Molecular and Immunological Characterization of Biliary Tract Cancers: A Paradigm Shift Towards a Personalized Medicine




Abstract
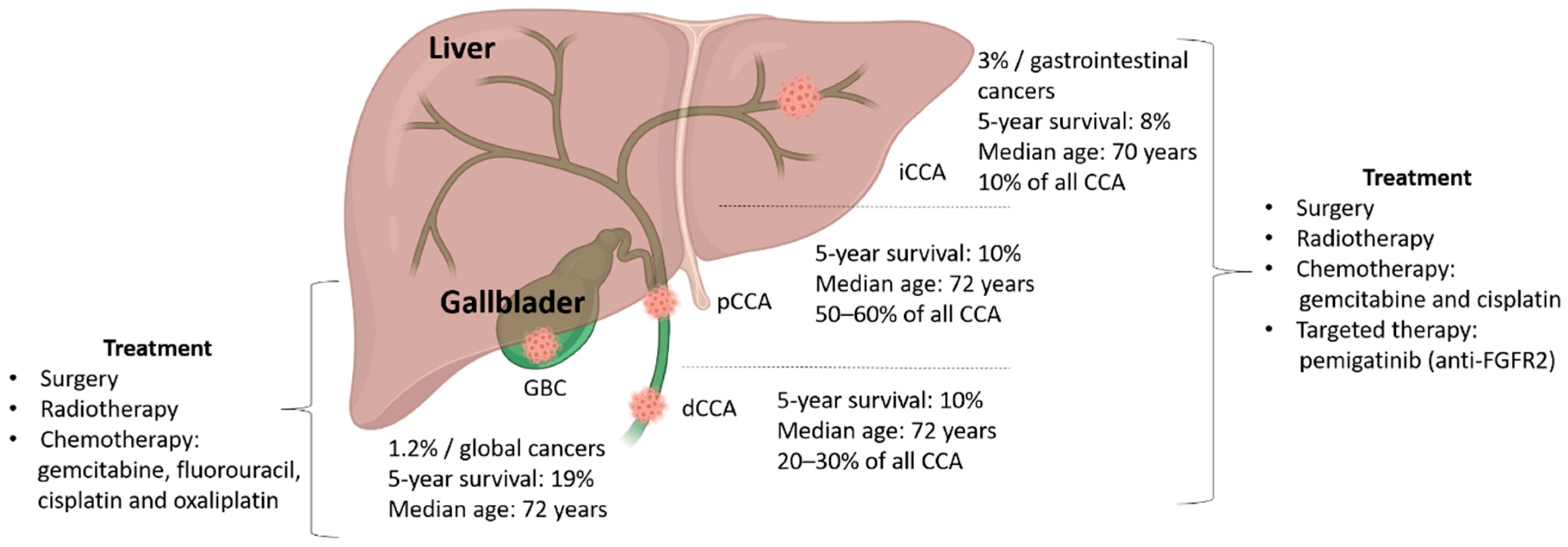
1. Introduction: Biliary Tract Cancers
2. Cholangiocarcinoma (CCA)
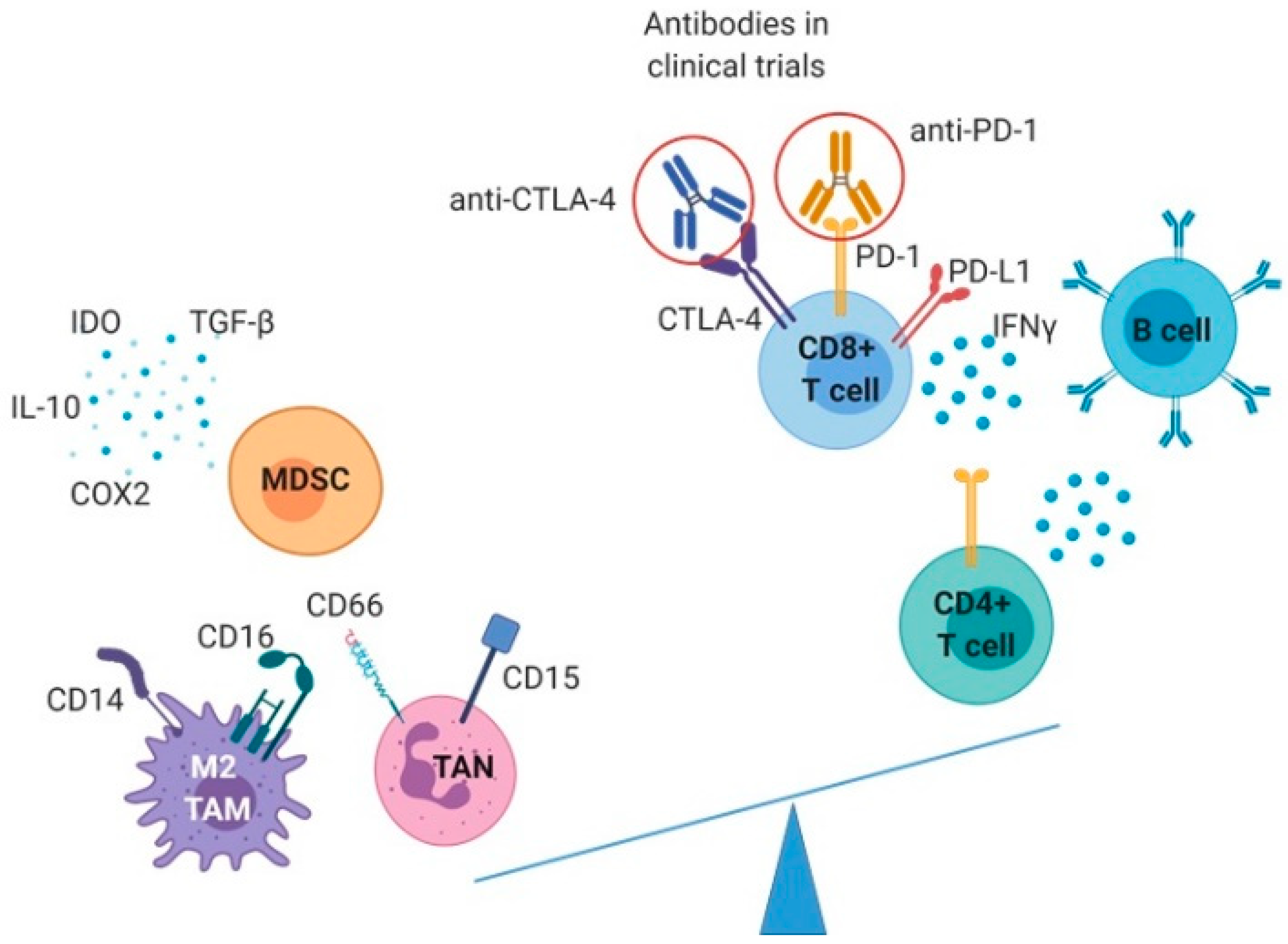
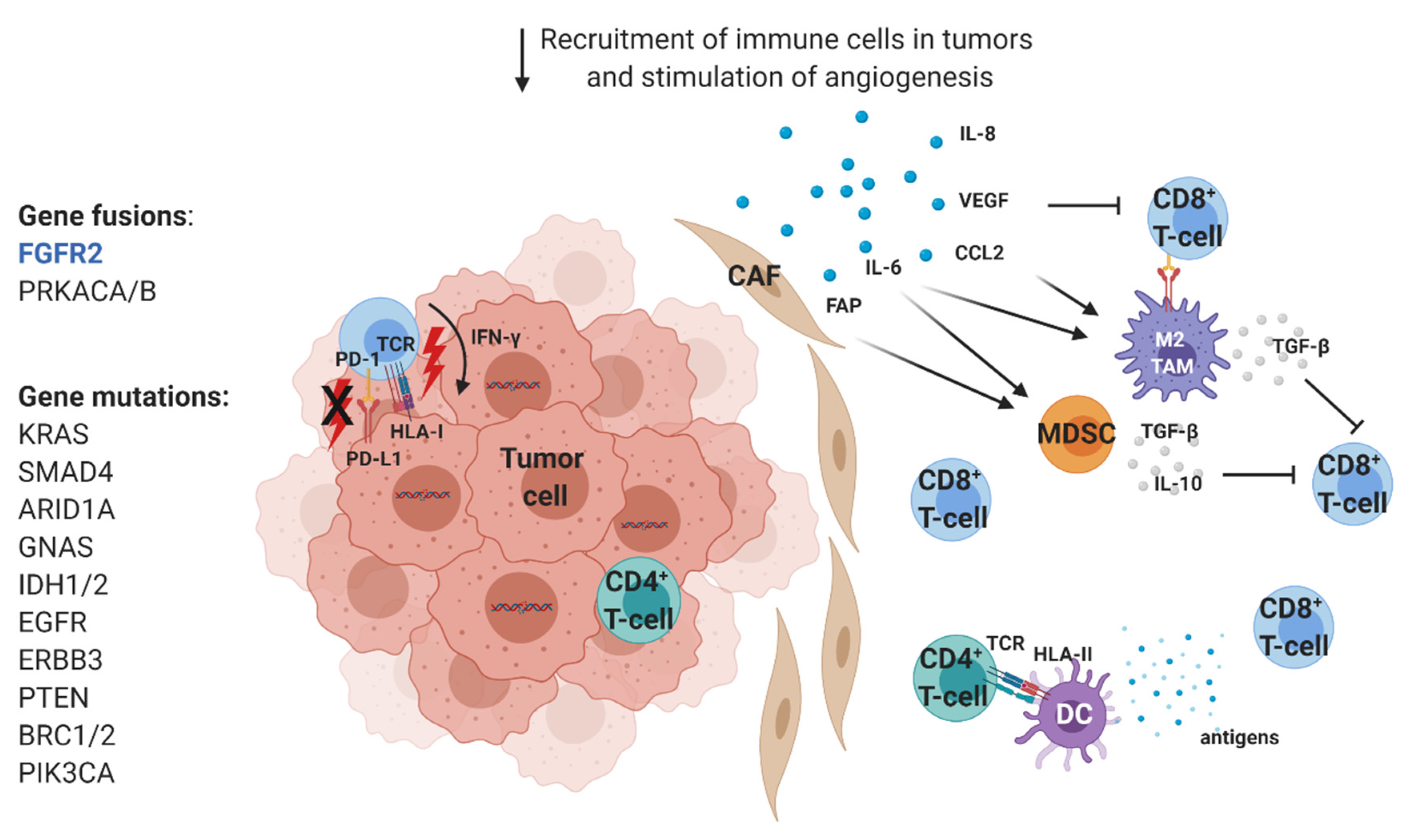
2.1. Immunological Characterization of CCA Infiltrates
2.1.1. T and B Cells in CCA
2.1.2. Innate Immune Cells
2.1.3. Immunotherapeutic Strategies in CCA
2.2. Molecular Characterization of CCA
Molecular Therapeutic Targets
3. Gallbladder Cancer
3.1. Molecular Characteristics and Immune Infiltrates in GBC
3.2. Recent Advances in Targeted Therapy and Immunotherapy of GBC
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tariq, N.U.; McNamara, M.G.; Valle, J.W. Biliary tract cancers: Current knowledge, clinical candidates and future challenges. Cancer Manag. Res. 2019, 11, 2623–2642. [Google Scholar] [CrossRef] [PubMed]
- Minicozzi, P.; Cassetti, T.; Vener, C.; Sant, M. Analysis of incidence, mortality and survival for pancreatic and biliary tract cancers across Europe, with assessment of influence of revised European age standardisation on estimates. Cancer Epidemiol. 2018, 55, 52–60. [Google Scholar] [CrossRef] [PubMed]
- McGee, E.E.; Jackson, S.S.; Petrick, J.L.; Van Dyke, A.L.; Adami, H.O.; Albanes, D.; Andreotti, G.; Beane-Freeman, L.E.; Berrington de Gonzalez, A.; Buring, J.E.; et al. Smoking, Alcohol, and Biliary Tract Cancer Risk: A Pooling Project of 26 Prospective Studies. J. Natl. Cancer Inst. 2019, 111, 1263–1278. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, F. Global burden of aflatoxin-induced hepatocellular carcinoma: A risk assessment. Environ. Health Perspect. 2010, 118, 818–824. [Google Scholar] [CrossRef]
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annu. Rev. Med. 2016, 67, 103–117. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of Hepatocellular Cancer in Patients with Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1828–1837.e2. [Google Scholar] [CrossRef]
- Takano, S.; Yokosuka, O.; Imazeki, F.; Tagawa, M.; Omata, M. Incidence of hepatocellular carcinoma in chronic hepatitis B and C: A prospective study of 251 patients. Hepatology 1995, 21, 650–655. [Google Scholar] [CrossRef]
- Forner, A.; Vidili, G.; Rengo, M.; Bujanda, L.; Ponz-Sarvise, M.; Lamarca, A. Clinical presentation, diagnosis and staging of cholangiocarcinoma. Liver Int. 2019, 39 (Suppl. 1), 98–107. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef]
- Primrose, J.N.; Fox, R.P.; Palmer, D.H.; Malik, H.Z.; Prasad, R.; Mirza, D.; Anthony, A.; Corrie, P.; Falk, S.; Finch-Jones, M.; et al. Capecitabine compared with observation in resected biliary tract cancer (BILCAP): A randomised, controlled, multicentre, phase 3 study. Lancet Oncol. 2019, 20, 663–673. [Google Scholar] [CrossRef]
- Patel, T. Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States. Hepatology 2001, 33, 1353–1357. [Google Scholar] [CrossRef] [PubMed]
- West, J.; Wood, H.; Logan, R.F.; Quinn, M.; Aithal, G.P. Trends in the incidence of primary liver and biliary tract cancers in England and Wales 1971–2001. Br. J. Cancer 2006, 94, 1751–1758. [Google Scholar] [CrossRef]
- Nakanuma, Y.; Kakuda, Y. Pathologic classification of cholangiocarcinoma: New concepts. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 277–293. [Google Scholar] [CrossRef]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [PubMed]
- Nakanuma, Y.; Sato, Y.; Harada, K.; Sasaki, M.; Xu, J.; Ikeda, H. Pathological classification of intrahepatic cholangiocarcinoma based on a new concept. World J. Hepatol. 2010, 2, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Deoliveira, M.L.; Schulick, R.D.; Nimura, Y.; Rosen, C.; Gores, G.; Neuhaus, P.; Clavien, P.A. New staging system and a registry for perihilar cholangiocarcinoma. Hepatology 2011, 53, 1363–1371. [Google Scholar] [CrossRef]
- Kendall, T.; Verheij, J.; Gaudio, E.; Evert, M.; Guido, M.; Goeppert, B.; Carpino, G. Anatomical, histomorphological and molecular classification of cholangiocarcinoma. Liver Int. 2019, 39 (Suppl. 1), 7–18. [Google Scholar] [CrossRef]
- Khan, S.A.; Tavolari, S.; Brandi, G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 2019, 39 (Suppl. 1), 19–31. [Google Scholar] [CrossRef]
- Bertuccio, P.; Bosetti, C.; Levi, F.; Decarli, A.; Negri, E.; La Vecchia, C. A comparison of trends in mortality from primary liver cancer and intrahepatic cholangiocarcinoma in Europe. Ann. Oncol. 2013, 24, 1667–1674. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Sirica, A.E.; Gores, G.J. Desmoplastic stroma and cholangiocarcinoma: Clinical implications and therapeutic targeting. Hepatology 2014, 59, 2397–2402. [Google Scholar] [CrossRef]
- Cadamuro, M.; Stecca, T.; Brivio, S.; Mariotti, V.; Fiorotto, R.; Spirli, C.; Strazzabosco, M.; Fabris, L. The deleterious interplay between tumor epithelia and stroma in cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Sirica, A.E. The role of cancer-associated myofibroblasts in intrahepatic cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 9, 44–54. [Google Scholar] [CrossRef]
- Hogdall, D.; Lewinska, M.; Andersen, J.B. Desmoplastic Tumor Microenvironment and Immunotherapy in Cholangiocarcinoma. Trends Cancer 2018, 4, 239–255. [Google Scholar] [CrossRef]
- Rimassa, L.; Personeni, N.; Aghemo, A.; Lleo, A. The immune milieu of cholangiocarcinoma: From molecular pathogenesis to precision medicine. J. Autoimmun. 2019, 100, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, J.; Aoudjehane, L.; Fouassier, L. Cancer-associated fibroblasts in cholangiocarcinoma. Curr. Opin. Gastroenterol. 2020, 36, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, H.; Wan, L.; Wang, Z.; Wang, H.; Ge, C.; Liu, Y.; Hao, Y.; Zhang, D.; Shi, G.; et al. Single cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J. Hepatol. 2020. [Google Scholar] [CrossRef]
- Job, S.; Rapoud, D.; Dos Santos, A.; Gonzalez, P.; Desterke, C.; Pascal, G.; Elarouci, N.; Ayadi, M.; Adam, R.; Azoulay, D.; et al. Identification of four immune subtypes characterized by distinct composition and functions of tumor microenvironment in intrahepatic cholangiocarcinoma. Hepatology 2019. [Google Scholar] [CrossRef]
- Wu, Y.; Li, J.; Jabbarzadeh Kaboli, P.; Shen, J.; Wu, X.; Zhao, Y.; Ji, H.; Du, F.; Zhou, Y.; Wang, Y.; et al. Natural killer cells as a double-edged sword in cancer immunotherapy: A comprehensive review from cytokine therapy to adoptive cell immunotherapy. Pharmacol. Res. 2020, 155, 104691. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef] [PubMed]
- Mrass, P.; Takano, H.; Ng, L.G.; Daxini, S.; Lasaro, M.O.; Iparraguirre, A.; Cavanagh, L.L.; von Andrian, U.H.; Ertl, H.C.; Haydon, P.G.; et al. Random migration precedes stable target cell interactions of tumor-infiltrating T cells. J. Exp. Med. 2006, 203, 2749–2761. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.F.; Bartumeus, F.; Gerard, A. T cell migration, search strategies and mechanisms. Nat. Rev. Immunol. 2016, 16, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Kasper, H.U.; Drebber, U.; Stippel, D.L.; Dienes, H.P.; Gillessen, A. Liver tumor infiltrating lymphocytes: Comparison of hepatocellular and cholangiolar carcinoma. World J. Gastroenterol. 2009, 15, 5053–5057. [Google Scholar] [CrossRef]
- Kim, R.; Coppola, D.; Wang, E.; Chang, Y.D.; Kim, Y.; Anaya, D.; Kim, D.W. Prognostic value of CD8CD45RO tumor infiltrating lymphocytes in patients with extrahepatic cholangiocarcinoma. Oncotarget 2018, 9, 23366–23372. [Google Scholar] [CrossRef] [PubMed]
- Goeppert, B.; Frauenschuh, L.; Zucknick, M.; Stenzinger, A.; Andrulis, M.; Klauschen, F.; Joehrens, K.; Warth, A.; Renner, M.; Mehrabi, A.; et al. Prognostic impact of tumour-infiltrating immune cells on biliary tract cancer. Br. J. Cancer 2013, 109, 2665–2674. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Yoshizawa, T.; Hirai, H.; Seino, H.; Morohashi, S.; Wu, Y.; Wakiya, T.; Kimura, N.; Kudo, D.; Ishido, K.; et al. Prognostic Impact of CD163+ Macrophages in Tumor Stroma and CD8+ T-Cells in Cancer Cell Nests in Invasive Extrahepatic Bile Duct Cancer. Anticancer Res. 2017, 37, 183–190. [Google Scholar] [CrossRef]
- Reuben, A.; Zhang, J.; Chiou, S.H.; Gittelman, R.M.; Li, J.; Lee, W.C.; Fujimoto, J.; Behrens, C.; Liu, X.; Wang, F.; et al. Comprehensive T cell repertoire characterization of non-small cell lung cancer. Nat. Commun. 2020, 11, 603. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Yarchoan, M.; Albacker, L.A.; Hopkins, A.C.; Montesion, M.; Murugesan, K.; Vithayathil, T.T.; Zaidi, N.; Azad, N.S.; Laheru, D.A.; Frampton, G.M.; et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Kida, A.; Mizukoshi, E.; Tamai, T.; Terashima, T.; Kitahara, M.; Arai, K.; Yamashita, T.; Fushimi, K.; Honda, M.; Kaneko, S. Immune responses against tumour-associated antigen-derived cytotoxic T lymphocyte epitopes in cholangiocarcinoma patients. Liver Int. 2018, 38, 2040–2050. [Google Scholar] [CrossRef]
- Ma, L.; Hernandez, M.O.; Zhao, Y.; Mehta, M.; Tran, B.; Kelly, M.; Rae, Z.; Hernandez, J.M.; Davis, J.L.; Martin, S.P.; et al. Tumor Cell Biodiversity Drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell 2019, 36, 418–430.e6. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A. HLA-mediated tumor escape mechanisms that may impair immunotherapy clinical outcomes via T-cell activation. Oncol. Lett. 2017, 14, 4415–4427. [Google Scholar] [CrossRef]
- Asahi, Y.; Hatanaka, K.C.; Hatanaka, Y.; Kamiyama, T.; Orimo, T.; Shimada, S.; Nagatsu, A.; Sakamoto, Y.; Kamachi, H.; Kobayashi, N.; et al. Prognostic impact of CD8+ T cell distribution and its association with the HLA class I expression in intrahepatic cholangiocarcinoma. Surg. Today 2020. [Google Scholar] [CrossRef] [PubMed]
- Sabbatino, F.; Villani, V.; Yearley, J.H.; Deshpande, V.; Cai, L.; Konstantinidis, I.T.; Moon, C.; Nota, S.; Wang, Y.; Al-Sukaini, A.; et al. PD-L1 and HLA Class I Antigen Expression and Clinical Course of the Disease in Intrahepatic Cholangiocarcinoma. Clin. Cancer Res. 2016, 22, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Gong, L.; Mo, Z.; Wang, W.; Wu, M.; Yang, J.; Zhang, Q.; Li, L.; Yao, J.; Dong, J. Programmed death ligand-1, tumor infiltrating lymphocytes and HLA expression in Chinese extrahepatic cholangiocarcinoma patients: Possible immunotherapy implications. Biosci. Trends 2019, 13, 58–69. [Google Scholar] [CrossRef]
- Fontugne, J.; Augustin, J.; Pujals, A.; Compagnon, P.; Rousseau, B.; Luciani, A.; Tournigand, C.; Cherqui, D.; Azoulay, D.; Pawlotsky, J.M.; et al. PD-L1 expression in perihilar and intrahepatic cholangiocarcinoma. Oncotarget 2017, 8, 24644–24651. [Google Scholar] [CrossRef]
- Gani, F.; Nagarajan, N.; Kim, Y.; Zhu, Q.; Luan, L.; Bhaijjee, F.; Anders, R.A.; Pawlik, T.M. Program Death 1 Immune Checkpoint and Tumor Microenvironment: Implications for Patients with Intrahepatic Cholangiocarcinoma. Ann. Surg. Oncol. 2016, 23, 2610–2617. [Google Scholar] [CrossRef]
- Montal, R.; Sia, D.; Montironi, C.; Leow, W.Q.; Esteban-Fabro, R.; Pinyol, R.; Torres-Martin, M.; Bassaganyas, L.; Moeini, A.; Peix, J.; et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J. Hepatol. 2020. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, X.Y.; Zhang, Y.; Xu, D.; Dong, J.; Zhang, Z.; Yi, C.H.; Jia, H.L.; Yang, X. Programmed death ligand 1 expression in human intrahepatic cholangiocarcinoma and its association with prognosis and CD8(+) T-cell immune responses. Cancer Manag. Res. 2018, 10, 4113–4123. [Google Scholar] [CrossRef] [PubMed]
- Jing, C.Y.; Fu, Y.P.; Yi, Y.; Zhang, M.X.; Zheng, S.S.; Huang, J.L.; Gan, W.; Xu, X.; Lin, J.J.; Zhang, J.; et al. HHLA2 in intrahepatic cholangiocarcinoma: An immune checkpoint with prognostic significance and wider expression compared with PD-L1. J. Immunother. Cancer 2019, 7, 77. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Miyagawa, S.; Ichikawa, E.; Soeda, J.; Miwa, S.; Miyagawa, Y.; Iijima, S.; Noike, T.; Kobayashi, A.; Kawasaki, S. Dendritic cells, T-cell infiltration, and Grp94 expression in cholangiocellular carcinoma. Hum. Pathol. 2004, 35, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Liu, Y.; Li, S.; Mao, Y.; Wang, J.; Shuang, Z.; Chen, J.; Li, S. Elevated neutrophil-to-lymphocyte ratio is an independent poor prognostic factor in patients with intrahepatic cholangiocarcinoma. Oncotarget 2016, 7, 50963–50971. [Google Scholar] [CrossRef]
- Najafi, M.; Hashemi Goradel, N.; Farhood, B.; Salehi, E.; Nashtaei, M.S.; Khanlarkhani, N.; Khezri, Z.; Majidpoor, J.; Abouzaripour, M.; Habibi, M.; et al. Macrophage polarity in cancer: A review. J. Cell. Biochem. 2019, 120, 2756–2765. [Google Scholar] [CrossRef]
- Thanee, M.; Loilome, W.; Techasen, A.; Namwat, N.; Boonmars, T.; