 Sensitizes PRC2-Dependent Tumors to Proteasome Inhibition. Clin. Cancer Res. 2017, 23, 4817–4830. [Google Scholar] [CrossRef]
- Guo, M.; Price, M.J.; Patterson, D.G.; Barwick, B.G.; Haines, R.R.; Kania, A.K.; Bradley, J.E.; Randall, T.D.; Boss, J.M.; Scharer, C.D. EZH2 Represses the B Cell Transcriptional Program and Regulates Antibody-Secreting Cell Metabolism and Antibody Production. J. Immunol. 2018, 200, 1039–1052. [Google Scholar] [CrossRef]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent Engagement of the Classical and Alternative NF-κB Pathways by Diverse Genetic Abnormalities in Multiple Myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, Y.N.; Glebov, O.K.; Zingone, A.; Keats, J.J.; Leif Bergsagel, P.; Michael Kuehl, W. Classical and/or alternative NF-κB pathway activation in multiple myeloma. Blood 2010, 115, 3541–3552. [Google Scholar] [CrossRef]
- Pires, B.R.B.; Silva, R.C.M.C.; Ferreira, G.M.; Abdelhay, E. NF-kappaB: Two sides of the same coin. Genes 2018, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Eluard, B.; Thieblemont, C.; Baud, V. NF-κB in the New Era of Cancer Therapy. Trends Cancer 2020, 6, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Dehghanifard, A.; Kaviani, S.; Abroun, S.; Mehdizadeh, M.; Saiedi, S.; Maali, A.; Ghaffari, S.; Azad, M. Various Signaling Pathways in Multiple Myeloma Cells and Effects of Treatment on These Pathways. Clin. Lymphoma Myeloma Leuk. 2018, 18, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-kB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Shih, V.F.S.; Tsui, R.; Caldwell, A.; Hoffmann, A. A single NFκB system for both canonical and non-canonical signaling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Li, Y.; Cheng, H.S.; Chng, W.J.; Tergaonkar, V.; Cleaver, J.E. Activation of mutant TERT promoter by RAS-ERK signaling is a key step in malignant progression of BRAF-mutant human melanomas. Proc. Natl. Acad. Sci. USA 2016, 113, 14402–14407. [Google Scholar] [CrossRef]
- Akincilar, S.C.; Low, K.C.; Liu, C.Y.; Yan, T.D.; Oji, A.; Ikawa, M.; Li, S.; Tergaonkar, V. Quantitative assessment of telomerase components in cancer cell lines. FEBS Lett. 2015, 589, 974–984. [Google Scholar] [CrossRef]
- Khattar, E.; Maung, K.Z.Y.; Chew, C.L.; Ghosh, A.; Mok, M.M.H.; Lee, P.; Zhang, J.; Chor, W.H.J.; Cildir, G.; Wang, C.Q.; et al. Rap1 regulates hematopoietic stem cell survival and affects oncogenesis and response to chemotherapy. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, M.B.; Li, Y.; Tergaonkar, V. Current insights to regulation and role of telomerase in human diseases. Antioxidants 2017, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Cildir, G.; Pant, H.; Lopez, A.F.; Tergaonkar, V. The transcriptional program, functional heterogeneity, and clinical targeting of mast cells. J. Exp. Med. 2017, 214, 2491–2506. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.S.; Nguyen, A.H.; Lee, J.H.; Li, F.; Singh, S.S.; Kumar, A.P.; Low, G.; Jha, S.; Tergaonkar, V.; Ahn, K.; et al. Negative regulation of signal transducer and activator of transcription-3 signalling cascade by lupeol inhibits growth and induces apoptosis in hepatocellular carcinoma cells. Br. J. Cancer 2014, 111, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the First Proteasome Inhibitor Anticancer Drug: Current Status and Future Perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef]
- Auphan, N.; DiDonato, J.A.; Rosette, C.; Helmberg, A.; Karin, M. Immunosuppression by glucocorticoids: Inhibition of NF-κB activity through induction of IκB synthesis. Science 1995, 270, 286–290. [Google Scholar] [CrossRef]
- Oh, K.-S.; Gottschalk, R.A.; Lounsbury, N.W.; Sun, J.; Dorrington, M.; Baek, S.; Sun, G.; Wang, Z.; Krauss, K.S.; Milner, J.D.; et al. Dual Roles for Ikaros in Regulation of Macrophage Chromatin State and Inflammatory Gene Expression. J. Immunol. 2018, 201, 757–771. [Google Scholar] [CrossRef]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef]
