Next Generation Sequencing in MPNs. Lessons from the Past and Prospects for Use as Predictors of Prognosis and Treatment Responses


Abstract
1. Introduction
2. Genomics in MPNs
2.1. NGS Analysis of Somatic Gene Mutations
2.2. Interaction of Somatic Gene Mutations Refined by NGS Analysis
3. NGS in the Diagnosis and Prognosis of MPNs
3.1. Development and Progression of Somatic Gene Mutations in Patients with MPNs
3.1.1. From CHIP to More Advanced Stages of MPNs
3.1.2. Implication of Mutation Order on MPN Phenotype
3.2. NGS in the Diagnostic Decision-Making in MPNs
3.3. Implication of Somatic Gene Mutations on Prognosis, Risk Stratification and Outcome Revealed by NGS
3.3.1. High Molecular Risk Mutations
3.3.2. Other Groups of Adverse Mutations
3.3.3. Fibrotic Progression
3.3.4. TET2 Mutations and Order of Mutations
3.3.5. TP53 and PPM1D Mutations
3.3.6. Prognostic Genomic Classification Models
3.3.7. Additional Mutations in Relation to Sex
3.3.8. Co-Occurring Non-Driver Somatic Mutations in Prognostication
3.3.9. Impact of the Number of Mutations on Prognosis and Outcome
3.3.10. NGS and Transplantation Outcome
4. Use of NGS to Decipher the Mutational Landscape in MPNs in Response to Therapy
4.1. Hydroxyurea
4.2. Interferon Alpha
4.3. Ruxolitinib
4.4. Combination Therapy with Interferon Alpha and Ruxolitinib
4.5. Using NGS in Early Treatment Decisions
4.6. Monitoring of Disease by NGS
5. Conclusions
Funding
Conflicts of Interest
References
- Spivak, J.L. Myeloproliferative Neoplasms. N. Engl. J. Med. 2017, 376, 2168–2181. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: Is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 2012, 119, 3219–3225. [Google Scholar] [CrossRef] [PubMed]
- Lussana, F.; Rambaldi, A. Inflammation and myeloproliferative neoplasms. J. Autoimmun. 2017, 85, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Hermouet, S.; Bigot-Corbel, E.; Gardie, B. Pathogenesis of Myeloproliferative Neoplasms: Role and Mechanisms of Chronic Inflammation. Mediators Inflamm. 2015, 2015, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, H.; Farkas, D.K.; Christiansen, C.F.; Hasselbalch, H.C.; Sørensen, H.T. Chronic myeloproliferative neoplasms and subsequent cancer risk: A Danish population-based cohort study. Blood 2011, 118, 6515–6520. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Perspectives on the increased risk of second cancer in patients with essential thrombocythemia, polycythemia vera and myelofibrosis. Eur. J. Haematol. 2015, 94, 96–98. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev. 2013, 24, 133–145. [Google Scholar] [CrossRef]
- Øbro, N.F.; Grinfeld, J.; Belmonte, M.; Irvine, M.; Shepherd, M.; Rao, T.N.; Karow, A.; Riedel, L.M.; Harris, O.; Baxter, E.J.; et al. Longitudinal Cytokine Profiling Identifies GRO-α and EGF as Potential Biomarkers of Disease Progression in Essential Thrombocythemia. HemaSphere 2020, 4. [Google Scholar] [CrossRef]
- Campbell, P.J.; Green, A.R. The myeloproliferative disorders. N. Engl. J. Med. 2006, 355, 2452–2466. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet (Lond. Engl.) 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Levine, R.L.; Belisle, C.; Wadleigh, M.; Zahrieh, D.; Lee, S.; Chagnon, P.; Gilliland, D.G.; Busque, L. X-inactivation-based clonality analysis and quantitative JAK2V617F assessment reveal a strong association between clonality and JAK2V617F in PV but not ET/MMM, and identifies a subset of JAK2V617F-negative ET and MMM patients with clonal hematopoiesis. Blood 2006, 107, 4139–4141. [Google Scholar] [CrossRef] [PubMed]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Pancrazzi, A.; Guerini, V.; Barosi, G.; Ruggeri, M.; Specchia, G.; Lo-Coco, F.; Delaini, F.; et al. Characteristics and clinical correlates of MPL 515W>L/K mutation in essential thrombocythemia. Blood 2008, 112, 844–847. [Google Scholar] [CrossRef]
- Chachoua, I.; Pecquet, C.; El-Khoury, M.; Nivarthi, H.; Albu, R.-I.; Marty, C.; Gryshkova, V.; Defour, J.-P.; Vertenoeil, G.; Ngo, A.; et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 2016, 127, 1325–1335. [Google Scholar] [CrossRef]
- Beer, P.A.; Campbell, P.J.; Scott, L.M.; Bench, A.J.; Erber, W.N.; Bareford, D.; Wilkins, B.S.; Reilly, J.T.; Hasselbalch, H.C.; Bowman, R.; et al. MPL mutations in myeloproliferative disorders: Analysis of the PT-1 cohort. Blood 2008, 112, 141–149. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Larsen, T.S.; Møller, M.B.; de Stricker, K.; Nørgaard, P.; Samuelsson, J.; Marcher, C.; Andersen, M.T.; Bjerrum, O.W.; Hasselbalch, H.C. Minimal residual disease and normalization of the bone marrow after long-term treatment with alpha-interferon2b in polycythemia vera. A report on molecular response patterns in seven patients in sustained complete hematological remission. Hematology 2009, 14, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.S.; Bjerrum, O.W.; Pallisgaard, N.; Andersen, M.T.; Møller, M.B.; Hasselbalch, H.C. Sustained major molecular response on interferon alpha-2b in two patients with polycythemia vera. Ann. Hematol. 2008, 87, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.N.; Vannucchi, A.M.; Kiladjian, J.-J.; Al-Ali, H.K.; Gisslinger, H.; Knoops, L.; Cervantes, F.; Jones, M.M.; Sun, K.; McQuitty, M.; et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 2016, 30, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef]
- Kiladjian, J.-J.; Cassinat, B.; Chevret, S.; Turlure, P.; Cambier, N.; Roussel, M.; Bellucci, S.; Grandchamp, B.; Chomienne, C.; Fenaux, P. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008, 112, 3065–3072. [Google Scholar] [CrossRef]
- Silver, R.T.; Kiladjian, J.-J.; Hasselbalch, H.C. Interferon and the treatment of polycythemia vera, essential thrombocythemia and myelofibrosis. Expert Rev. Hematol. 2013, 6, 49–58. [Google Scholar] [CrossRef]
- Kiladjian, J.; Cassinat, B.; Turlure, P.; Cambier, N.; Roussel, M.; Bellucci, S.; Menot, M.; Massonnet, G.; Dutel, J.; Ghomari, K.; et al. High molecular response rate of polycythemia vera patients treated with pegylated interferon alpha–2a. Blood 2006, 108, 2037–2041. [Google Scholar] [CrossRef]
- Bose, P.; Verstovsek, S. Updates in the management of polycythemia vera and essential thrombocythemia. Ther. Adv. Hematol. 2019, 10, 2040620719870052. [Google Scholar] [CrossRef]
- Kiladjian, J.-J.; Zachee, P.; Hino, M.; Pane, F.; Masszi, T.; Harrison, C.N.; Mesa, R.; Miller, C.B.; Passamonti, F.; Durrant, S.; et al. Long-term efficacy and safety of ruxolitinib versus best available therapy in polycythaemia vera (RESPONSE): 5-year follow up of a phase 3 study. Lancet. Haematol. 2020, 7, e226–e237. [Google Scholar] [CrossRef]
- Gisslinger, H.; Klade, C.; Georgiev, P.; Krochmalczyk, D.; Gercheva-Kyuchukova, L.; Egyed, M.; Rossiev, V.; Dulicek, P.; Illes, A.; Pylypenko, H.; et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): A randomised, non-inferiority, phase 3 trial and its extension study. Lancet. Haematol. 2020, 7, e196–e208. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.W.N.; Miller, C.B.; Silver, R.T.; Talpaz, M.; et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J. Hematol. Oncol. 2017, 10, 55. [Google Scholar] [CrossRef]
- Knudsen, T.A.; Hansen, D.L.; Ocias, L.F.; Bjerrum, O.W.; Brabrand, M.; El Fassi, D.; Frederiksen, M.; Kjær, L.; Kristensen, T.K.; Kruse, T.A.; et al. Long-term efficacy and safety of recombinant interferon alpha-2 vs. hydroxyures in polycythemia vera: Preliminary results from the three-year analysis of the DALIAH trial - a randomized controlled phase III clinical trial. Blood 2018, 132. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. A new era for IFN-α in the treatment of Philadelphia-negative chronic myeloproliferative neoplasms. Expert Rev. Hematol. 2011, 4, 637–655. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C.; Holmstrom, M.O. Perspectives on interferon-alpha in the treatment of polycythemia vera and related myeloproliferative neoplasms: Minimal residual disease and cure? Semin. Immunopathol. 2019, 41, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C.; Silver, R.T. Interferon in polycythemia vera and related neoplasms. Can it become the treatment of choice without a randomized trial? Expert Rev. Hem 2015, 8, 1–7. [Google Scholar] [CrossRef]
- Pedersen, R.K.; Andersen, M.; Knudsen, T.A.; Sajid, Z.; Gudmand-Hoeyer, J.; Dam, M.J.B.; Skov, V.; Kjaer, L.; Ellervik, C.; Larsen, T.S.; et al. Data-driven analysis of JAK2V617F kinetics during interferon-alpha2 treatment of patients with polycythemia vera and related neoplasms. Cancer Med. 2020, 9, 2039–2051. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couedic, J.-P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- Miklossy, G.; Hilliard, T.S.; Turkson, J. Therapeutic modulators of STAT signalling for human diseases. Nat. Rev. Drug Discov. 2013, 12, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.P.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Pietra, D.; Li, S.; Brisci, A.; Passamonti, F.; Rumi, E.; Theocharides, A.; Ferrari, M.; Gisslinger, H.; Kralovics, R.; Cremonesi, L.; et al. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood 2008, 111, 1686–1689. [Google Scholar] [CrossRef]
- Pardanani, A.; Guglielmelli, P.; Lasho, T.L.; Pancrazzi, A.; Finke, C.M.; Vannucchi, A.M.; Tefferi, A. Primary myelofibrosis with or without mutant MPL: Comparison of survival and clinical features involving 603 patients. Leukemia 2011, 25, 1834–1839. [Google Scholar] [CrossRef][Green Version]
- Schnittger, S.; Bacher, U.; Eder, C.; Dicker, F.; Alpermann, T.; Grossmann, V.; Kohlmann, A.; Kern, W.; Haferlach, C.; Haferlach, T. Molecular analyses of 15,542 patients with suspected BCR-ABL1-negative myeloproliferative disorders allow to develop a stepwise diagnostic workflow. Haematologica 2012, 97, 1582–1585. [Google Scholar] [CrossRef]
- Bridgford, J.L.; Lee, S.M.; Lee, C.M.M.; Guglielmelli, P.; Rumi, E.; Pietra, D.; Wilcox, S.; Chhabra, Y.; Rubin, A.F.; Cazzola, M.; et al. Novel drivers and modifiers of MPL-dependent oncogenic transformation identified by deep mutational scanning. Blood 2020, 135, 287–292. [Google Scholar] [CrossRef]
- Pietra, D.; Rumi, E.; Ferretti, V.V.; Di Buduo, C.A.; Milanesi, C.; Cavalloni, C.; Sant’Antonio, E.; Abbonante, V.; Moccia, F.; Casetti, I.C.; et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016, 30, 431–438. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Green, A.R. Molecular determinants of pathogenesis and clinical phenotype in myeloproliferative neoplasms. Haematologica 2017, 102, 7–17. [Google Scholar] [CrossRef]
- Patel, U.; Luthra, R.; Medeiros, L.J.; Patel, K.P. Diagnostic, Prognostic, and Predictive Utility of Recurrent Somatic Mutations in Myeloid Neoplasms. Clin. Lymphoma, Myeloma Leuk. 2017, 17, S62–S74. [Google Scholar] [CrossRef]
- Oz Puyan, F.; Alkan, S. The Progress of Next Generation Sequencing in the Assessment of Myeloid Malignancies. Balkan Med. J. 2019, 36, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Skoda, R.C.; Duek, A.; Grisouard, J. Pathogenesis of myeloproliferative neoplasms. Exp. Hematol. 2015, 43, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Kuo, F.C.; Dong, F. Next-generation sequencing-based panel testing for myeloid neoplasms. Curr. Hematol. Malig. Rep. 2015, 10, 104–111. [Google Scholar] [CrossRef]
- Murati, A.; Brecqueville, M.; Devillier, R.; Mozziconacci, M.-J.; Gelsi-Boyer, V.; Birnbaum, D. Myeloid malignancies: Mutations, models and management. BMC Cancer 2012, 12, 304. [Google Scholar] [CrossRef]
- Matynia, A.P.; Szankasi, P.; Shen, W.; Kelley, T.W. Molecular genetic biomarkers in myeloid malignancies. Arch. Pathol. Lab. Med. 2015, 139, 594–601. [Google Scholar] [CrossRef]
- Hsu, J.I.; Dayaram, T.; Tovy, A.; De Braekeleer, E.; Jeong, M.; Wang, F.; Zhang, J.; Heffernan, T.P.; Gera, S.; Kovacs, J.J.; et al. PPM1D Mutations Drive Clonal Hematopoiesis in Response to Cytotoxic Chemotherapy. Cell Stem Cell 2018, 23, 700–713. [Google Scholar] [CrossRef]
- Coltro, G.; Rotunno, G.; Mannelli, L.; Mannarelli, C.; Fiaccabrino, S.; Romagnoli, S.; Sant’Antonio, E.; Guglielmelli, P.; Vannucchi, A.M. RAS/MAPK Pathway Mutations are Associated with Adverse Survival Outcomes and may Predict Resistance to JAK Inhibitors in Myelofibrosis. EHA Abstr. 2020. [Google Scholar]
- Delic, S.; Rose, D.; Kern, W.; Nadarajah, N.; Haferlach, C.; Haferlach, T.; Meggendorfer, M. Application of an NGS-based 28-gene panel in myeloproliferative neoplasms reveals distinct mutation patterns in essential thrombocythaemia, primary myelofibrosis and polycythaemia vera. Br. J. Haematol. 2016, 175, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016, 1, 105–111. [Google Scholar] [CrossRef]
- Wanquet, A.; Courtier, F.; Guille, A.; Carbuccia, N.; Garnier, S.; Adelaide, J.; Gelsi-Boyer, V.; Mozziconacci, M.J.; Rey, J.; Vey, N.; et al. Mutation patterns in essential thrombocythemia, polycythemia vera and secondary myelofibrosis. Leuk. Lymphoma 2019, 60, 1289–1293. [Google Scholar] [CrossRef]
- Courtier, F.; Garnier, S.; Carbuccia, N.; Guille, A.; Adelaide, J.; Chaffanet, M.; Hirsch, P.; Paz, D.L.; Slama, B.; Vey, N.; et al. Targeted molecular characterization shows differences between primary and secondary myelofibrosis. Genes. Chromosomes Cancer 2020, 59, 30–39. [Google Scholar] [CrossRef]
- Andreasson, B.; Pettersson, H.; Wasslavik, C.; Johansson, P.; Palmqvist, L.; Asp, J. ASXL1 mutations, previous vascular complications and age at diagnosis predict survival in 85 WHO-defined polycythaemia vera patients. Br. J. Haematol. 2020, 189, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.P.; Newberry, K.J.; Luthra, R.; Jabbour, E.; Pierce, S.; Cortes, J.; Singh, R.; Mehrotra, M.; Routbort, M.J.; Luthra, M.; et al. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood 2015, 126, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Alduaij, W.; McNamara, C.J.; Schuh, A.; Arruda, A.; Sukhai, M.; Kanwar, N.; Thomas, M.; Spiegel, J.; Kennedy, J.A.; Stockley, T.; et al. Clinical Utility of Next-generation Sequencing in the Management of Myeloproliferative Neoplasms: A Single-Center Experience. HemaSphere 2018, 4, e44. [Google Scholar] [CrossRef]
- Agarwal, R.; Blombery, P.; McBean, M.; Jones, K.; Fellowes, A.; Doig, K.; Forsyth, C.; Westerman, D.A. Clinicopathological differences exist between CALR- and JAK2-mutated myeloproliferative neoplasms despite a similar molecular landscape: Data from targeted next-generation sequencing in the diagnostic laboratory. Ann. Hematol. 2017, 96, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Rotunno, G.; Pacilli, A.; Artusi, V.; Rumi, E.; Maffioli, M.; Delaini, F.; Brogi, G.; Fanelli, T.; Pancrazzi, A.; Pietra, D.; et al. Epidemiology and clinical relevance of mutations in postpolycythemia vera and postessential thrombocythemia myelofibrosis: A study on 359 patients of the AGIMM group. Am. J. Hematol. 2016, 91, 681–686. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients With Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef]
- Boiocchi, L.; Hasserjian, R.P.; Pozdnyakova, O.; Wong, W.J.; Lennerz, J.K.; Le, L.P.; Dias-Santagata, D.; Iafrate, A.J.; Hobbs, G.S.; Nardi, V. Clinicopathological and molecular features of SF3B1-mutated myeloproliferative neoplasms. Hum. Pathol. 2019, 86, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pronier, E.; Delhommeau, F. Role of TET2 mutations in myeloproliferative neoplasms. Curr. Hematol. Malig. Rep. 2012, 7, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Marneth, A.E.; Mullally, A. The Molecular Genetics of Myeloproliferative Neoplasms. Cold Spring Harb. Perspect. Med. 2020, 10, a034876. [Google Scholar] [CrossRef] [PubMed]
- Langabeer, S.E.; Andrikovics, H.; Asp, J.; Bellosillo, B.; Carillo, S.; Haslam, K.; Kjaer, L.; Lippert, E.; Mansier, O.; Oppliger Leibundgut, E.; et al. Molecular diagnostics of myeloproliferative neoplasms. Eur. J. Haematol. 2015, 95, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Angona, A.; Fernández-Rodríguez, C.; Alvarez-Larrán, A.; Camacho, L.; Longarón, R.; Torres, E.; Pairet, S.; Besses, C.; Bellosillo, B. Molecular characterisation of triple negative essential thrombocythaemia patients by platelet analysis and targeted sequencing. Blood Cancer J. 2016, 6, e463. [Google Scholar] [CrossRef]
- Inoue, D.; Matsumoto, M.; Nagase, R.; Saika, M.; Fujino, T.; Nakayama, K.I.; Kitamura, T. Truncation mutants of ASXL1 observed in myeloid malignancies are expressed at detectable protein levels. Exp. Hematol. 2016, 44, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Verger, E.; Cassinat, B.; Chauveau, A.; Dosquet, C.; Giraudier, S.; Schlageter, M.H.; Ianotto, J.C.; Yassin, M.A.; Al-Dewik, N.; Carillo, S.; et al. Clinical and molecular response to interferon-α therapy in essential thrombocythemia patients with CALR mutations. Blood 2015, 126, 2585–2591. [Google Scholar] [CrossRef] [PubMed]
- Holz-Schietinger, C.; Matje, D.M.; Reich, N.O. Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J. Biol. Chem. 2012, 287, 30941–30951. [Google Scholar] [CrossRef]
- Cottin, L.; Riou, J.; Orvain, C.; Ianotto, J.C.; Boyer, F.; Renard, M.; Truchan-Graczyk, M.; Murati, A.; Jouanneau-Courville, R.; Allangba, O.; et al. Sequential mutational evaluation of CALR -mutated myeloproliferative neoplasms with thrombocytosis reveals an association between CALR allele burden evolution and disease progression. Br. J. Haematol. 2020, 188, 935–944. [Google Scholar] [CrossRef]
- Khan, S.N.; Jankowska, A.M.; Mahfouz, R.; Dunbar, A.J.; Sugimoto, Y.; Hosono, N.; Hu, Z.; Cheriyath, V.; Vatolin, S.; Przychodzen, B.; et al. Multiple mechanisms deregulate EZH2 and histone H3 lysine 27 epigenetic changes in myeloid malignancies. Leukemia 2013, 27, 1301–1309. [Google Scholar] [CrossRef]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef]
- Shirai, C.L.; Ley, J.N.; White, B.S.; Kim, S.; Tibbitts, J.; Shao, J.; Ndonwi, M.; Wadugu, B.; Duncavage, E.J.; Okeyo-Owuor, T.; et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell 2015, 27, 631–643. [Google Scholar] [CrossRef]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.-W.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.-B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, J.Y.; McNamara, C.; Kennedy, J.A.; Panzarella, T.; Arruda, A.; Stockley, T.; Sukhai, M.; Thomas, M.; Bartoszko, J.; Ho, J.; et al. Impact of genomic alterations on outcomes in myelofibrosis patients undergoing JAK1/2 inhibitor therapy. Blood Adv. 2017, 1, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Szuber, N.; Lasho, T.L.; Finke, C.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Gangat, N.; Tefferi, A. Determinants of long-term outcome in type 1 calreticulin-mutated myelofibrosis. Leukemia 2019, 33, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Bartels, S.; Faisal, M.; Büsche, G.; Schlue, J.; Hasemeier, B.; Schipper, E.; Vogtmann, J.; Westphal, L.; Lehmann, U.; Kreipe, H. Mutations associated with age-related clonal hematopoiesis in PMF patients with rapid progression to myelofibrosis. Leukemia 2020, 34, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Hussaini, M.; Zhang, H.; Shao, H.; Qin, D.; Zhang, X.; Ma, Z.; Hussnain Naqvi, S.M.; Zhang, L.; Moscinski, L.C. Comparison of the Mutational Profiles of Primary Myelofibrosis, Polycythemia Vera, and Essential Thrombocytosis. Am. J. Clin. Pathol. 2017, 147, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Luque Paz, D.; Chauveau, A.; Boyer, F.; Buors, C.; Samaison, L.; Cottin, L.; Seegers, V.; Ferec, C.; Le Marechal, C.; Gueguen, P.; et al. Sequential analysis of 18 genes in polycythemia vera and essential thrombocythemia reveals an association between mutational status and clinical outcome. Genes. Chromosomes Cancer 2017, 56, 354–362. [Google Scholar] [CrossRef]
- Byun, J.M.; Song, S.; Koh, Y.; Yoon, S.-S.; Kim, D. The Temporal Sequence and the Differences in Somatic Mutation Acquisition Determines Clinical Behaviors of JAK2-Positive Myeloproliferative Neoplasms. Anticancer Res. 2019, 39, 6273–6282. [Google Scholar] [CrossRef]
- Gill, H.; Ip, H.W.; Yim, R.; Tang, W.F.; Pang, H.H.; Lee, P.; Leung, G.M.K.; Li, J.; Tang, K.; So, J.C.C.; et al. Next-generation sequencing with a 54-gene panel identified unique mutational profile and prognostic markers in Chinese patients with myelofibrosis. Ann. Hematol. 2019, 98, 869–879. [Google Scholar] [CrossRef]
- Bartels, S.; Faisal, M.; Busche, G.; Schlue, J.; Kreipe, H.; Lehmann, U. Fibrotic progression in Polycythemia vera is associated with early concomitant driver-mutations besides JAK2. Leukemia 2018, 32, 556–558. [Google Scholar] [CrossRef]
- Stevens, E.A.; Jenkins, I.C.; Beppu, L.W.; Zhang, Q.; Salit, R.; Loeb, K.R.; Deeg, H.J.; Radich, J.P. Targeted Sequencing Improves DIPSS-Plus Prognostic Scoring in Myelofibrosis Patients Undergoing Allogeneic Transplant. Biol. Blood Marrow Transplant. 2020, 26, 1371–1374. [Google Scholar] [CrossRef]
- Stengel, A.; Jeromin, S.; Haferlach, T.; Meggendorfer, M.; Kern, W.; Haferlach, C. Detection and characterization of homozygosity of mutated CALR by copy neutral loss of heterozygosity in myeloproliferative neoplasms among cases with high CALR mutation loads or with progressive disease. Haematologica 2019, 104, e187–e190. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Functional and cancer genomics of ASXL family members. Br. J. Cancer 2013, 109, 299–306. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Biamonte, F.; Score, J.; Hidalgo-Curtis, C.; Cervantes, F.; Maffioli, M.; Fanelli, T.; Ernst, T.; Winkelman, N.; Jones, A.V.; et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood 2011, 118, 5227–5234. [Google Scholar] [CrossRef] [PubMed]
- Gelsi-Boyer, V.; Brecqueville, M.; Devillier, R.; Murati, A.; Mozziconacci, M.-J.; Birnbaum, D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J. Hematol. Oncol. 2012, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Lasho, T.L.; Pardanani, A.; McClure, R.F.; Mesa, R.A.; Levine, R.L.; Gary Gilliland, D.; Tefferi, A. Concurrent MPL515 and JAK2V617F mutations in myelofibrosis: Chronology of clonal emergence and changes in mutant allele burden over time. Br. J. Haematol. 2006, 135, 683–687. [Google Scholar] [CrossRef]
- Nussenzveig, R.H.; Pham, H.T.; Perkins, S.L.; Prchal, J.T.; Agarwal, A.M.; Salama, M.E. Increased frequency of co-existing JAK2 exon-12 or MPL exon-10 mutations in patients with low JAK2(V617F) allelic burden. Leuk. Lymphoma 2016, 57, 1429–1435. [Google Scholar] [CrossRef]
- Pardanani, A.; Lasho, T.L.; Finke, C.M.; Tefferi, A. Infrequent occurrence of MPL exon 10 mutations in polycythemia vera and post-polycythemia vera myelofibrosis. Am. J. Hematol. 2011, 86, 701–702. [Google Scholar] [CrossRef]
- Usseglio, F.; Beaufils, N.; Calleja, A.; Raynaud, S.; Gabert, J. Detection of CALR and MPL Mutations in Low Allelic Burden JAK2 V617F Essential Thrombocythemia. J. Mol. Diagn. 2017, 19, 92–98. [Google Scholar] [CrossRef]
- Holmström, M.O.; Novotny, G.W.; Petersen, J.; Aaboe-Jørgensen, M.; Hasselbalch, H.C.; Andersen, M.H.; Nielsen, S.L.; El Fassi, D.; Schöllkopf, C. Progression of JAK2-mutant polycythemia vera to CALR-mutant myelofibrosis severely impacts on disease phenotype and response to therapy. Leuk. Lymphoma 2019, 60, 3296–3299. [Google Scholar] [CrossRef]
- Beucher, A.; Dib, M.; Orvain, C.; Bouvier, A.; Jouanneau-Courville, R.; Dobo, I.; Cottin, L.; Guardiola, P.; Rousselet, M.-C.; Blanchet, O.; et al. Next generation sequencing redefines a triple negative essential thrombocythaemia as double-positive with rare mutations on JAK2 V617 and MPL W515 hotspots. Br. J. Haematol. 2019, 186, 785–788. [Google Scholar] [CrossRef]
- Pardanani, A.; Lasho, T.; Finke, C.; Oh, S.T.; Gotlib, J.; Tefferi, A. LNK mutation studies in blast-phase myeloproliferative neoplasms, and in chronic-phase disease with TET2, IDH, JAK2 or MPL mutations. Leukemia 2010, 24, 1713–1718. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Busque, L.; Levine, R.L. Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Cell Stem Cell 2018, 22, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Jan, M.; Ebert, B.L.; Jaiswal, S. Clonal hematopoiesis. Semin. Hematol. 2017, 54, 43–50. [Google Scholar] [CrossRef]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef]
- Busque, L.; Patel, J.P.; Figueroa, M.E.; Vasanthakumar, A.; Provost, S.; Hamilou, Z.; Mollica, L.; Li, J.; Viale, A.; Heguy, A.; et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012, 44, 1179–1181. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- McKerrell, T.; Park, N.; Moreno, T.; Grove, C.S.; Ponstingl, H.; Stephens, J.; Crawley, C.; Craig, J.; Scott, M.A.; Hodkinson, C.; et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015, 10, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Craver, B.M.; El Alaoui, K.; Scherber, R.M.; Fleischman, A.G. The critical role of inflammation in the pathogenesis and progression of myeloid malignancies. Cancers (Basel). 2018, 10, 104. [Google Scholar] [CrossRef] [PubMed]
- Ferrone, C.K.; Blydt-Hansen, M.; Rauh, M.J. Age-Associated TET2 Mutations: Common Drivers of Myeloid Dysfunction, Cancer and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Abegunde, S.O.; Buckstein, R.; Wells, R.A.; Rauh, M.J. An inflammatory environment containing TNFalpha favors Tet2-mutant clonal hematopoiesis. Exp. Hematol. 2018, 59, 60–65. [Google Scholar] [CrossRef]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; LaTocha, D.H.; Yang, F.; Press, R.D.; et al. TNFalpha facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef]
- Coombs, C.C.; Zehir, A.; Devlin, S.M.; Kishtagari, A.; Syed, A.; Jonsson, P.; Hyman, D.M.; Solit, D.B.; Robson, M.E.; Baselga, J.; et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017, 21, 374–382. [Google Scholar] [CrossRef]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef]
- Zoi, K.; Cross, N.C.P. Genomics of Myeloproliferative Neoplasms. J. Clin. Oncol. 2017, 35, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Song, L.; Zhu, P.; Zhang, B.; Tao, Y.; Xu, X.; Li, F.; Wu, K.; Liang, J.; Shao, D.; et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell 2012, 148, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Engle, E.; Fisher, D.; Miller, C.; McLellan, M.; Fulton, R.; Moore, D.; Wilson, R.; Ley, T.; Oh, S. Clonal Evolution Revealed by Whole Genome Sequencing in a Case of Primary Myelofibrosis Transformed to Secondary Acute Myeloid Leukemia. Leukemia 2015, 29, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Silver, R.T.; Vandris, K.; Wang, Y.L.; Adriano, F.; Jones, A.V.; Christos, P.J.; Cross, N.C.P. JAK2(V617F) allele burden in polycythemia vera correlates with grade of myelofibrosis, but is not substantially affected by therapy. Leuk. Res. 2011, 35, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Rambaldi, A.; Barosi, G.; Marchioli, R.; Marfisi, R.M.; Finazzi, G.; Guerini, V.; Fabris, F.; et al. Clinical profile of homozygous JAK2V617F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007, 110, 840–846. [Google Scholar] [CrossRef]
- Larsen, T.S.; Pallisgaard, N.; M??ller, M.B.; Hasselbalch, H.C.; Møller, M.B.; Hasselbalch, H.C. The JAK2 V617F allele burden in essential thrombocythemia, polycythemia vera and primary myelofibrosis - Impact on disease phenotype. Eur. J. Haematol. 2007, 79, 508–515. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Huang, J.; Finke, C.; Mesa, R.A.; Li, C.Y.; Wu, W.; Hanson, C.A.; Pardanani, A. Low JAK2V617F allele burden in primary myelofibrosis, compared to either a higher allele burden or unmutated status, is associated with inferior overall and leukemia-free survival. Leukemia 2008, 22, 756–761. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Barosi, G.; Specchia, G.; Rambaldi, A.; Lo Coco, F.; Antonioli, E.; Pieri, L.; Pancrazzi, A.; Ponziani, V.; Delaini, F.; et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood 2009, 114, 1477–1483. [Google Scholar] [CrossRef]
- Bose, P.; Verstovsek, S. Prognosis of Primary Myelofibrosis in the Genomic Era. Clin. Lymphoma. Myeloma Leuk. 2016, 16 Suppl., S105–S113. [Google Scholar] [CrossRef]
- Theocharides, A.; Boissinot, M.; Girodon, F.; Garand, R.; Teo, S.-S.; Lippert, E.; Talmant, P.; Tichelli, A.; Hermouet, S.; Skoda, R.C. Leukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood 2007, 110, 375–379. [Google Scholar] [CrossRef]
- Beer, P.A.; Delhommeau, F.; LeCouédic, J.P.; Dawson, M.A.; Chen, E.; Bareford, D.; Kušec, R.; McMullin, M.F.; Harrison, C.N.; Vannucchi, A.M.; et al. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood 2010, 115, 2891–2900. [Google Scholar] [CrossRef] [PubMed]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of Mutation Order on Myeloproliferative Neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef]
- Nangalia, J.; Nice, F.L.; Wedge, D.C.; Godfrey, A.L.; Grinfeld, J.; Thakker, C.; Massie, C.E.; Baxter, J.; Sewell, D.; Silber, Y.; et al. DNMT3A mutations occur early or late in patients with myeloproliferative neoplasms and mutation order influences phenotype. Haematologica 2015, 100, e438–e442. [Google Scholar] [CrossRef]
- Rumi, E.; Cazzola, M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood 2017, 129, 680–692. [Google Scholar] [CrossRef]
- Barbui, T.; Tefferi, A.; Vannucchi, A.M.; Passamonti, F.; Silver, R.T.; Hoffman, R.; Verstovsek, S.; Mesa, R.; Kiladjian, J.-J.; Hehlmann, R.; et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: Revised management recommendations from European LeukemiaNet. Leukemia 2018, 32, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Pietra, D.; Ferretti, V.; Klampfl, T.; Harutyunyan, A.S.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014, 123, 1544–1551. [Google Scholar] [CrossRef]
- Rotunno, G.; Mannarelli, C.; Guglielmelli, P.; Pacilli, A.; Pancrazzi, A.; Pieri, L.; Fanelli, T.; Bosi, A.; Vannucchi, A.M. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood 2014, 123, 1552–1555. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Biamonte, F.; Rotunno, G.; Artusi, V.; Artuso, L.; Bernardis, I.; Tenedini, E.; Pieri, L.; Paoli, C.; Mannarelli, C.; et al. Impact of mutational status on outcomes in myelofibrosis patients treated with ruxolitinib in the COMFORT-II study. Blood 2014, 123, 2157–2160. [Google Scholar] [CrossRef]
- Rumi, E.; Pietra, D.; Pascutto, C.; Guglielmelli, P.; Martinez-Trillos, A.; Casetti, I.; Colomer, D.; Pieri, L.; Pratcorona, M.; Rotunno, G.; et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood 2014, 124, 1062–1069. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; Ketterling, R.; Hanson, C.H.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: Clinical, cytogenetic and molecular comparisons. Leukemia 2014, 28, 1472–1477. [Google Scholar] [CrossRef]
- Merker, J.D.; Roskin, K.M.; Ng, D.; Pan, C.; Fisk, D.G.; King, J.J.; Hoh, R.; Stadler, M.; Okumoto, L.M.; Abidi, P.; et al. Comprehensive whole-genome sequencing of an early-stage primary myelofibrosis patient defines low mutational burden and non-recurrent candidate genes. Haematologica 2013, 98, 1689–1696. [Google Scholar] [CrossRef][Green Version]
- Tenedini, E.; Bernardis, I.; Artusi, V.; Artuso, L.; Roncaglia, E.; Guglielmelli, P.; Pieri, L.; Bogani, C.; Biamonte, F.; Rotunno, G.; et al. Targeted cancer exome sequencing reveals recurrent mutations in myeloproliferative neoplasms. Leukemia 2014, 28, 1052–1059. [Google Scholar] [CrossRef]
- Wang, L.; Swierczek, S.I.; Drummond, J.; Hickman, K.; Kim, S.J.; Walker, K.; Doddapaneni, H.; Muzny, D.M.; Gibbs, R.A.; Wheeler, D.A.; et al. Whole-exome sequencing of polycythemia vera revealed novel driver genes and somatic mutation shared by T cells and granulocytes. Leukemia 2014, 28, 935–938. [Google Scholar] [CrossRef]
- Angona, A.; Alvarez-Larrán, A.; Bellosillo, B.; Martínez-Avilés, L.; Camacho, L.; Fernández-Rodríguez, C.; Pairet, S.; Longarón, R.; Ancochea, Á.; Senín, A.; et al. Hematopoietic clonal dominance, stem cell mutations, and evolutionary pattern of JAK2V617F allele burden in polycythemia vera. Eur. J. Haematol. 2015, 94, 251–257. [Google Scholar] [CrossRef]
- Kirschner, M.M.J.; Schemionek, M.; Schubert, C.; Chatain, N.; Sontag, S.; Isfort, S.; Ortiz-Bruchle, N.; Schmitt, K.; Kruger, L.; Zerres, K.; et al. Dissecting Genomic Aberrations in Myeloproliferative Neoplasms by Multiplex-PCR and Next Generation Sequencing. PLoS ONE 2015, 10, e0123476. [Google Scholar] [CrossRef]
- Asp, J.; Andreasson, B.; Hansson, U.; Wasslavik, C.; Abelsson, J.; Johansson, P.; Palmqvist, L. Mutation status of essential thrombocythemia and primary myelofibrosis defines clinical outcome. Haematologica 2016, 101, e129–e132. [Google Scholar] [CrossRef]
- Cabagnols, X.; Favale, F.; Pasquier, F.; Messaoudi, K.; Defour, J.P.; Ianotto, J.C.; Marzac, C.; Le Couedic, J.P.; Droin, N.; Chachoua, I.; et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood 2016, 127, 333–342. [Google Scholar] [CrossRef]
- Jeromin, S.; Kohlmann, A.; Meggendorfer, M.; Schindela, S.; Perglerová, K.; Nadarajah, N.; Kern, W.; Haferlach, C.; Haferlach, T.; Schnittger, S. Next-generation deep-sequencing detects multiple clones of CALR mutations in patients with BCR-ABL1 negative MPN. Leukemia 2016, 30, 973–976. [Google Scholar] [CrossRef]
- Magor, G.W.; Tallack, M.R.; Klose, N.M.; Taylor, D.; Korbie, D.; Mollee, P.; Trau, M.; Perkins, A.C. Rapid Molecular Profiling of Myeloproliferative Neoplasms Using Targeted Exon Resequencing of 86 Genes Involved in JAK-STAT Signaling and Epigenetic Regulation. J. Mol. Diagnostics 2016, 18, 707–718. [Google Scholar] [CrossRef]
- Milosevic Feenstra, J.D.; Nivarthi, H.; Gisslinger, H.; Leroy, E.; Rumi, E.; Chachoua, I.; Bagienski, K.; Kubesova, B.; Pietra, D.; Gisslinger, B.; et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 2016, 127, 325–332. [Google Scholar] [CrossRef]
- Casolari, D.A.; Nguyen, T.; Butcher, C.M.; Iarossi, D.G.; Hahn, C.N.; Bray, S.C.; Neufing, P.; Parker, W.T.; Feng, J.; Maung, K.Z.Y.; et al. A novel, somatic, transforming mutation in the extracellular domain of Epidermal Growth Factor Receptor identified in myeloproliferative neoplasm. Sci. Rep. 2017, 7, 2467. [Google Scholar] [CrossRef]
- Chang, Y.C.; Lin, H.C.; Chiang, Y.H.; Chen, C.G.S.; Huang, L.; Wang, W.T.; Cheng, C.C.; Lin, J.; Chang, Y.F.; Chang, M.C.; et al. Targeted next-generation sequencing identified novel mutations in triple-negative myeloproliferative neoplasms. Med. Oncol. 2017, 34, 1–6. [Google Scholar] [CrossRef]
- Courtier, F.; Carbuccia, N.; Garnier, S.; Guille, A.; Adelaide, J.; Cervera, N.; Gelsi-Boyer, V.; Mozziconacci, M.-J.; Rey, J.; Vey, N.; et al. Genomic analysis of myeloproliferative neoplasms in chronic and acute phases. Haematologica 2017, 102, e11–e14. [Google Scholar] [CrossRef]
- Kröger, N.; Panagiota, V.; Badbaran, A.; Zabelina, T.; Triviai, I.; Araujo Cruz, M.M.; Shahswar, R.; Ayuk, F.; Gehlhaar, M.; Wolschke, C.; et al. Impact of Molecular Genetics on Outcome in Myelofibrosis Patients after Allogeneic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2017, 23, 1095–1101. [Google Scholar] [CrossRef]
- Masarova, L.; Patel, K.P.; Newberry, K.J.; Cortes, J.; Borthakur, G.; Konopleva, M.; Estrov, Z.; Kantarjian, H.; Verstovsek, S. Pegylated interferon alfa-2a in patients with essential thrombocythaemia or polycythaemia vera: A post-hoc, median 83 month follow-up of an open-label, phase 2 trial. Lancet. Haematol. 2017, 4, e165–e175. [Google Scholar] [CrossRef]
- Newberry, K.J.; Patel, K.; Masarova, L.; Luthra, R.; Manshouri, T.; Jabbour, E.; Bose, P.; Daver, N.; Cortes, J.; Kantarjian, H.; et al. Clonal evolution and outcomes in myelofibrosis after ruxolitinib discontinuation. Blood 2017, 130, 1125–1131. [Google Scholar] [CrossRef]
- Silver, R.T.; Barel, A.C.; Lascu, E.; Ritchie, E.K.; Roboz, G.J.; Christos, P.J.; Orazi, A.; Hassane, D.C.; Tam, W.; Cross, N.C.P. The effect of initial molecular profile on response to recombinant interferon-alpha (rIFNalpha) treatment in early myelofibrosis. Cancer 2017, 123, 2680–2687. [Google Scholar] [CrossRef]
- Zaidi, U.; Shahid, S.; Fatima, N.; Ahmed, S.; Sufaida, G.; Nadeem, M.; Shamsi, T. Genomic profile of a patient with triple negative essential thrombocythemia, unresponsive to therapy: A case report and literature review. J. Adv. Res. 2017, 8, 375–378. [Google Scholar] [CrossRef]
- Ayres-Silva, J.P.; Bonamino, M.H.; Gouveia, M.E.; Monte-Mor, B.C.R.; Coutinho, D.F.; Daumas, A.H.; Solza, C.; Braggio, E.; Zalcberg, I.R. Genetic Alterations in Essential Thrombocythemia Progression to Acute Myeloid Leukemia: A Case Series and Review of the Literature. Front. Oncol. 2018, 8, 32. [Google Scholar] [CrossRef]
- Ianotto, J.-C.; Chauveau, A.; Boyer-Perrard, F.; Gyan, E.; Laribi, K.; Cony-Makhoul, P.; Demory, J.-L.; de Renzis, B.; Dosquet, C.; Rey, J.; et al. Benefits and pitfalls of pegylated interferon-α2a therapy in patients with myeloproliferative neoplasm-associated myelofibrosis: A French Intergroup of Myeloproliferative neoplasms (FIM) study. Haematologica 2018, 103, 438–446. [Google Scholar] [CrossRef]
- Ju, M.; Fu, R.; Li, H.; Liu, X.; Xue, F.; Chen, Y.; Liu, W.; Huang, Y.; Zhang, L.L.L.L.; Yang, R.; et al. Mutation profiling by targeted sequencing of “triple-negative” essential thrombocythaemia patients - supplemental. Br. J. Haematol. 2018, 181, 857–860. [Google Scholar] [CrossRef]
- Kubesova, B.; Pavlova, S.; Malcikova, J.; Kabathova, J.; Radova, L.; Tom, N.; Tichy, B.; Plevova, K.; Kantorova, B.; Fiedorova, K.; et al. Low-burden TP53 mutations in chronic phase of myeloproliferative neoplasms: Association with age, hydroxyurea administration, disease type and JAK2 mutational status. Leukemia 2018, 32, 450–461. [Google Scholar] [CrossRef]
- Pacilli, A.; Rotunno, G.; Mannarelli, C.; Fanelli, T.; Pancrazzi, A.; Contini, E.; Mannelli, F.; Gesullo, F.; Bartalucci, N.; Fattori, G.C.; et al. Mutation landscape in patients with myelofibrosis receiving ruxolitinib or hydroxyurea. Blood Cancer J. 2018, 8, 122. [Google Scholar] [CrossRef]
- Senín, A.; Fernández-Rodríguez, C.; Bellosillo, B.; Camacho, L.; Longarón, R.; Angona, A.; Besses, C.; Álvarez-Larrán, A. Non-driver mutations in patients with JAK2V617F-mutated polycythemia vera or essential thrombocythemia with long-term molecular follow-up. Ann. Hematol. 2018, 97, 443–451. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef]
- Tefferi, A.; Barraco, D.; Lasho, T.L.; Shah, S.; Begna, K.H.; Al-Kali, A.; Hogan, W.J.; Litzow, M.R.; Hanson, C.A.; Ketterling, R.P.; et al. Momelotinib therapy for myelofibrosis: A 7-year follow-up. Blood Cancer J. 2018, 8, 29. [Google Scholar] [CrossRef]
- Acha, P.; Xandri, M.; Fuster-Tormo, F.; Palomo, L.; Xicoy, B.; Cabezon, M.; Marce, S.; Granada, I.; Vela, D.; Sagues, M.; et al. Diagnostic and prognostic contribution of targeted NGS in patients with triple-negative myeloproliferative neoplasms. Am. J. Hematol. 2019, 94, E264–E267. [Google Scholar] [CrossRef]
- Gagelmann, N.; Ditschkowski, M.; Bogdanov, R.; Bredin, S.; Robin, M.; Cassinat, B.; Shahswar, R.; Thol, F.; Heuser, M.; Socié, G.; et al. Comprehensive clinical-molecular transplant scoring system for myelofibrosis undergoing stem cell transplantation. Blood 2019, 133, 2233–2242. [Google Scholar] [CrossRef]
- Knudsen, T.A.; Skov, V.; Werner, L.; Duke, W.; Gibson, C.J.; Nag, A.; Thorner, A.R.; Wollison, B.; Hansen, D.L.; Stauffer Larsen, T.; et al. Genomic Profiling of a Phase III Clinical Trial of Interferon Versus Hydroxyurea in MPN Patients Reveals Mutation-Specific and Treatment-Specific Patterns of Response. Blood (ASH Annu. Meet. Abstr. 2019, 134, 4202. [Google Scholar] [CrossRef]
- Luque Paz, D.L.; Mansier, O.; Riou, J.; Conejero, C.; Roy, L.; Belkhodja, C.; Ugo, V.; Giraudier, S. Positive impact of molecular analysis on prognostic scores in essential thrombocythemia: A single center prospective cohort experience. Haematologica 2019, 104, e134–e137. [Google Scholar]
- Mannina, D.; Gagelmann, N.; Badbaran, A.; Ditschkowski, M.; Bogdanov, R.; Robin, M.; Cassinat, B.; Heuser, M.; Shahswar, R.; Thol, F.; et al. Allogeneic stem cell transplantation in patients with myelofibrosis harboring the MPL mutation. Eur. J. Haematol. 2019, 103, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Nam, A.S.; Kim, K.-T.; Chaligne, R.; Izzo, F.; Ang, C.; Taylor, J.; Myers, R.M.; Abu-Zeinah, G.; Brand, R.; Omans, N.D.; et al. Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature 2019, 571, 355–360. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.M.; Hamblin, A.; Yap, C.; Fox, S.; Boucher, R.; Panchal, A.; Alimam, S.; Dreau, H.; Howard, K.; Ware, P.; et al. The poor outcome in high molecular risk, hydroxycarbamide-resistant/intolerant ET is not ameliorated by ruxolitinib. Blood 2019, 134, 2107–2111. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Meira, A.; Buck, G.; Clark, S.-A.; Povinelli, B.J.; Alcolea, V.; Louka, E.; McGowan, S.; Hamblin, A.; Sousos, N.; Barkas, N.; et al. Unravelling Intratumoral Heterogeneity through High-Sensitivity Single-Cell Mutational Analysis and Parallel RNA Sequencing. Mol. Cell 2019, 73, 1292–1305. [Google Scholar] [CrossRef] [PubMed]
- Schischlik, F.; Jäger, R.; Rosebrock, F.; Hug, E.; Schuster, M.; Holly, R.; Fuchs, E.; Milosevic Feenstra, J.D.; Bogner, E.; Gisslinger, B.; et al. Mutational landscape of the transcriptome offers putative targets for immunotherapy of myeloproliferative neoplasms. Blood 2019, 134, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Tamari, R.; Rapaport, F.; Zhang, N.; McNamara, C.; Kuykendall, A.; Sallman, D.A.; Komrokji, R.; Arruda, A.; Najfeld, V.; Sandy, L.; et al. Impact of High-Molecular-Risk Mutations on Transplantation Outcomes in Patients with Myelofibrosis. Biol. Blood Marrow Transplant. 2019, 25, 1142–1151. [Google Scholar] [CrossRef]
- Yacoub, A.; Mascarenhas, J.; Kosiorek, H.; Prchal, J.T.; Berenzon, D.; Baer, M.R.; Ritchie, E.; Silver, R.T.; Kessler, C.; Winton, E.; et al. Pegylated interferon alfa-2a for polycythemia vera or essential thrombocythemia resistant or intolerant to hydroxyurea. Blood 2019, 134, 1498–1509. [Google Scholar] [CrossRef]
- Cassinat, B.; Verger, E.; Maslah, N.; Gauthier, N.; Vainchenker, W.; Giraudier, S.; Kiladjian, J.-J. CCND2 mutations are infrequent events in BCR-ABL1 negative myeloproliferative neoplasm patients. Haematologica 2020. [Google Scholar] [CrossRef]
- Gill, H.; Leung, G.M.K.; Yim, R.; Lee, P.; Pang, H.H.; Ip, H.-W.; Leung, R.Y.Y.; Li, J.; Panagiotou, G.; Ma, E.S.K.; et al. Myeloproliferative neoplasms treated with hydroxyurea, pegylated interferon alpha-2A or ruxolitinib: Clinicohematologic responses, quality-of-life changes and safety in the real-world setting. Hematology 2020, 25, 247–257. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Carobbio, A.; Rumi, E.; De Stefano, V.; Mannelli, L.; Mannelli, F.; Rotunno, G.; Coltro, G.; Betti, S.; Cavalloni, C.; et al. Validation of the IPSET score for thrombosis in patients with prefibrotic myelofibrosis. Blood Cancer J. 2020, 10, 21. [Google Scholar] [CrossRef]
- Karantanos, T.; Chaturvedi, S.; Braunstein, E.M.; Spivak, J.; Resar, L.; Karanika, S.; Williams, D.M.; Rogers, O.; Gocke, C.D.; Moliterno, A.R. Sex determines the presentation and outcomes in MPN and is related to sex-specific differences in the mutational burden. Blood Adv. 2020, 4, 2567–2576. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Klade, C.; Georgiev, P.; Krochmalczyk, D.; Gercheva-Kyuchukova, L.; Egyed, M.; Rossiev, V.; Dulicek, P.; Illes, A.; Pylypenko, H.; et al. Ropeginterferon Alpha-2b is Efficacious and Reduces Variant TET2 Allele Burden in Patients with Polycythaemia Vera and TET2 Mutation: Genetic Analysis of Phase III PROUD-PV/CONTINUATION-PV Studies. EHA Abstr. 2020. [Google Scholar]
- Mylonas, E.; Yoshida, K.; Frick, M.; Hoyer, K.; Christen, F.; Kaeda, J.; Obenaus, M.; Noerenberg, D.; Hennch, C.; Chan, W.; et al. Single-cell analysis based dissection of clonality in myelofibrosis. Nat. Commun. 2020, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Nonino, A.; Campregher, P.V.; de Souza Santos, F.P.; Mazzeu, J.F.; Pereira, R.W. Genomic characterization and prognostication applied to a Brazilian cohort of patients with myelofibrosis. Int. J. Hematol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Segura-Diaz, A.; Stuckey, R.; Florido, Y.; Gonzalez-Martin, J.M.; Lopez-Rodriguez, J.F.; Sanchez-Sosa, S.; Gonzalez-Perez, E.; Saez Perdomo, M.N.; Perera, M.D.M.; de la Iglesia, S.; et al. Thrombotic Risk Detection in Patients with Polycythemia Vera: The Predictive Role of DNMT3A/TET2/ASXL1 Mutations. Cancers (Basel) 2020, 12, 934. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Screening for ASXL1 and SRSF2 mutations is imperative for treatment decision-making in otherwise low or intermediate-1 risk patients with myelofibrosis. Br. J. Haematol. 2018, 183, 678–681. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Finke, C.; Gangat, N.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A. Prognostic significance of ASXL1 mutation types and allele burden in myelofibrosis. Leukemia 2018, 32, 837–839. [Google Scholar] [CrossRef]
- Lasho, T.L.; Jimma, T.; Finke, C.M.; Patnaik, M.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. SRSF2 mutations in primary myelofibrosis: Significant clustering with IDH mutations and independent association with inferior overall and leukemia-free survival. Blood 2012, 120, 4168–4171. [Google Scholar] [CrossRef]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Finke, C.; Mannarelli, C.; Belachew, A.A.; Pancrazzi, A.; Wassie, E.A.; Ketterling, R.P.; et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: An international study of 570 patients. Leukemia 2014, 28, 1494–1500. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Nicolosi, M.; Mudireddy, M.; Szuber, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Gangat, N.; et al. Driver mutations and prognosis in primary myelofibrosis: Mayo-Careggi MPN alliance study of 1,095 patients. Am. J. Hematol. 2018, 93, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Yonal-Hindilerden, I.; Daglar-Aday, A.; Akadam-Teker, B.; Yilmaz, C.; Nalcaci, M.; Yavuz, A.S.; Sargin, D. Prognostic significance of ASXL1, JAK2V617F mutations and JAK2V617F allele burden in Philadelphia-negative myeloproliferative neoplasms. J. Blood Med. 2015, 6, 157–175. [Google Scholar] [CrossRef]
- Tefferi, A.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. U2AF1 mutation types in primary myelofibrosis: Phenotypic and prognostic distinctions. Leukemia 2018, 32, 2274–2278. [Google Scholar] [CrossRef]
- Tefferi, A.; Pardanani, A.; Lim, K.H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Gangat, N.; Finke, C.M.; Schwager, S.; Mullally, A.; et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia 2009, 23, 905–911. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Manshouri, T.; Patel, J.; Harris, K.; Yao, J.; Hedvat, C.; Heguy, A.; Bueso-Ramos, C.; Kantarjian, H.; Levine, R.L.; et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010, 70, 447–452. [Google Scholar] [CrossRef]
- Quintas-Cardama, A.; Abdel-Wahab, O.; Manshouri, T.; Kilpivaara, O.; Cortes, J.; Roupie, A.-L.A.L.; Zhang, S.J.; Harris, D.; Estrov, Z.; Kantarjian, H.; et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon a -2a. Blood 2013, 122, 893–901. [Google Scholar] [CrossRef]
- Harutyunyan, A.; Klampfl, T.; Cazzola, M.; Kralovics, R. p53 lesions in leukemic transformation. N. Engl. J. Med. 2011, 364, 488–490. [Google Scholar] [CrossRef]
- Ruark, E.; Snape, K.; Humburg, P.; Loveday, C.; Bajrami, I.; Brough, R.; Rodrigues, D.N.; Renwick, A.; Seal, S.; Ramsay, E.; et al. Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature 2013, 493, 406–410. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, L.H.; Wan, H.; Yang, R.; Wang, Z.; Feng, J.; Yang, S.; Jones, S.; Wang, S.; Zhou, W.; et al. Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas. Nat. Genet. 2014, 46, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Bian, E.-B.; Zong, G.; Xie, Y.-S.; Meng, X.-M.; Huang, C.; Li, J.; Zhao, B. TET family proteins: New players in gliomas. J. Neurooncol. 2014, 116, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Sclafani, F.; Wilson, S.H.; Cunningham, D.; Gonzalez De Castro, D.; Kalaitzaki, E.; Begum, R.; Wotherspoon, A.; Capdevila, J.; Glimelius, B.; Rosello, S.; et al. Analysis of KRAS, NRAS, BRAF, PIK3CA and TP53 mutations in a large prospective series of locally advanced rectal cancer patients. Int. J. cancer 2020, 146, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Esfahani, M.S.; Lee, L.J.; Jeon, Y.-J.; Flynn, R.A.; Stehr, H.; Hui, A.B.; Ishisoko, N.; Kildebeck, E.; Newman, A.M.; Bratman, S.V.; et al. Functional significance of U2AF1 S34F mutations in lung adenocarcinomas. Nat. Commun. 2019, 10, 5712. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, C.S.; Amici, M.; Bortolotto, Z.A.; Doherty, A.; Csaba, Z.; Fafouri, A.; Dournaud, P.; Gressens, P.; Collingridge, G.L.; Peineau, S. The role of JAK-STAT signaling within the CNS. JAK-STAT 2013, 2, e22925. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, H.; Farkas, D.K.; Christiansen, C.F.; Larsen, T.S.; Hasselbalch, H.C.; Stentoft, J.; Sørensen, H.T. Survival of patients with chronic myeloproliferative neoplasms and new primary cancers: A population-based cohort study. Lancet. Haematol. 2015, 2, e289–e296. [Google Scholar] [CrossRef][Green Version]
- Landtblom, A.R.; Bower, H.; Andersson, T.M.L.; Dickman, P.W.; Samuelsson, J.; Björkholm, M.; Kristinsson, S.Y.; Hultcrantz, M. Second malignancies in patients with myeloproliferative neoplasms: A population-based cohort study of 9379 patients. Leukemia 2018, 32, 2203–2210. [Google Scholar] [CrossRef]
- Pettersson, H.; Knutsen, H.; Holmberg, E.; Andréasson, B. Increased incidence of another cancer in myeloproliferative neoplasms patients at the time of diagnosis. Eur. J. Haematol. 2015, 94, 152–156. [Google Scholar] [CrossRef]
- Cervantes, F.; Dupriez, B.; Pereira, A.; Passamonti, F.; Reilly, J.T.; Morra, E.; Vannucchi, A.M.; Mesa, R.A.; Demory, J.-L.; Barosi, G.; et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009, 113, 2895–2901. [Google Scholar] [CrossRef]
- Passamonti, F.; Cervantes, F.; Vannucchi, A.M.; Morra, E.; Rumi, E.; Pereira, A.; Guglielmelli, P.; Pungolino, E.; Caramella, M.; Maffioli, M.; et al. A dynamic prognostic model to predict survival in primary myelofibrosis: A study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010, 115, 1703–1708. [Google Scholar] [CrossRef]
- Passamonti, F.; Giorgino, T.; Mora, B.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia 2017, 31, 2726–2731. [Google Scholar] [CrossRef]
- Yuan, Y.; Liu, L.; Chen, H.; Wang, Y.; Xu, Y.; Mao, H.; Li, J.; Mills, G.B.; Shu, Y.; Li, L.; et al. Comprehensive Characterization of Molecular Differences in Cancer between Male and Female Patients. Cancer Cell 2016, 29, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Score, J.; Mannarelli, C.; Pancrazzi, A.; Biamonte, F.; Pardanani, A.; Zoi, K.; Reiter, A.; et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: An international study of 797 patients. Leukemia 2014, 28, 1804–1810. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, I.; Hasselbalch, H.C. Acute leukemia and myelodysplasia in patients with a Philadelphia chromosome negative chronic myeloproliferative disorder treated with hydroxyurea alone or with hydroxyurea after busulphan. Am. J. Hematol. 2003, 74, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Spivak, J.L.; Hasselbalch, H. Hydroxycarbamide: A user ’ s guide for chronic myeloproliferative disorders. Expert Rev. Anticancer Ther. 2011, 11, 403–414. [Google Scholar] [CrossRef]
- Kiladjian, J.J.; Chevret, S.; Dosquet, C.; Chomienne, C.; Rain, J.D. Treatment of polycythemia vera with hydroxyurea and pipobroman: Final results of a randomized trial initiated in 1980. J. Clin. Oncol. 2011, 29, 3907–3913. [Google Scholar] [CrossRef]
- Kissova, J.; Ovesna, P.; Penka, M.; Bulikova, A.; Kiss, I. Second Malignancies in Philadelphia-negative Myeloproliferative Neoplasms − Single-center Experience. Anticancer Res. 2014, 34, 2489–2496. [Google Scholar]
- Esteve, E.; Georgescu, V.; Heitzmann, P.; Martin, L. [Multiple skin and mouth squamous cell carcinomas related to long-term treatment with hydroxyurea]. Ann. Dermatol. Venereol. 2001, 128, 919–921. [Google Scholar]
- Spanoudakis, E.; Bazdiara, I.; Kotsianidis, I.; Margaritis, D.; Goutzouvelidis, A.; Christoforidou, A.; Tsatalas, C.; Bourikas, G. Hydroxyurea (HU) is effective in reducing JAK2V617F mutated clone size in the peripheral blood of essential thrombocythemia (ET) and polycythemia vera (PV) patients. Ann. Hematol. 2009, 88, 629–632. [Google Scholar] [CrossRef]
- Larsen, T.S.; Pallisgaard, N.; de Stricker, K.; Møller, M.B.; Hasselbalch, H.C. Limited efficacy of hydroxyurea in lowering of the JAK2 V617F allele burden. Hematology 2009, 14, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, E.; Carobbio, A.; Pieri, L.; Pancrazzi, A.; Guglielmelli, P.; Delaini, F.; Ponziani, V.; Bartalucci, N.; Tozzi, L.; Bosi, A.; et al. Hydroxyurea does not appreciably reduce JAK2 v617F allele burden in patients with polycythemia vera or essential thrombocythemia. Haematologica 2010, 95, 1435–1438. [Google Scholar] [CrossRef]
- Campbell, P.J.; Scott, L.M.; Buck, G.; Wheatley, K.; East, C.L.; Marsden, J.T.; Duffy, A.; Boyd, E.M.; Bench, A.J.; Scott, M.A.; et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: A prospective study. Lancet (Lond. Engl.) 2005, 366, 1945–1953. [Google Scholar] [CrossRef]
- Harrison, C.N.; Campbell, P.J.; Buck, G.; Wheatley, K.; East, C.L.; Bareford, D.; Wilkins, B.S.; van der Walt, J.D.; Reilly, J.T.; Grigg, A.P.; et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N. Engl. J. Med. 2005, 353, 33–45. [Google Scholar] [CrossRef]
- King, K.Y.; Matatall, K.A.; Shen, C.-C.; Goodell, M.A.; Swierczek, S.I.; Prchal, J.T. Comparative long-term effects of interferon alpha and hydroxyurea on human hematopoietic progenitor cells. Exp. Hematol. 2015, 43, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Larrán, A.; Pereira, A.; Cervantes, F.; Arellano-Rodrigo, E.; Hernández-Boluda, J.C.; Ferrer-Marín, F.; Angona, A.; Gómez, M.; Muiña, B.; Guillén, H.; et al. Assessment and prognostic value of the European leukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood 2012, 119, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Guryanova, O.A.; Shank, K.; Spitzer, B.; Luciani, L.; Koche, R.P.; Garrett-Bakelman, F.E.; Ganzel, C.; Durham, B.H.; Mohanty, A.; Hoermann, G.; et al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat. Med. 2016, 22, 1488–1495. [Google Scholar] [CrossRef] [PubMed]
- Velu, T.; Delwiche, F.; Gangji, D.; Monsieur, R.; Flament, J.; Stryckmans, P.; Wybran, J.; Bellens, R. Therapeutic effect of human recombinant interferon-alpha-2C in essential thrombocythaemia. Oncology 1985, 42 Suppl. 1, 10–14. [Google Scholar] [CrossRef]
- Ludwig, H.; Linkesch, W.; Gisslinger, H.; Fritz, E.; Sinzinger, H.; Radaszkiewicz, T.; Chott, A.; Flener, R.; Micksche, M. Interferon-alfa corrects thrombocytosis in patients with myeloproliferative disorders. Cancer Immunol. Immunother. 1987, 25, 266–273. [Google Scholar] [CrossRef]
- Hasselbalch, H. Interferon in myelofibrosis. Lancet (Lond. Engl.) 1988, 1, 355. [Google Scholar] [CrossRef]
- Silver, R.T. Recombinant interferon-alpha for treatment of polycythaemia vera. Lancet (Lond. Engl.) 1988, 2, 403. [Google Scholar] [CrossRef]
- Foa, P.; Massaro, P.; Caldiera, S.; LaTargia, M.L.; Iurlo, A.; Clerici, C.; Fornier, M.; Bertoni, F.; Maiolo, A.T. Long-term therapeutic efficacy and toxicity of recombinant interferon-alpha 2a in polycythaemia vera. Eur. J. Haematol. 1998, 60, 273–277. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Kantarjian, H.; Manshouri, T.; Luthra, R.; Estrov, Z.; Pierce, S.; Richie, M.A.; Borthakur, G.; Konopleva, M.; Cortes, J.; et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J. Clin. Oncol. 2009, 27, 5418–5424. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, J.; Hasselbalch, H.; Bruserud, O.; Temerinac, S.; Brandberg, Y.; Merup, M.; Linder, O.; Bjorkholm, M.; Pahl, H.L.; Birgegard, G. A phase II trial of pegylated interferon alpha-2b therapy for polycythemia vera and essential thrombocythemia: Feasibility, clinical and biologic effects, and impact on quality of life. Cancer 2006, 106, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Stauffer Larsen, T.; Iversen, K.F.; Hansen, E.; Mathiasen, A.B.; Marcher, C.; Frederiksen, M.; Larsen, H.; Helleberg, I.; Riley, C.H.; Bjerrum, O.W.; et al. Long term molecular responses in a cohort of Danish patients with essential thrombocythemia, polycythemia vera and myelofibrosis treated with recombinant interferon alpha. Leuk. Res. 2013, 37, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Mondello, P.; Di Mirto, C.; Cuzzocrea, S.; Arrigo, C.; Mian, M.; Pitini, V. Interferon Alpha Has a Strong Anti-tumor Effect in Philadelphia-negative Myeloproliferative Neoplasms. Clin. Lymphoma. Myeloma Leuk. 2019, 19, e489–e495. [Google Scholar] [CrossRef]
- Utke Rank, C.; Weis Bjerrum, O.; Larsen, T.S.; Kjær, L.; de Stricker, K.; Riley, C.H.; Hasselbalch, H.C. Minimal residual disease after long-term interferon-alpha2 treatment: A report on hematological, molecular and histomorphological response patterns in 10 patients with essential thrombocythemia and polycythemia vera. Leuk. Lymphoma 2016, 57, 348–354. [Google Scholar] [CrossRef]
- Hasan, S.; Lacout, C.; Marty, C.; Cuingnet, M.; Solary, E.; Vainchenker, W.; Villeval, J.-L. JAK2V617F expression in mice amplifies early hematopoietic cells and gives them a competitive advantage that is hampered by IFNalpha. Blood 2013, 122, 1464–1477. [Google Scholar] [CrossRef]
- Kiladjian, J.J.; Giraudier, S.; Cassinat, B. Interferon-alpha for the therapy of myeloproliferative neoplasms: Targeting the malignant clone. Leukemia 2016, 30, 776–781. [Google Scholar] [CrossRef]
- Riley, C.H.; Jensen, M.K.; Brimnes, M.K.; Hasselbalch, H.C.; Bjerrum, O.W.; Straten, P.T.; Svane, I.M.; Hansen, M.; Brimnes, M.K.; Hasselbalch, H.C.; et al. Increase in circulating CD4+CD25+Foxp3+ T cells in patients with Philadelphia-negative chronic myeloproliferative neoplasms during treatment with IFN-α. Eur. J. Haematol. 2011, 118, 2170–2173. [Google Scholar] [CrossRef][Green Version]
- Riley, C.H.; Hansen, M.; Brimnes, M.K.; Hasselbalch, H.C.; Bjerrum, O.W.; Straten, P.T.; Svane, I.M.; Jensen, M.K. Expansion of circulating CD56bright natural killer cells in patients with JAK2-positive chronic myeloproliferative neoplasms during treatment with interferon-α. Eur. J. Haematol. 2015, 94, 227–234. [Google Scholar] [CrossRef]
- Riley, C.H.; Brimnes, M.K.; Hansen, M.; Jensen, M.K.; Hasselbalch, H.C.; Kjaer, L.; Straten, P.T.; Svane, I.M. Interferon-α induces marked alterations in circulating regulatory T cells, NK cell subsets, and dendritic cells in patients with JAK2V617F-positive essential thrombocythemia and polycythemia vera. Eur. J. Haematol. 2016, 97, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Kovacsovics-Bankowski, M.; Kelley, T.W.; Efimova, O.; Kim, S.J.; Wilson, A.; Swierczek, S.; Prchal, J. Changes in peripheral blood lymphocytes in polycythemia vera and essential thrombocythemia patients treated with pegylated-interferon alpha and correlation with JAK2(V617F) allelic burden. Exp. Hematol. Oncol. 2015, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zhang, W.; Li, Y.; Berenzon, D.; Wang, X.; Wang, J.; Mascarenhas, J.; Xu, M.; Hoffman, R. Interferon-alpha targets JAK2V617F-positive hematopoietic progenitor cells and acts through the p38 MAPK pathway. Exp. Hematol. 2010, 38, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Kiladjian, J.J.; Massé, A.; Cassinat, B.; Mokrani, H.; Teyssandier, I.; Le Couédic, J.P.; Cambier, N.; Almire, C.; Pronier, E.; Casadevall, N.; et al. Clonal analysis of erythroid progenitors suggests that pegylated interferon α-2a treatment targets JAK2 V617F clones without affecting TET2 mutant cells. Leukemia 2010, 24, 1519–1523. [Google Scholar] [CrossRef]
- Harrison, C.; Kiladjian, J.-J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef]
- Mullally, A.; Bruedigam, C.; Poveromo, L.; Heidel, F.H.; Purdon, A.; Vu, T.; Austin, R.; Heckl, D.; Breyfogle, L.J.; Kuhn, C.P.; et al. Depletion of Jak2V617F myeloproliferative neoplasm-propagating stem cells by interferon-α in a murine model of polycythemia vera. Blood 2013, 121, 3692–3702. [Google Scholar] [CrossRef]
- Hasan, S.; Cassinat, B.; Droin, N.; Le Couedic, J.P.; Favale, F.; Monte-Mor, B.; Lacout, C.; Fontenay, M.; Dosquet, C.; Chomienne, C.; et al. Use of the 46/1 haplotype to model JAK2(V617F) clonal architecture in PV patients: Clonal evolution and impact of IFNalpha treatment. Leukemia 2014, 28, 460–463. [Google Scholar] [CrossRef]
- Them, N.C.C.; Bagienski, K.; Berg, T.; Gisslinger, B.; Schalling, M.; Chen, D.; Buxhofer-Ausch, V.; Thaler, J.; Schloegl, E.; Gastl, G.A.; et al. Molecular responses and chromosomal aberrations in patients with polycythemia vera treated with peg-proline-interferon alpha-2b. Am. J. Hematol. 2015, 90, 288–294. [Google Scholar] [CrossRef]
- Passegué, E.; Ernst, P.A. IFN-alpha wakes up sleeping hematopoietic stem cells. Nat. Med. 2009, 15, 612–613. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Kiladjian, J.J.; Silver, R.T. Interferon alfa in the treatment of philadelphia-negative chronic myeloproliferative neoplasms. J. Clin. Oncol. 2011, 29, 2011–2012. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Massaro, P.; Foa, P.; Pomati, M.; LaTargia, M.L.; Iurlo, A.; Clerici, C.; Caldiera, S.; Fornier, M.; Maiolo, A.T. Polycythemia vera treated with recombinant interferon-alpha 2a: Evidence of a selective effect on the malignant clone. Am. J. Hematol. 1997, 56, 126–128. [Google Scholar] [CrossRef]
- Essers, M.A.G.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009, 458, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Trumpp, A.; Essers, M.; Wilson, A. Awakening dormant haematopoietic stem cells. Nat. Rev. Immunol. 2010, 10, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.W.N.; Miller, C.B.; Silver, R.T.; et al. Efficacy, safety and survival with ruxolitinib in patients with myelofibrosis: Results of a median 2-year follow-up of COMFORT-I. Haematologica 2013, 98, 1865–1871. [Google Scholar] [CrossRef]
- Cervantes, F.; Vannucchi, A.M.; Kiladjian, J.-J.; Al-Ali, H.K.; Sirulnik, A.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.S.; Passamonti, F.; et al. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood 2013, 122, 4047–4053. [Google Scholar] [CrossRef]
- Al-Ali, H.K.; Griesshammer, M.; le Coutre, P.; Waller, C.F.; Liberati, A.M.; Schafhausen, P.; Tavares, R.; Giraldo, P.; Foltz, L.; Raanani, P.; et al. Safety and efficacy of ruxolitinib in an open-label, multicenter, single-arm phase 3b expanded-access study in patients with myelofibrosis: A snapshot of 1144 patients in the JUMP trial. Haematologica 2016, 101, 1065–1073. [Google Scholar] [CrossRef]
- Verstovsek, S.; Passamonti, F.; Rambaldi, A.; Barosi, G. A Phase 2 Study of Ruxolitinib, an Oral JAK1 and JAK2 Inhibitor, in Patients With Advanced Polycythemia Vera Who Are Refractory or Intolerant to Hydroxyurea. Cancer 2014, 120, 513–520. [Google Scholar] [CrossRef]
- Harrison, C.N.; Mead, A.J.; Panchal, A.; Fox, S.; Yap, C.; Gbandi, E.; Houlton, A.; Alimam, S.; Ewing, J.; Wood, M.; et al. Ruxolitinib vs best available therapy for ET intolerant or resistant to hydroxycarbamide. Blood 2017, 130, 1889–1897. [Google Scholar] [CrossRef]
- Verstovsek, S.; Passamonti, F.; Rambaldi, A.; Barosi, G.; Rumi, E.; Gattoni, E.; Pieri, L.; Zhen, H.; Granier, M.; Assad, A.; et al. Ruxolitinib for essential thrombocythemia refractory to or intolerant of hydroxyurea: Long-term phase 2 study results. Blood 2017, 130, 1768–1771. [Google Scholar] [CrossRef]
- Gunawan, A.; Harrington, P.; Garcia-Curto, N.; McLornan, D.; Radia, D.; Harrison, C. Ruxolitinib for the Treatment of Essential Thrombocythemia. HemaSphere 2018, 2, e56. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Verstovsek, S. Ruxolitinib for essential thrombocythemia? Oncoscience 2017, 4, 148–149. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C. JAK inhibitors and myelofibrosis, Einstein and ruxolitinib. Haematologica 2015, 100, 409–411. [Google Scholar] [CrossRef] [PubMed]
- Elli, E.M.; Baratè, C.; Mendicino, F.; Palandri, F.; Palumbo, G.A. Mechanisms Underlying the Anti-inflammatory and Immunosuppressive Activity of Ruxolitinib. Front. Oncol. 2019, 9, 1186. [Google Scholar] [CrossRef] [PubMed]
- Polverelli, N.; Palumbo, G.A.; Binotto, G.; Abruzzese, E.; Benevolo, G.; Bergamaschi, M.; Tieghi, A.; Bonifacio, M.; Breccia, M.; Catani, L.; et al. Epidemiology, outcome, and risk factors for infectious complications in myelofibrosis patients receiving ruxolitinib: A multicenter study on 446 patients. Hematol. Oncol. 2018. [Google Scholar] [CrossRef]
- Deininger, M.; Radich, J.; Burn, T.C.; Huber, R.; Paranagama, D.; Verstovsek, S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood 2015, 126, 1551–1555. [Google Scholar] [CrossRef]
- Verstovsek, S.; Vannucchi, A.M.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Kirito, K.; et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica 2016, 101, 821–829. [Google Scholar] [CrossRef]
- Pardanani, A.; Vannucchi, A.M.; Passamonti, F.; Cervantes, F.; Barbui, T.; Tefferi, A. JAK inhibitor therapy for myelofibrosis: Critical assessment of value and limitations. Leukemia 2011, 25, 218–225. [Google Scholar] [CrossRef]
- Tefferi, A. JAK inhibitors for myeloproliferative neoplasms: Clarifying facts from myths. Blood 2012, 119, 2721–2730. [Google Scholar] [CrossRef]
- Griner, L.N.; Mcgraw, K.L.; Johnson, J.O.; List, A.F.; Reuther, G.W. JAK2-V617F-mediated signalling is dependent on lipid rafts and statins inhibit JAK2-V617F-dependent cell growth. Br. J. Haematol. 2013, 160, 177–187. [Google Scholar] [CrossRef]
- Pieri, L.; Pancrazzi, A.; Pacilli, A.; Rabuzzi, C.; Rotunno, G.; Fanelli, T.; Guglielmelli, P.; Fjerza, R.; Paoli, C.; Verstovsek, S.; et al. JAK2V617F complete molecular remission in polycythemia vera/essential thrombocythemia patients treated with ruxolitinib. Blood 2015, 125, 3352–3353. [Google Scholar] [CrossRef] [PubMed]
- Lane, S.W.; Mullally, A. Jak2V617F myeloproliferative neoplasm stem cells and interferon-alpha. Oncotarget 2013, 4, 500–501. [Google Scholar] [CrossRef][Green Version]
- Mullally, A.; Lane, S.W.; Ball, B.; Megerdichian, C.; Okabe, R.; Al-Shahrour, F.; Paktinat, M.; Haydu, J.E.; Housman, E.; Lord, A.M.; et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell 2010, 17, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, S.U.; Kjaer, L.; Bjørn, M.E.; Knudsen, T.A.; Sørensen, A.L.; Andersen, C.B.L.; Bjerrum, O.W.; Brochmann, N.; El Fassi, D.; Kruse, T.A.; et al. Safety and efficacy of combination therapy of interferon-α2 and ruxolitinib in polycythemia vera and myelofibrosis. Cancer Med. 2018, 7, 3571–3581. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, A.L.; Mikkelsen, S.U.; Knudsen, T.A.; Bjorn, M.E.; Andersen, C.L.; Bjerrum, O.W.; Brochmann, N.; Patel, D.A.; Gjerdrum, L.M.R.; El Fassi, D.; et al. Ruxolitinib and interferon-alpha2 combination therapy for patients with polycythemia vera or myelofibrosis: A phase II study. Haematologica 2019. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Perspectives on the impact of JAK-inhibitor therapy upon inflammation-mediated comorbidities in myelofibrosis and related neoplasms. Expert Rev. Hematol. 2014, 7, 203–216. [Google Scholar] [CrossRef]
- Bjorn, M.E.; Hasselbalch, H.C. Minimal residual disease or cure in MPNs? Rationales and perspectives on combination therapy with interferon-alpha2 and ruxolitinib. Expert Rev. Hematol. 2017, 10, 393–404. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Bjørn, M.E. MPNs as Inflammatory Diseases: The Evidence, Consequences, and Perspectives. Mediat. Inflamm. 2015, 2015, 102476. [Google Scholar] [CrossRef]
- Griner, L.N.; McGraw, K.L.; Johnson, J.O.; List, A.F.; Reuther, G.W. A mechanistic rationale for the use of statins to enhance JAK inhibitor therapy in MPNs. Blood 2011, 118, 53. [Google Scholar] [CrossRef]
- Kaelin, W.G.J. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Nijman, S.M.B. Synthetic lethality: General principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011, 585, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Myelofibrosis with myeloid metaplasia: The advanced phase of an untreated disseminated hematological cancer. Time to change our therapeutic attitude with early upfront treatment? Leuk. Res. 2009, 33, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Skov, V.; Dam, M.J.B.; Pedersen, R.K.; Andersen, M.; Knudsen, T.A.; Sajid, Z.; Kjær, L.; Ellervik, C.; Hasselbalch, H.C.; Ottesen, J.T. Superiority of IFN Versus HU Using a Novel Biomarker-Based Tool for Assessment of Disease Burden in MPNs. Blood (ASH Annu. Meet. Abstr.) 2019, 134, 2972. [Google Scholar] [CrossRef]
- Bjørn, M.; de Stricker, K.; Kjær, L.; Ellemann, K.; Hasselbalch, H. Combination therapy with interferon and JAK1-2 inhibitor is feasible: Proof of concept with rapid reduction in JAK2V617F-allele burden in polycythemia vera. Leuk. Res. reports 2014, 3, 73–75. [Google Scholar]
- Goldie, J.H.; Coldman, A.J. The genetic origin of drug resistance in neoplasms: Implications for systemic therapy. Cancer Res. 1984, 44, 3643–3653. [Google Scholar]



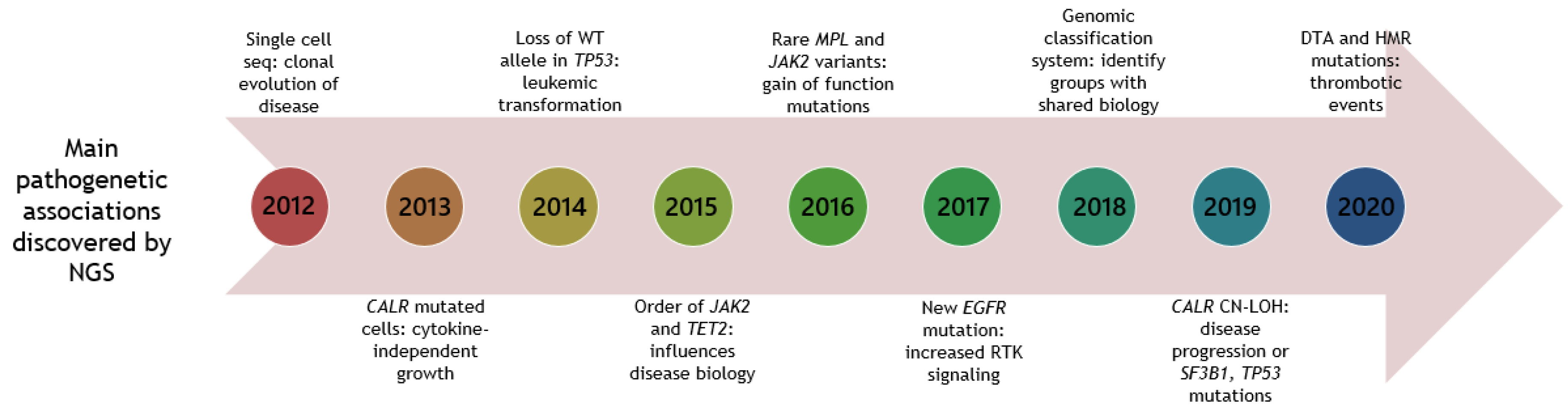{kind=link}
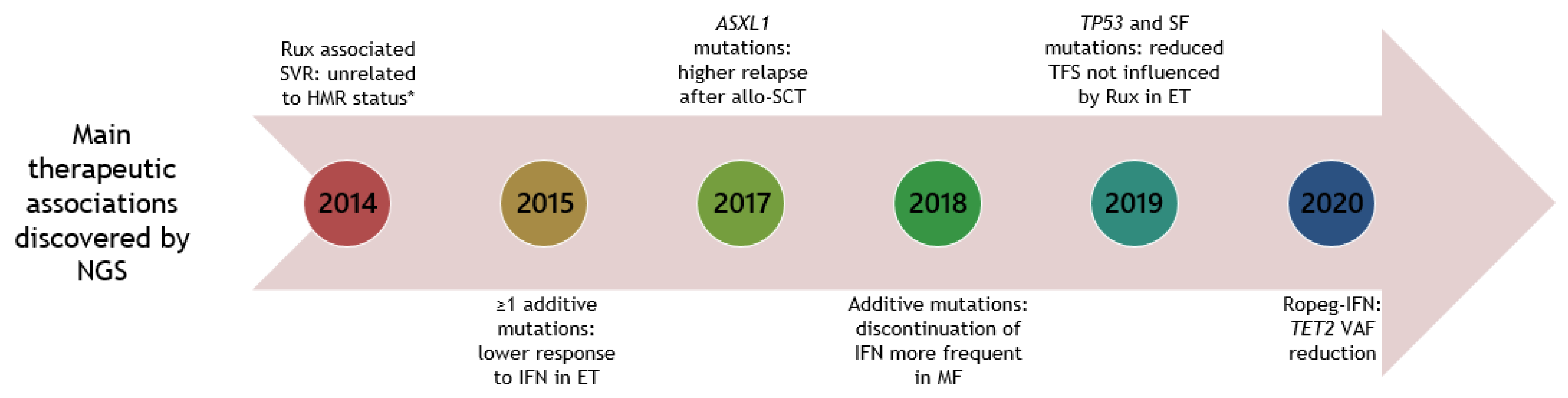{kind=link}
| Gene | ET % | PV % | SMF % | PMF % | Ref |
|---|---|---|---|---|---|
| DNA methylation | |||||
| DNMT3A | <10 | 3–15 | <5 | 5–15 | [58,59,60,61,62,63,64,65,66] |
| IDH1/2 | <2 | <2 | <2 | <5 | [60,61,63,64,65,67,68] |
| TET2 | 10–20 | 15–30 | 20–40 | 10–15 | [58,59,60,62,64,65,66] |
| Chromatin modifiers | |||||
| ASXL1 | 5–10 | 5–10 | 10–25 | 20–45 | [58,59,60,61,62,63,64,65,66,67,68] |
| EZH2 | <5 | <5 | 5–15 | 3–12 | [60,61,62,63,64,65,66,67,68] |
| RNA splicing | |||||
| SF3B1 | <7 | <6 | 2–14 | 3–18 | [58,59,60,61,62,63,65,69] |
| SRSF2 | <5 | <5 | <5 | 10–35 | [58,59,60,61,62,63,65,66,67,68] |
| ZRSR2 | <3 | <5 | 5–10 | 1–10 | [59,60,61,62,63] |
| U2AF1 | <5 | <5 | 5–20 | 5–20 | [58,59,60,61,62,63,65,66] |
| Signaling | |||||
| CBL | <7 | <7 | <5 | <7 | [57,58,60,61,65] |
| KIT | 2 | 3 | NA | <2 | [59,60] |
| NRAS | <2 | <2 | <3 | <5 | [57,59,60,61,63,64,65] |
| SH2B3 | 3–5 | <9 | 5 | 6 | [60,61] |
| Transcription factors | |||||
| CEBPA | 4 | 2–6 | NA | 9 | [59,60,63] |
| RUNX1 | <3 | <2 | <3 | <5 | [59,60,61,63,64,65] |
| Tumor suppressors | |||||
| TP53 | <9 | <5 | <14 | <7 | [58,59,60,61,62,63,64,65,66] |
| DNA damage | |||||
| PPM1D | 3 | 1 | <1 | <1 | [20] |
| Study (Author) | Patients (No) and Disease | Type of Study | Method (Gen No) | Main Prognostic Findings | Ref |
|---|---|---|---|---|---|
| Hou et al., 2012 | 1 ET | Baseline single cell | Illumina (WES) | NA | [122] |
| Klampfl et al., 2013 | 6 PMF | Baseline | Illumina (WES) | NA | [12] |
| Merker et al., 2013 | WGS: 1 PMF Targeted seq: 40 ET, 42 PV, 96 MF | Baseline | CompleteGenomics (WGS), Illumina (6) | NA | [141] |
| Nangalia et al., 2013 | 62 ET, 48 PV, 39 PMF, 2 MPN-U | Baseline and follow-up | Illumina (WES) | CALR mut ET higher risk of MF | [13] |
| Guglielmelli et al., 2014 | 27 PET-MF, 54 PPV-MF, 85 PMF | Baseline and Rux | Roche 454 GS or PGM (14) | Spleen response unrelated to HMR or LMR mutations during rux | [138] |
| Lundberg et al., 2014 | 69 ET, 94 PV, 34 PMF | Serial follow-up | Illumina (104) | TET2, TP53, ≥2 mutations: shorter OS, LFS | [120] |
| Tenedini et al., 2014 | Study 1: 9 PV, 5 PPV-MF, 11 PMF Validation:50 PV, 48 SMF, 91 PMF | Baseline and follow-up | Roche 454 GS (WES), PGM (121) | NRAS in PMF: shorter OS | [142] |
| Wang et al., 2014 | 31 PV | Baseline | Illumina (WES) and PGM (42) | NA | [143] |
| Angona et al., 2015 | 36 PV, 9 PPV-MF | Baseline | Roche 454 GS (4) | NA | [144] |
| Engle et al., 2015 | 1 PMF | Baseline and follow-up | Illumina WGS, targeted seq (58) | NA | [123] |
| Kirschner et al., 2015 | 10 ET, 9 SMF, 17 PV, 10 PMF | Baseline | Illumina (48) | NA | [145] |
| Ortmann et al., 2015 | First cohort: 92 ET, 107 PV, 47 MF Follow-up cohort: 918 MPN | Baseline and Rux | Exome or Illumina (111/65) | JAK2 first vs. TET2 first: younger, higher risk of thrombosis | [132] |
| Patel et al., 2015 | 10 PET-MF, 31 PPV-MF, 54 PMF | Baseline and Rux | Illumina (28) | ≥1 mutations in ASXL1, EZH2, IDH1/2: shorter OS and TTD during rux. | [64] |
| Verger et al., 2015 | 31 ET, Control group: 12 ET(aspirin only), 14 ET–HU only | Baseline and IFN | Illumina (7) | ≥1 additional mutation: higher rate of no response to IFN | [75] |
| Angona et al., 2016 | 29 ET, all triple negative | Baseline | Roche 454 GS (NA) | NA | [73] |
| Asp et al., 2016 | ET: 8 TN, 18 CALR, 18 JAK2V617F, 7 MPL. PMF: 7 TN, 1 MPL | Baseline and follow-up | Illumina (54) | TN (PMF), MPL (ET): shorter OS ASXL1, SRSF2 in ET: shorter OS | [146] |
| Cabagnols et al., 2016 | 17 ET, all triple negative | Baseline | Illumina (WES) | NA | [147] |
| Delic et al., 2016 | 40 ET, 30 PV, 30 PMF | Baseline | Illumina (28) | ASXL1, EZH2, SF3B1, SRSF2, U2AF1 more mutated in PMF than ET | [58] |
| Jeromin et al.,2016 | 88 MPN | Baseline | Roche 454 GS (1) | NA | [148] |
| Magor et al., 2016 | 16 ET, 8 PV, 6 SMF, 11 PMF | Baseline | Ion torrent PGM (86) | NA | [149] |
| M Feenstra et al., 2016 | 4 ET, 4 PMF | Baseline | Illumina WES | NA | [150] |
| Rotunno et al., 2016 | 165 PET-MF, 194 PPV-MF | Baseline and follow-up | PGM (5) | TN, SRSF2 (PET-MF): shorter OS | [67] |
| Tefferi et al., 2016 | Mayo cohort: 183 ET, 133 PV Italian cohort: 174 ET, 215 PV | Baseline and follow-up | PGM (5), Illumina (27) | PV: SRSF2: shorter OS, LFS, MFS ET: IDH2, SH2B3: shorter OS. | [59] |
| Tefferi et al., 2016 | 182 PMF | Baseline and follow-up | PGM (5), Illumina (27) | ASXL1, SRSF2, CBL, KIT: shorter OS SRSF2, RUNX1, SH2B3, CEBPA: shorter LFS | [60] |
| Agarwal et al., 2017 | 114 ET, 3 PET-MF, 5 PPV-MF, 44 PMF | Baseline | Illumina (26) | CALR type 1: more common in PMF than CALR type 2 | [66] |
| Casolari et al., 2017 | 15 PV | Baseline | Solid (657) | NA | [151] |
| Chang et al., 2017 | 7 ET, 8 PV, 1 PMF, all triple negative | Baseline | Ion Proton (409) | NA | [152] |
| Courtier et al., 2017 | 57 Chronic phase MPN, 38 Post-MPN AML | NGS in chronic phase during disease | Illumina (79) | Acute phase an average gain of 1 mutation compared to chronic phase. | [153] |
| Kröger et al., 2017 | 101 PMF, 46 SMF, 13 MF transformed | Baseline and follow-up | Solid and PGM (5 and 18) | ASXL1: relapse. IDH2: worse PFS. CALR: improved PFS and OS | [154] |
| Luque Paz et al., 2017 | 22 ET, 28 PV, 50% selected with disease progression | Baseline and follow-up after 3 years | PGM (18) | ≥2 mutations or ASXL1, IDH1/2, or SRSF2: disease progression | [86] |
| Masarova et al., 2017 | 6 ET/PV | Baseline and IFN | NA (44) | DNMT3A, ASXL1 acquired at transformation | [155] |
| Newberry et al., 2017 | 62 MF | Baseline and Rux | Illumina (28) | Clonal evolution after Rux: shorter OS | [156] |
| Silver et al., 2017 | 2 PET-MF, 7 PPV-MF, 21 PMF | Baseline and IFN | Illumina (45) | ≥3 mut, ASXL1, SRSR2: adverse events | [157] |
| Song et al., 2017 | 27 ET, 33 PV, 75 PMF | Baseline | Illumina (32) | ASXL1, SRSF2 more frequent in PMF | [85] |
| Spiegel et al., 2017 | 23 PET-MF, 27 PPV-MF, 50 PMF | Baseline and Rux and MMB | Illumina (54) | ≥3 mut, HMR, ASXL1, EZH2: shorter OS | [82] |
| Zaidi et al., 2017 | 1 ET | Baseline and follow-up | Illumina (54) | NA | [158] |
| Alduaij et al., 2018 | 21 ET, 26 PV, 28 PET-MF, 15 PPV-MF, 64 PMF, 12 Post-MPN-AML | Baseline and follow-up | Illumina (54) | HMR mutations versus no HMR mutations: early HCT vs. delayed HCT | [65] |
| Ayres-Silva et al., 2018 | 3 ET and 3 paired Post-ET-AML | Baseline and transformation | Illumina (WES) | TP53 mutations during transformation, all HU | [159] |
| Bartels et al., 2018 | 36 PV with stable disease, 28 PV with fibrotic progression | Baseline and follow-up | NA (23) | Additional mutations (not TET2): increased risk of fibrotic progression | [89] |
| Grinfeld et al., 2018 | 1321 ET,356 PV,309 MF,14 MPN-U, 35 other. Serial:290 ET,30PV,10MF | Baseline and follow-up | Illumina (69 and WES) | Developed a prognostic classification system | [20] |
| Guglielmelli et al., 2018 | 490 PMF and 315 PMF | Baseline and follow-up | PGM 5, Illumina 27 | ≥1 HMR mutations: shorter OS | [68] |
| Ianotto et al., 2018 | 49 MF | Baseline and IFN | PGM (26) | ≥1 mutation: shorter LFS | [160] |
| Ju et al., 2018 | 68 ET | Baseline | NA (360) | NA | [161] |
| Kubesova et al., 2018 | Untreated: 22 ET, 22 PV, 36 PMF Treated: 80 ET, 116 PV, 53 PMF | Baseline and follow-up (HU, IFN, ANA) | Illumina (1) | TP53 mutations are associated with age | [162] |
| Pacilli et al., 2018 | Rux: 7 PET-MF, 16 PPV-MF, 23 PMF. HU: 6 SMF, 19 PMF | Baseline and Rux or HU | PGM (27) | ASXL1: shorter duration of spleen volume reduction | [163] |
| Senin et al., 2018 | Baseline: 37 ET, 63 PV. Follow-up: 50 No progression, 24 MF, 12 AML | Baseline and ANA, BUS, P3S2, IFN, HU | Illumina and Roche 454 GS (50) | SF3B1, IDH1/2: higher MT. ASXL1, TP53, SRSF2, IDH1/2, RUNX1: higher LT | [164] |
| Tefferi et al., 2018 | 641 PMF | Baseline and follow-up | PGM 5, Illumina 27 | ASXL1, SRSF2: shorter OS, LFS | [165] |
| Tefferi et al., 2018 | 100 MF | Baseline and MMB | Illumina (27) | ASXL1, SRSF2: shorter OS, SRSF2 shorter LFS | [166] |
| Acha et al., 2019 | 35 ET (TN), 8 PMF (TN) | Baseline and follow-up | Illumina (17) | ≥1 mutation: shorter OS | [167] |
| Beucher et al., 2019 | 1 triple negative ET with rare JAK2 and MPL mutations | Baseline and HU | Illumina (69) | SF3B1 VAF increased and TET2, JAK2, MPL decreased during HU | [101] |
| Boiocchi et al., 2019 | 29 ET, 21 PV, 51 PMF, 21 SMF, 21 MPNU | Baseline | Illumina (101) | MPN: SF3B1: lower hemoglobin | [69] |
| Byun et al., 2019 | 16 ET, 17 PV, 8 PMF | Baseline and follow-up | Illumina (47) | ASXL1: higher risk of LT. Splicing gene mutations: shorter OS, higher LT | [87] |
| Courtier et al., 2019 | 31 PET-MF, 28 PPV-MF, 86 PMF | Baseline and follow-up | Illumina (79–106) | SMF: ASXL1, TP53: shorter OS. PMF: SRSF2, TP53: shorter OS | [62] |
| Gagelmann et al., 2019 | 55 PET-MF, 46 PPV-MF, 260 PMF | Baseline and HCT | Solid and PGM (20) | ASXL1: shorter OS after HCT | [168] |
| Gill et al., 2019 | 17 PET-MF, 14 PPV-MF, 70 PMF | Baseline and follow-up | Illumina (54) | SMF + PMF: CUX1, TP53: shorter OS. SRSF2: shorter LFS | [88] |
| Knudsen et al., 2019 | 72 ET, 89 PV, 16 Pre-PMF, 25 PMF | Baseline and IFN, HU | Illumina (100) | DNMT3A acquired during IFN | [169] |
| Luque Paz et al., 2019 | 1190 ET | Baseline and follow-up | Illumina (16) | ≥1 mutation: shorter OS | [170] |
| Mannina et al., 2019 | 14 PMF, 4 PET-MF (all MPL positive) | Baseline and HCT | PGM (20) | MPL: favorable OS after HCT | [171] |
| Nam et al., 2019 | 6 ET, 5 MF | Baseline | Illumina (45) | NA | [172] |
| O’Sullivan et al., 2019 | 110 ET | Baseline and Rux | Illumina (32) | SF3B1, TP53: shorter TRFS | [173] |
| Rodriguez-Meira et al., 2019 | 1 ET, 1 PV, 3 SMF, 5 PMF | Baseline | Illumina single cell | NA | [174] |
| Schischlik et al., 2019 | 30 ET, 1 PV, 46 PMF | Baseline | Illumina (54) | NA | [175] |
| Stengel et al., 2019 | 50 CALRpos | Baseline | Illumina (14) | SF3B1 associated with CN-LOHpos TP53 associated with del5q. | [91] |
| Szuber et al., 2019 | PMF (99–120 patients) | Baseline and follow-up | NA (6) | ≥1 HMR: shorter LFS, OS, TFS | [83] |
| Tamari et al., 2019 | 62 PMF, 20 Post-ET-MF, 18 Post-PV-MF, 1 MPN-U | Baseline and follow-up (HCT) | NA (585) | DNMT3A, U2AF1: shorter RFS. HMR mutations: no impact on OS or RFS | [176] |
| Wanquet et al., 2019 | 35 ET, 14 PV, 31 PET-MF, 28 PPV-MF. Paired: 2 ET, 6 PV | Baseline and follow-up | Illumina (33) | TP53: shorter OS | [61] |
| Yacoub et al., 2019 | 110 ET/PV | Baseline + IFN | Illumina (156) | CALR mutation: higher CR | [177] |
| Andreasson et al., 2020 | 85 PV | Baseline and follow-up | Illumina (54) | ASXL1, vascular complication, ≥3 mutations: shorter OS | [63] |
| Bartels et al., 2020 | PMF without (27) or with (77) development of fibrosis | Baseline and follow-up | PGM (23) | SRSF2, U2AF1, SF3B1, IDH1/2, EZH2 risk factors for fibrotic progression | [84] |
| Cassinat et al., 2020 | 233 ET, 187 PV, 169 MF | Baseline | Illumina (36) | NA | [178] |
| Coltro et al., 2020 | 132 PF-PMF, 155 PMF, 177 SMF | Baseline and Rux | CBL, KRAS, NRAS: shorter OS and LFS | [57] | |
| Cottin et al., 2020 | 45 ET | Baseline and follow-up | Illumina (52) | 8 TET2 and 1 DNMT3A: higher VAF at follow-up | [77] |
| Gill et al., 2020 | 56 ET, 23 PV, 46 MF | Baseline, IFN, HU, Rux | Illumina (69) | PV: CREBBP: inferior response rate | [179] |
| Guglielmelli et al., 2020 | 132 pre-PMF | Baseline and follow-up | PGM (5) | HMR: associated with arterial thrombosis | [180] |
| Karantanos et al., 2020 | 66 ET, 31 PV, 64 PMF, 49 SMF, 9 AML | Baseline and follow-up | NA (63) | Higher number of additional mutations in men compared to women | [181] |
| Kralovics et al., 2020 | 163 PV | Baseline and IFN | Illumina (54) | JAK2, TET2 decrease during ropeg-IFN | [182] |
| Mylonas et al., 2020 | WES: 8 PMF, 7 PET/PPV-MF Targeted seq: 7 MF | Baseline and Rux | Illumina (WES) and targeted seq | Mutations were acquired in BRAF, CBL, KRAS, NRAS, and RIT1 | [183] |
| Nonino et al., 2020 | 27 MF | Baseline | Illumina (255) | NA | [184] |
| Segura-Diaz et al., 2020 | 25 ET, 16 PV, 16 PMF, 11 SMF and PV case-control cohort (55) | Baseline and follow-up | Illumina (30) | DTA mutations: associated with vascular events in PV | [185] |
| Stevens et al., 2020 | 22 PMF, 33 SMF | Pre and post-HCT | Illumina (75) | ≥3 additional mutations pre-transplant: higher PTR and NRM | [90] |
| Tefferi et al., 2020 | 502 ET, 404 PV | Baseline and follow-up | PGM (5), Illumina (27) | ET: SF3B1, SRSF2, EZH2: shorter OS PV: SRSF2, IDH2: shorter OS | [186] |
| Gene | ET | PV | SMF | PMF | Comments | Ref |
|---|---|---|---|---|---|---|
| DNA methylation | ||||||
| DNMT3A | Risk of HU-cytopenia. | Higher risk of MT. Higher risk of HU-cytopenia | Reduced RFS after HCT. | Reduced RFS after HCT. | [89,164,176] | |
| IDH1/2 | Shorter OS (IDH2). Higher risk of MT, LT. Higher risk of HU-cytopenia. | Shorter OS (IDH2). Higher risk of MT, LT. Higher risk of HU-cytopenia. | Shorter OS. Higher risk of LT. Lower SVR during Rux. Worse PFS after HCT (IDH2). | Shorter OS. Higher risk of LT. Lower SVR during Rux. Worse PFS after HCT (IDH2). | [59,64,86,89,154,164,186] | |
| TET2 | Associated with older age and thrombosis. | Associated with thrombosis. | NA | Associated with older age. | Shorter OS and higher LT in MPNs | [59,60,120,185] |
| Chromatin modifiers | ||||||
| ASXL1 | Shorter OS. Higher risk of LT. Associated with splenomegaly. | Shorter OS. Higher risk of LT. Associated with thrombosis and older age. | Shorter OS. Higher risk of LT. Shorter TTF and duration of SVR during Rux. Higher relapse and shorter OS after HCT. | Shorter OS. Higher risk of LT. Shorter TTF and duration of SVR during Rux. Higher relapse and shorter OS after HCT. | Lower Hb in MPNs. | [59,60,62,63,64,82,86,120,147,154,155,164,165,166,169] |
| EZH2 | Shorter OS. Higher risk of LT, MT. | NA | Shorter OS. Higher risk of LT. Shorter TTF during Rux. | Shorter OS. Higher risk of LT. Shorter TTF during Rux. | Higher leukocyte counts in MPNs. | [59,61,64,82,120,186] |
| RNA splicing | ||||||
| SF3B1 | Shorter OS. Higher risk of MT, LT. Higher platelets. | Higher risk of MT. | NA | NA | Lower Hb in MPNs. | [59,69,164,186] |
| SRSF2 | Shorter OS. Higher risk of LT. Higher risk of HU-cytopenia. | Shorter OS. Higher risk of MT, LT. Higher risk of HU-cytopenia. | Shorter OS. | Shorter OS. Higher risk of LT. Associated with anemia. | [60,67,146,164,165,186] | |
| ZRSR2 | NA | NA | Worse prognosis | NA | [61,62] | |
| U2AF1 | Higher risk of MT. | Higher risk of MT. | Reduced RFS and OS after HCT. | Shorter OS. Associated with anemia and thrombocythemia. Reduced RFS, OS after HCT. | [59,60,165,176,186] | |
| Signaling | ||||||
| CBL | NA | NA | Shorter TTF during Rux. | Shorter OS. Shorter TTF during Rux. | [60,82] | |
| KIT | NA | NA | NA | Shorter OS. | [60] | |
| NRAS | NA | NA | Higher risk of LT. | Higher risk of MT, LT. | [142,183] | |
| SH2B3 | Shorter OS. | Associated with splenomegaly. | NA | Higher risk of LT. | [59,60] | |
| Transcription factors | ||||||
| CEBPA | NA | NA | NA | Higher risk of LT | [60] | |
| RUNX1 | Higher risk of LT. Higher risk of HU-cytopenia. | Higher risk of LT. Higher risk of HU-cytopenia. | NA | Higher risk of LT | [60,164,186] | |
| Tumor suppressors | ||||||
| TP53 | Higher risk of LT. | Higher risk of LT. | Shorter OS. Higher risk of LT. | Shorter OS. Higher risk of LT. | Shorter OS and higher LT in MPNs | [59,61,62,120,153,164,186] |
| DNA damage | ||||||
| PPM1D | Unclear | Unclear | Unclear | Unclear | [20] | |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skov, V. Next Generation Sequencing in MPNs. Lessons from the Past and Prospects for Use as Predictors of Prognosis and Treatment Responses. Cancers 2020, 12, 2194. https://doi.org/10.3390/cancers12082194
Skov V. Next Generation Sequencing in MPNs. Lessons from the Past and Prospects for Use as Predictors of Prognosis and Treatment Responses. Cancers. 2020; 12(8):2194. https://doi.org/10.3390/cancers12082194
Chicago/Turabian StyleSkov, Vibe. 2020. "Next Generation Sequencing in MPNs. Lessons from the Past and Prospects for Use as Predictors of Prognosis and Treatment Responses" Cancers 12, no. 8: 2194. https://doi.org/10.3390/cancers12082194
APA StyleSkov, V. (2020). Next Generation Sequencing in MPNs. Lessons from the Past and Prospects for Use as Predictors of Prognosis and Treatment Responses. Cancers, 12(8), 2194. https://doi.org/10.3390/cancers12082194