- Shannon, E.; Sandoval, F.; Greig, N.; Stagg, P. Lenalidomide alone or lenalidomide plus dexamethasone significantly inhibit IgG and IgM in vitro⋯A possible explanation for their mechanism of action in treating multiple myeloma. Int. Immunopharmacol. 2012, 12, 441–446. [Google Scholar] [CrossRef][Green Version]
- Moreaux, J.; Legouffe, E.; Jourdan, E.; Quittet, P.; Rème, T.; Lugagne, C.; Moine, P.; Rossi, J.-F.; Klein, B.; Tarte, K. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood 2004, 103, 3148–3157. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Li, X.F.; Breitkreutz, I.; Song, W.; Neri, P.; Catley, L.; Podar, K.; Hideshima, T.; Chauhan, D.; Raje, N.; et al. Role of B-cell-activating factor in adhesion and growth of human multiple myeloma cells in the bone marrow microenvironment. Cancer Res. 2006, 66, 6675–6682. [Google Scholar] [CrossRef] [PubMed]
- Mondello, P.; Cuzzocrea, S.; Navarra, M.; Mian, M. Bone marrow micro-environment is a crucial player for myelomagenesis and disease progression. Oncotarget 2017, 8, 20394–20409. [Google Scholar] [CrossRef] [PubMed]
- Pinzone, J.J.; Hall, B.M.; Thudi, N.K.; Vonau, M.; Qiang, Y.-W.; Rosol, T.J.; Shaughnessy, J.D. The role of Dickkopf-1 in bone development, homeostasis, and disease. Blood 2009, 113, 517–525. [Google Scholar] [CrossRef]
- Hata, H. Bone lesions and macrophage inflammatory protein-1 alpha (MIP-1α) in human multiple myeloma. Leuk. Lymphoma 2005, 46, 967–972. [Google Scholar] [CrossRef]
- Chng, W.J.; Glebov, O.; Bergsagel, P.L.; Kuehl, W.M. Genetic events in the pathogenesis of multiple myeloma. Best Pract. Res. Clin. Haematol. 2007, 20, 571–596. [Google Scholar] [CrossRef]
- de Weers, M.; Tai, Y.-T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.H.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef]
- Kaku, H.; Horikawa, K.; Obata, Y.; Kato, I.; Okamoto, H.; Sakaguchi, N.; Gerondakis, S.; Takatsu, K. NF-kB is required for CD38-mediated induction of Cg1 germline transcripts in murine B. lymphocytes. Int. Immunol. 2002, 14, 1055–1064. [Google Scholar] [CrossRef]
- Qian, Y.; Chen, C.; Ma, L.; Wang, Z.; Wang, L.-F.; Zuo, L.; Yang, Y.; Huang, X.; Jiang, M.; Wang, X.; et al. CD38 Deficiency Promotes Inflammatory Response Through Activating Sirt1/NF-κB-Mediated Inhibition of TLR2 Expression in Macrophages. Mediat. Inflamm. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Neri, P.; Kumar, S.; Fulciniti, M.T.; Vallet, S.; Chhetri, S.; Mukherjee, S.; Tai, Y.; Chauhan, D.; Tassone, P.; Venuta, S.; et al. Neutralizing B-cell-activating factor antibody improves survival and inhibits osteoclastogenesis in a severe combined immunodeficient human multiple myeloma model. Clin. Cancer Res. 2007, 13, 5903–5909. [Google Scholar] [CrossRef]
- Blimark, C.; Holmberg, E.; Mellqvist, U.H.; Landgren, O.; Björkholm, M.; Hultcrantz, M.; Kjellander, C.; Turesson, I.; Kristinsson, S.Y. Multiple myeloma and infections: A population-based study on 9253 multiple myeloma patients. Haematologica 2015, 100, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Nucci, M.; Anaissie, E. Infections in Patients with Multiple Myeloma in the Era of High-Dose Therapy and Novel Agents. Clin. Infect. Dis. 2009, 49, 1211–1225. [Google Scholar] [CrossRef] [PubMed]
- Teh, B.W.; Harrison, S.J.; Worth, L.J.; Thursky, K.A.; Slavin, M.A. Infection risk with immunomodulatory and proteasome inhibitor–based therapies across treatment phases for multiple myeloma: A systematic review and meta-analysis. Eur. J. Cancer 2016, 67, 21–37. [Google Scholar] [CrossRef]
- Li, J.; Li, Y.; Huang, B.; Zheng, D.; Chen, M.; Zhou, Z. Drug-Induced Modulation of T Lymphocytes as a Potential Mechanism of Susceptibility to Infections in Patients with Multiple Myeloma During Bortezomib Therapy. Cell Biochem. Biophys. 2014, 71, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Offidani, M.; Corvatta, L.; Bringhen, S.; Gentili, S.; Gay, F.; Maracci, L.; Boccadoro, M.; Leoni, P.; Palumbo, A. Infection Complications in 476 Patients with Newly Diagnosed Multiple Myeloma Treated with Lenalidomide or Bortezomib Combinations. Blood 2015, 126, 5365. [Google Scholar] [CrossRef]
- Prescott, J.; Cook, S. Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors. Cells 2018, 7, 115. [Google Scholar] [CrossRef]
- Goldstein, D.A. Denosumab for bone lesions in multiple myeloma—What is its value? Haematologica 2018, 103, 753–754. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-B, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef]
- Raje, N.S.; Moreau, P.; Terpos, E.; Benboubker, L.; Grząśko, N.; Holstein, S.A.; Oriol, A.; Huang, S.-Y.; Beksac, M.; Kuliczkowski, K.; et al. Phase 2 study of tabalumab, a human anti-B-cell activating factor antibody, with bortezomib and dexamethasone in patients with previously treated multiple myeloma. Br. J. Haematol. 2017, 176, 783–795. [Google Scholar] [CrossRef]
- Liu, J.; Sudom, A.; Min, X.; Cao, Z.; Gao, X.; Ayres, M.; Lee, F.; Cao, P.; Johnstone, S.; Plotnikova, O.; et al. Structure of the nuclear factor κB-inducing kinase (NIK) kinase domain reveals a constitutively active conformation. J. Biol. Chem. 2012, 287, 27326–27334. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, Y.N.; Brents, L.A.; Li, Z.; Bergsagel, L.P.; McGee, L.R.; Kuehl, M.W. Novel inhibitors are cytotoxic for myeloma cells with NFkB inducing kinase-dependent activation of NFkB. Oncotarget 2014, 5, 4554–4566. [Google Scholar] [CrossRef] [PubMed]
- Rushe, M.; Silvian, L.; Bixler, S.; Chen, L.L.; Cheung, A.; Bowes, S.; Cuervo, H.; Berkowitz, S.; Zheng, T.; Guckian, K.; et al. Structure of a NEMO/IKK-Associating Domain Reveals Architecture of the Interaction Site. Structure 2008, 16, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Habineza Ndikuyeze, G.; Gaurnier-Hausser, A.; Patel, R.; Baldwin, A.S.; May, M.J.; Flood, P.; Krick, E.; Propert, K.J.; Mason, N.J. A Phase I Clinical Trial of Systemically Delivered NEMO Binding Domain Peptide in Dogs with Spontaneous Activated B-Cell like Diffuse Large B-Cell Lymphoma. PLoS ONE 2014, 9, e95404. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Li, Y.; Bharath, S.R.; Ozturk, M.B.; Bowler, M.W.; Loo, B.Z.L.; Tergaonkar, V.; Song, H. Structural basis for reactivating the mutant TERT promoter by cooperative binding of p52 and ETS1. Nat. Commun. 2018, 9, 3138. [Google Scholar] [CrossRef]
- Greil, C.; Engelhardt, M.; Ihorst, G.; Schoeller, K.; Bertz, H.; Marks, R.; Zeiser, R.; Duyster, J.; Einsele, H.; Finke, J.; et al. Allogeneic transplantation of multiple myeloma patients may allow long-term survival in carefully selected patients with acceptable toxicity and preserved quality of life. Haematologica 2019, 104, 370–379. [Google Scholar] [CrossRef]
- Esma, F.; Salvini, M.; Troia, R.; Boccadoro, M.; Larocca, A.; Pautasso, C. Melphalan hydrochloride for the treatment of multiple myeloma. Expert Opin. Pharmacother. 2017, 18, 1127–1136. [Google Scholar] [CrossRef]
- Kyle, R.A.; Rajkumar, S.V. Multiple myeloma. Blood 2008, 111, 2962–2972. [Google Scholar] [CrossRef]
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S.; et al. Oral Selinexor–Dexamethasone for Triple-Class Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 381, 727–738. [Google Scholar] [CrossRef]
- Bahlis, N.J.; Sutherland, H.; White, D.; Sebag, M.; Lentzsch, S.; Kotb, R.; Venner, C.P.; Gasparetto, C.; Del Col, A.; Neri, P.; et al. Selinexor plus low-dose bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma. Blood 2018, 132, 2546–2554. [Google Scholar] [CrossRef]
- Van Rhee, F.; Szmania, S.M.; Dillon, M.; Van Abbema, A.M.; Li, X.; Stone, M.K.; Garg, T.K.; Shi, J.; Moreno-Bost, A.M.; Yun, R.; et al. Combinatorial efficacy of anti-CS1 monoclonal antibody elotuzumab (HuLuc63) and bortezomib against multiple myeloma. Mol. Cancer Ther. 2009, 8, 2616–2624. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.F.; Anderson, K.C.; Tai, Y.T. Targeting B cell maturation antigen (BCMA) in multiple myeloma: Potential uses of BCMA-based immunotherapy. Front. Immunol. 2018, 9, 1821. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.S.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Drent, E.; Themeli, M.; Poels, R.; De Jong-Korlaar, R.; Yuan, H.; De Bruijn, J.; Martens, A.C.; Zweegman, S.; Van De Donk, N.W.; Groen, R.; et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol. Ther. 2017, 25, 1946–1958. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; He, S.; Deng, Y.; Zhang, J.; Peng, Y.; Hughes, T.; Yi, L.; Kwon, C.-H.; Wang, Q.-E.; Devine, S.M.; et al. Genetic modification of T cells redirected toward CS1 enhances eradication of myeloma cells. Clin. Cancer Res. 2014, 20, 3989–4000. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, M.; Raje, N. Anti-BCMA CAR T-cell therapy in multiple myeloma: Can we do better? Leukemia 2020, 34, 21–34. [Google Scholar] [CrossRef]
- Jelinek, T.; Hajek, R. PD-1/PD-L1 inhibitors in multiple myeloma: The present and the future. Oncoimmunology 2016, 5, e1254856. [Google Scholar] [CrossRef]
- Gowrishankar, K.; Gunatilake, D.; Gallagher, S.J.; Tiffen, J.; Rizos, H.; Hersey, P. Inducible but Not Constitutive Expression of PD-L1 in Human Melanoma Cells Is Dependent on Activation of NF-κB. PLoS ONE 2015, 10, e0123410. [Google Scholar] [CrossRef]
- Blaquiere, N.; Castanedo, G.M.; Burch, J.D.; Berezhkovskiy, L.M.; Brightbill, H.; Brown, S.; Chan, C.; Chiang, P.C.; Crawford, J.J.; Dong, T.; et al. Scaffold-Hopping Approach to Discover Potent, Selective, and Efficacious Inhibitors of NF-ÎB Inducing Kinase. J. Med. Chem. 2018, 61, 6801–6813. [Google Scholar]
- Cheng, G.; Mei, X.B.; Yan, Y.; Chen, J.; Zhang, B.; Li, J.; Dong, X.; Lin, N.; Zhou, Y. Identification of new NIK inhibitors by discriminatory analysis-based molecular docking and biological evaluation. Arch. Pharm. (Weinheim) 2019, 352, e1800374. [Google Scholar] [CrossRef]
- Takakura, N.; Matsuda, M.; Khan, M.; Hiura, F.; Aoki, K.; Hirohashi, Y.; Mori, K.; Yasuda, H.; Hirata, M.; Kitamura, C.; et al. A novel inhibitor of NF-κB-inducing kinase prevents bone loss by inhibiting osteoclastic bone resorption in ovariectomized mice. Bone 2020, 135, 115316. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, X.; Su, M.-B.; Gao, L.-X.; Zhou, Y.-B.; Yuan, B.; Lyu, X.; Yan, Z.; Hu, C.; Zhang, H.; et al. Discovery of a Potent and Selective NF-κB-Inducing Kinase (NIK) Inhibitor That Has Anti-inflammatory Effects in Vitro and in Vivo. J. Med. Chem. 2020, 63, 4388–4407. [Google Scholar] [PubMed]
- Fulciniti, M.; Tassone, P.; Hideshima, T.; Vallet, S.; Nanjappa, P.; Ettenberg, S.A.; Shen, Z.; Patel, N.; Tai, Y.; Chauhan, D.; et al. Anti-DKK1 mAb (BHQ880) as a potential therapeutic agent for multiple myeloma. Blood 2009, 114, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Oliva, L.; Cascio, P.; Pengo, N.; Fontana, F.; Cerruti, F.; Orsi, A.; Pasqualetto, E.; Mezghrani, A.; Calbi, V.; et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009, 113, 3040–3049. [Google Scholar] [CrossRef]
- Scott, K.; Hayden, P.J.; Will, A.; Wheatley, K.; Coyne, I. Bortezomib for the treatment of multiple myeloma. Cochrane Database Syst. Rev. 2016, 4, CD010816. [Google Scholar] [CrossRef]
- Chari, A.; Richardson, P.G.; Romanus, D.; Dimopoulos, M.A.; Sonneveld, P.; Terpos, E.; Hajek, R.; Raju, A.; Palumbo, A.; Cain, L.E.; et al. Real-world outcomes and factors impacting treatment choice in relapsed and/or refractory multiple myeloma (RRMM): A comparison of VRd, KRd, and IRd. Expert Rev. Hematol. 2020, 13, 421–433. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Siegel, D.; White, D.J.; Boccia, R.; Iskander, K.S.; Yang, Z.; Kimball, A.S.; Mezzi, K.; Ludwig, H.; Niesvizky, R. Carfilzomib vs bortezomib in patients with multiple myeloma and renal failure: A subgroup analysis of ENDEAVOR. Blood 2019, 133, 147–155. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Goldschmidt, H.; Niesvizky, R.; Joshua, D.; Chng, W.-J.; Oriol, A.; Orlowski, R.Z.; Ludwig, H.; Facon, T.; Hájek, R.; et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): An interim overall survival analysis of an open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 1327–1337. [Google Scholar] [CrossRef]
- van de Donk, N.W. Carfilzomib versus bortezomib: No longer an ENDEAVOR. Lancet Oncol. 2017, 18, 1288–1290. [Google Scholar] [CrossRef]
- Siegel, D.S.; Dimopoulos, M.A.; Ludwig, H.; Facon, T.; Goldschmidt, H.; Jakubowiak, A.; San-Miguel, J.; Obreja, M.; Blaedel, J.; Stewart, A.K. Improvement in overall survival with carfilzomib, lenalidomide, and dexamethasone in patients with relapsed or refractory multiple myeloma. J. Clin. Oncol. 2018, 36, 728–734. [Google Scholar] [CrossRef]
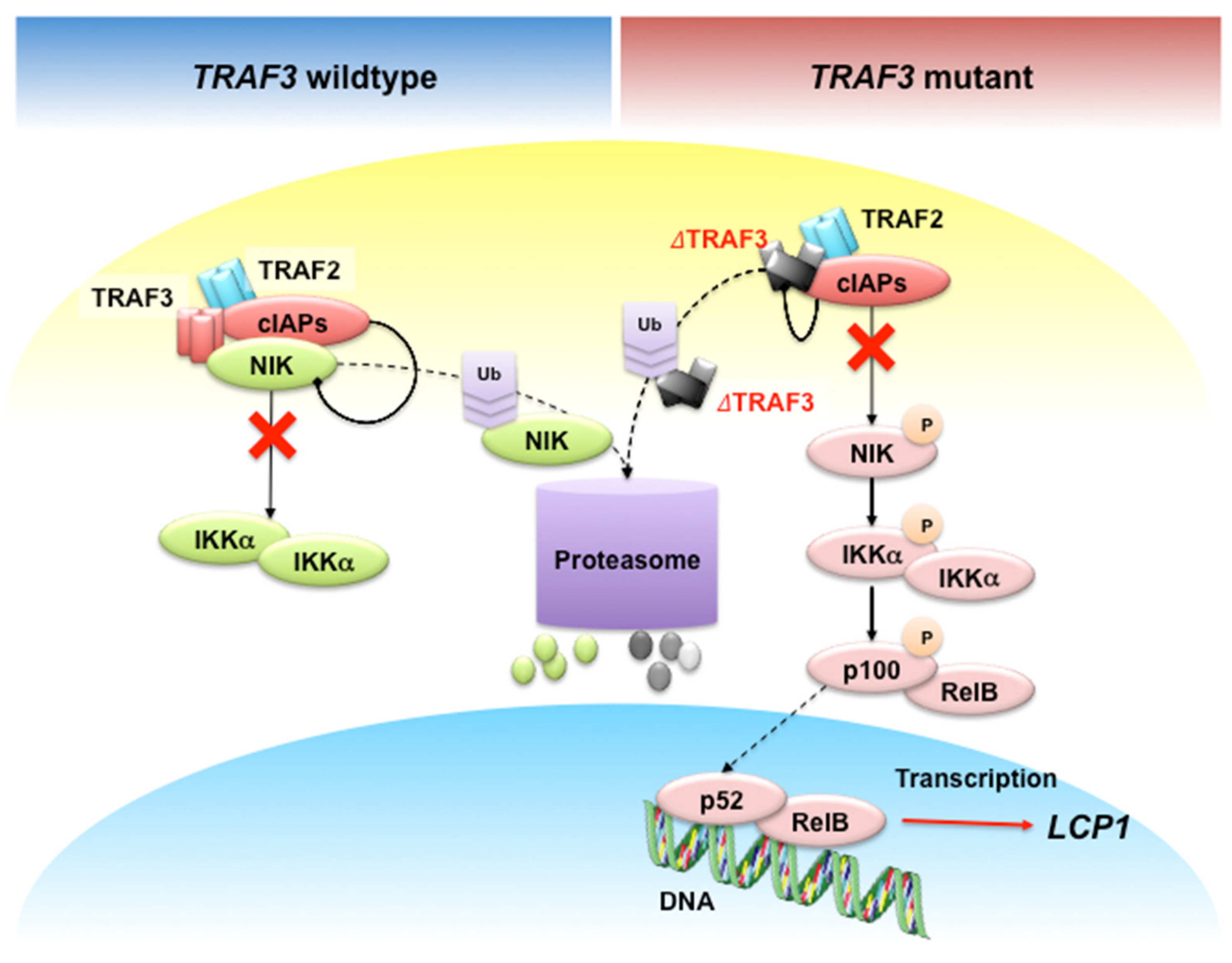
- Shin, E.M.; Neja, S.A.; Fidan, K.; Chua, J.Y.H.; Chung, T.; Bertin, N.; Tergaonkar, V.; Chng, W.; Ooi, M.G. Lymphocyte cytosolic protein 1 (LCP1) is a novel TRAF3 dysregulation biomarker with potential prognostic value in multiple myeloma. Genome Instab. Dis. 2020. [Google Scholar] [CrossRef]
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.C.; Mak, T.W.; et al. Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 2008, 9, 1371–1378. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Matsuzawa, A.; Zhang, W.Z.; Tseng, P.H.; Keats, J.J.; Wang, H.; Vignali, D.A.A.; Bergsagel, P.L.; Karin, M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat. Immunol. 2008, 9, 1364–1370. [Google Scholar] [CrossRef]
- Lin, W.W.; Yi, Z.; Stunz, L.L.; Maine, C.J.; Sherman, L.A.; Bishop, G.A. The adaptor protein TRAF3 inhibits interleukin-6 receptor signaling in B cells to limit plasma cell development. Sci. Signal. 2015, 8, ra88. [Google Scholar] [CrossRef] [PubMed]
- Mambetsariev, N.; Lin, W.W.; Wallis, A.M.; Stunz, L.L.; Bishop, G.A. TRAF3 deficiency promotes metabolic reprogramming in B cells. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mambetsarieva, N.; Lin, W.W.; Stunza, L.L.; Hansona, B.M.; Hildebranda, J.M.; Bishop, G.A. Nuclear TRAF3 is a negative regulator of CREB in B cells. Proc. Natl. Acad. Sci. USA 2016, 113, 1032–1037. [Google Scholar] [CrossRef]
- Xie, P.; Stunz, L.L.; Larison, K.D.; Yang, B.; Bishop, G.A. Tumor Necrosis Factor Receptor-Associated Factor 3 Is a Critical Regulator of B Cell Homeostasis in Secondary Lymphoid Organs. Immunity 2007, 27, 253–267. [Google Scholar] [CrossRef]
- Ma, T.; Sadashivaiah, K.; Chellaiah, M.A. Regulation of sealing ring formation by L-plastin and cortactin in osteoclasts. J. Biol. Chem. 2010, 285, 29911–29924. [Google Scholar] [CrossRef]
- Chellaiah, M.A.; Majumdar, S.; Aljohani, H. Peptidomimetic inhibitors of L-plastin reduce the resorptive activity of osteoclast but not the bone forming activity of osteoblasts in vitro. PLoS ONE 2018, 13, e0204209. [Google Scholar] [CrossRef]
- Tiedemann, K.; Sadvakassova, G.; Mikolajewicz, N.; Juhas, M.; Sabirova, Z.; Tabariès, S.; Gettemans, J.; Siegel, P.M.; Komarova, S.V. Exosomal Release of L-Plastin by Breast Cancer Cells Facilitates Metastatic Bone Osteolysis. Transl. Oncol. 2019, 12, 462–474. [Google Scholar] [CrossRef]
- Klemke, M.; Rafael, M.T.; Wabnitz, G.H.; Weschenfelder, T.; Konstandin, M.H.; Garbi, N.; Autschbach, F.; Hartschuh, W.; Samstag, Y. Phosphorylation of ectopically expressed L-plastin enhances invasiveness of human melanoma cells. Int. J. Cancer 2007, 120, 2590–2599. [Google Scholar] [CrossRef] [PubMed]
- Dun, M.D.; Chalkley, R.J.; Faulkner, S.; Keene, S.; Avery-Kiejda, K.A.; Scott, R.J.; Falkenby, L.S.; Cairns, M.J.; Larsen, M.R.; Bradshaw, L.A.; et al. Proteotranscriptomic profiling of 231-BR breast cancer cells: Identification of potential biomarkers and therapeutic targets for brain metastasis. Mol. Cell. Proteom. 2015, 14, 2316–2330. [Google Scholar] [CrossRef] [PubMed]
- Inaguma, S.; Riku, M.; Ito, H.; Tsunoda, T.; Ikeda, H.; Kasai, K. GLI1 orchestrates CXCR4/CXCR7 signaling to enhance migration and metastasis of breast cancer cells. Oncotarget 2015, 6, 33648–33657. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Kato, M.; Yoshikawa, T.; Chen, H.; Brown, E.J.; Masuho, Y.; Omata, M.; Seki, N. Differential expression of the L-plastin gene in human colorectal cancer progression and metastasis. Biochem. Biophys. Res. Commun. 2001, 289, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Foran, E.; McWilliam, P.; Kelleher, D.; Croke, D.T.; Long, A. The leukocyte protein L-plastin induces proliferation, invasion and loss of E-cadherin expression in colon cancer cells. Int. J. Cancer 2006, 118, 2098–2104. [Google Scholar] [CrossRef]
- Ning, Y.; Gerger, A.; Zhang, W.; Hanna, D.L.; Yang, D.; Winder, T.; Wakatsuki, T.; Labonte, M.J.; Stintzing, S.; Volz, N.; et al. Plastin polymorphisms predict gender- and stage-specific colon cancer recurrence after adjuvant chemotherapy. Mol. Cancer Ther. 2014, 13, 528–539. [Google Scholar] [CrossRef]
- Fang, Z.-Q.; Zang, W.-D.; Chen, R.; Ye, B.; Wang, X.; Yi, S.; Chen, W.; He, F.; Ye, G. Gene expression profile and enrichment pathways in different stages of bladder cancer. Genet. Mol. Res. 2013, 12, 1479–1489. [Google Scholar] [CrossRef]
- Kim, D.S.; Choi, Y.D.; Moon, M.; Kang, S.; Lim, J.B.; Kim, K.Y.; Park, K.M.; Cho, N.H. Composite three-marker assay for early detection of kidney cancer. Cancer Epidemiol. Biomark. Prev. 2013, 22, 390–398. [Google Scholar] [CrossRef]
- Dubovsky, J.A.; Chappell, D.L.; Harrington, B.K.; Agrawal, K.; Andritsos, L.A.; Flynn, J.M.; Jones, J.A.; Paulaitis, M.E.; Bolon, B.; Johnson, A.J.; et al. Lymphocyte cytosolic protein 1 is a chronic lymphocytic leukemia membrane-associated antigen critical to niche homing. Blood 2013, 122, 3308–3316. [Google Scholar] [CrossRef]
- Freudenmann, L.K.; Mayer, C.; Rodemann, H.P.; Dittmann, K. Reduced exosomal L-Plastin is responsible for radiation-induced bystander effect. Exp. Cell Res. 2019, 383, 111498. [Google Scholar] [CrossRef]
- Bosseler, M.; Marani, V.; Broukou, A.; Lequeux, A.; Kaoma, T.; Schlesser, V.; François, J.H.; Palissot, V.; Berchem, G.J.; Aouali, N.; et al. Inhibition of HIF1α-dependent upregulation of phospho-L-Plastin resensitizes multiple myeloma cells to frontline therapy. Int. J. Mol. Sci. 2018, 19, 1551. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Wadajkar, A.S.; Aljohani, H.; Reynolds, M.A.; Kim, A.J.; Chellaiah, M. Engineering of L-Plastin Peptide-Loaded Biodegradable Nanoparticles for Sustained Delivery and Suppression of Osteoclast Function In Vitro. Int. J. Cell Biol. 2019, 2019, 6943986. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, H.; Yasuda, T.; Aiba, Y.; Sanjo, H.; Hamadate, M.; Watarai, H.; Sakurai, H.; Kurosaki, T. PKCβ regulates BCR-mediated IKK activation by facilitating the interaction between TAK1 and CARMA1. J. Exp. Med. 2005, 202, 1423–1431. [Google Scholar] [CrossRef]
- Stadanlick, J.E.; Kaileh, M.; Karnell, F.G.; Scholz, J.L.; Miller, J.P.; Quinn, W.J.; Brezski, R.J.; Treml, L.S.; Jordan, K.A.; Monroe, J.G.; et al. Tonic B cell antigen receptor signals supply an NF-κB substrate for prosurvival BLyS signaling. Nat. Immunol. 2008, 9, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Shinzawa, M.; Konno, H.; Qin, J.; Akiyama, N.; Miyauchi, M.; Ohashi, H.; Miyamoto-Sato, H.; Yanagawa, H.; Akiyama, T.; Inoue, J.-i. Catalytic subunits of the phosphatase calcineurin interact with NF-κB-inducing kinase (NIK) and attenuate NIK-dependent gene expression. Sci. Rep. 2015, 5, 10758. [Google Scholar] [CrossRef] [PubMed]
- Wicker, L.S.; Boltz, R.C.; Matt, V.; Nichols, E.A.; Peterson, L.B.; Sigal, N.H. Suppression of B cell activation by cyclosporin A, FK506 and rapamycin. Eur. J. Immunol. 1990, 20, 2277–2283. [Google Scholar] [CrossRef]
- Tan, D.; Chng, W.J.; Chou, T.; Nawarawong, W.; Hwang, S.Y.; Chim, C.S.; Chen, W.; Durie, B.G.M.; Lee, J.H. Management of multiple myeloma in Asia: Resource-stratified guidelines. Lancet Oncol. 2013, 14, e571–e581. [Google Scholar] [CrossRef]
- Fonseca, R.; Bergsagel, P.L.; Drach, J.; Shaughnessy, J.; Gutierrez, N.; Stewart, A.K.; Morgan, G.; Van Ness, B.; Chesi, M.; Minvielle, S.; et al. International Myeloma Working Group molecular classification of multiple myeloma: Spotlight review. Leukemia 2009, 23, 2210–2221. [Google Scholar] [CrossRef]
- Varga, G.; Mikala, G.; Andrikovics, H.; Koszarska, M.; Balassa, K.; Ádám, E.; Kozma, A.; Tordai, A.; Masszi, T. NFKB1 -94ins/delATTG polymorphism is a novel prognostic marker in first line-treated multiple myeloma. Br. J. Haematol. 2015, 168, 679–688. [Google Scholar] [CrossRef]
- Kumar, S.; Kaufman, J.L.; Gasparetto, C.; Mikhael, J.; Vig, R.; Pegouri, B.; Benboubker, R.; Facon, T.; Amiot, M.; Moreau, P.; et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 2017, 130, 2401–2409. [Google Scholar] [CrossRef]
- Martinez-Garcia, E.; Popovic, R.; Min, D.J.; Sweet, S.M.M.; Thomas, P.M.; Zamdborg, L.; Heffner, A.; Will, C.; Lamy, L.; Staudt, L.M.; et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t (4;14) multiple myeloma cells. Blood 2011, 117, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Coussens, N.P.; Kales, S.C.; Henderson, M.J.; Lee, O.W.; Horiuchi, K.Y.; Wang, Y.; Chen, Q.; Kuznetsova, E.; Wu, J.; Chakka, S.; et al. High-throughput screening with nucleosome substrate identifies small-molecule inhibitors of the human histone lysine methyltransferase NSD2. J. Biol. Chem. 2018, 293, 13750–13765. [Google Scholar] [CrossRef]
- Brighton, T.A.; Khot, A.; Harrison, S.J.; Ghez, D.; Weiss, B.M.; Kirsch, A.; Magen, H.; Gironella, M.; Oriol, A.; Streetly, M.; et al. Randomized, double-blind, placebo-controlled, multicenter study of siltuximab in high-risk smoldering multiple myeloma. Clin. Cancer Res. 2019, 25, 3772–3775. [Google Scholar] [CrossRef]
- Jonsson, B.; Nilsson, K.; Nygren, P.; Larsson, R. SDZ PSC-833—A novel potent in vitro chemosensitizer in multiple myeloma. Anticancer Drugs 1992, 3, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Friedenberg, W.R.; Rue, M.; Blood, E.A.; Dalton, W.S.; Shustik, C.; Larson, R.A.; Sonneveld, P.; Greipp, P.R. Phase III study of PSC-833 (valspodar) in combination with vincristine, doxorubicin, and dexamethasone (valspodar/VAD) versus VAD alone in patients with recurring or refractory multiple myeloma (E1A95): A trial of the Eastern Cooperative Oncology Group. Cancer 2006, 106, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Fassmannová, D.; Sedlák, F.; Sedláček, J.; Špička, I.; Grantz Šašková, K. Nelfinavir Inhibits the TCF11/Nrf1-Mediated Proteasome Recovery Pathway in Multiple Myeloma. Cancers 2020, 12, 1065. [Google Scholar] [CrossRef]
- Reddi, D.M.; Lu, C.M.; Fedoriw, G.; Liu, Y.; Wang, F.F.; Ely, S.; Boswell, E.L.; Louissaint, A.; Arcasoy, M.O.; Goodman, B.K.; et al. Myeloid neoplasms secondary to plasma cell myeloma: An intrinsic predisposition or therapy-related phenomenon? A clinicopathologic study of 41 cases and correlation of cytogenetic features with treatment regimens. Am. J. Clin. Pathol. 2012, 138, 855–866. [Google Scholar] [CrossRef]
- Klimek, V.M. Recent advances in the management of therapy-related myelodysplastic syndromes and acute myeloid leukemia. Curr. Opin. Hematol. 2013, 20, 137–143. [Google Scholar] [CrossRef]
- Pemmaraju, N.; Shah, D.; Kantarjian, H.; Orlowski, R.Z.; Nogueras González, G.M.; Baladanayuthapani, V.; Jain, N.; Wagner, V.; Garcia-Manero, G.; Shah, J.; et al. Characteristics and outcomes of patients with multiple myeloma who develop therapy-related myelodysplastic syndrome, chronic myelomonocytic leukemia, or acute myeloid leukemia. Clin. Lymphoma Myeloma Leuk. 2015, 15, 110–114. [Google Scholar] [CrossRef]
- Fernández-Caballero, M.; Salmerón, D.; Dolores Chirlaque, M.; Chen-Liang, T.H.; Hurtado, A.M.; García Malo, M.D.; Ortuño, F.J.; Roldán, V.; Vicente, V.; Jerez, A.; et al. Increasing therapy-related myeloid neoplasms in multiple myeloma. Eur. J. Clin. Investig. 2018, 49, e13050. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Line of Therapy | Inflammation | Angiogenesis | DNA & RNA | Proteasome | Cell Death | Cell Cycle | Other Mechanism | Drug Target |
|---|---|---|---|---|---|---|---|---|---|
| Bortezomib | 1 | NF-κB | |||||||
| Dexamethasone | 1 | ||||||||
| Lenalidomide | 1 | VEGF, bFGF | |||||||
| Cyclophosphamide | 1 | DNA | |||||||
| Carfilzomib | 2 | Proteasome | |||||||
| Daratumumab | 2 | CD38 | |||||||
| Doxorubicin | 2 | Topoisomerase | |||||||
| Ixazomib | 2 | Proteasome | |||||||
| Melphalan | 2 | DNA | |||||||
| Prednisone | 2 | ||||||||
| Bendamustine | 3 | DNA | |||||||
| Cisplatin | 3 | DNA | |||||||
| Elotuzumab | 3 | CS1 | |||||||
| Etoposide | 3 | Topoisomerase | |||||||
| Panobinostat | 3 | HDAC | |||||||
| Pomalidomide | 3 | VEGF, bFGF | |||||||
| Thalidomide | 3 | VEGF, bFGF | |||||||
| Abatacept | CT | CD80, CD86 | |||||||
| Abemaciclib | CT | CDK4, CDK6 | |||||||
| Acalabrutinib | CT | BTK | |||||||
| ACP-319 | CT | PI3K | |||||||
| ALT-803 | CT | IL-15 | |||||||
| ASA | CT | COX-1/2 | |||||||
| Atezolizumab | CT | PD-L1 | |||||||
| Avelumab | CT | PD-L1 | |||||||
| Azacitadine | CT | DNA methylation | |||||||
| AZD5991 | CT | Mcl-1 | |||||||
| Binimetinib | CT | MEK-1/2 | |||||||
| Busulfan | CT | DNA | |||||||
| Carmustin | CT | DNA | |||||||
| CC-92480 | CT | CRBN | |||||||
| CCS1477 | CT | p300, CBP | |||||||
| Cetrelimab | CT | PD-1 | |||||||
| Clarithromycin | CT | Antibiotic | |||||||
| CLR131 | CT | ||||||||
| Cobimetinib | CT | MEK1 | |||||||
| CT-011 | CT | PD-1 | |||||||
| CYT-0851 | CT | RAD51 | |||||||
| Cytarabine | CT | DNA | |||||||
| Dabrafenib | CT | BRAF | |||||||
| Denosumab | CT | RANKL | |||||||
| Depsipeptide | CT | HDAC | |||||||
| Durvalumab | CT | PD-L1 | |||||||
| Enasidenib | CT | IDH2 | |||||||
| Encorafenib | CT | BRAF | |||||||
| Erdafitinib | CT | pan-FGFR | |||||||
| Fludarabin | CT | DNA | |||||||
| GBR1342 | CT | CD38, CD3 | |||||||
| Gemcitabine | CT | DNA | |||||||
| GSK2857916 | CT | BCMA | |||||||
| GSK3174998 | CT | OX40 | |||||||
| GSK3359609 | CT | ICOS | |||||||
| Idasanutlin | CT | MDM2 | |||||||
| Ipilimumab | CT | CTLA4 | |||||||
| Isatuximab | CT | CD38 | |||||||
| JNJ-42756493 | CT | pan-FGFR | |||||||
| Leflunomide | CT | PKC | |||||||
| Melflufen | CT | DNA | |||||||
| Metformin | CT | Complex I | |||||||
| Nelfinavir | CT | Antiviral, Akt | |||||||
| Nirogacestat | CT | γ-secretase | |||||||
| Nivolumab | CT | PD-1 | |||||||
| MP0250 | CT | VEGF, HGF | |||||||
| ONC201 | CT | ERK-1/2 | |||||||
| Osalmid | CT | ||||||||
| PD-L1 peptide | CT | PD-1 | |||||||
| Pembrolizumab | CT | PD-1 | |||||||
| Pralatrexate | CT | RFC-1 | |||||||
| Cemiplimab | CT | PD-1 | |||||||
| REGN5458 | CT | BCMA, CD3 | |||||||
| Ricolinostat | CT | HDAC6 | |||||||
| Rituximab | CT | CD20 | |||||||
| Romidepsin | CT | HDAC | |||||||
| Ruxolitinib | CT | JAK-1/2 | |||||||
| Selinexor | CT | Exportin | |||||||
| Siltuximab | CT | IL-6 | |||||||
| Sonidegib | CT | Smo | |||||||
| TAK-573 | CT | CD38 | |||||||
| TJ202 | CT | CD38 | |||||||
| Tositumomab | CT | CD20 | |||||||
| Trametinib | CT | MEK-1/2 | |||||||
| Venetoclax | CT | Bcl-2 | |||||||
| Vorinostat | CT | HDAC | |||||||
| Aspirin | NA | COX-1/2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, A.H.-H.; Shin, E.M.; Tergaonkar, V.; Chng, W.-J. Targeting NF-κB Signaling for Multiple Myeloma. Cancers 2020, 12, 2203. https://doi.org/10.3390/cancers12082203
Wong AH-H, Shin EM, Tergaonkar V, Chng W-J. Targeting NF-κB Signaling for Multiple Myeloma. Cancers. 2020; 12(8):2203. https://doi.org/10.3390/cancers12082203
Chicago/Turabian StyleWong, Ada Hang-Heng, Eun Myoung Shin, Vinay Tergaonkar, and Wee-Joo Chng. 2020. "Targeting NF-κB Signaling for Multiple Myeloma" Cancers 12, no. 8: 2203. https://doi.org/10.3390/cancers12082203
APA StyleWong, A. H.-H., Shin, E. M., Tergaonkar, V., & Chng, W.-J. (2020). Targeting NF-κB Signaling for Multiple Myeloma. Cancers, 12(8), 2203. https://doi.org/10.3390/cancers12082203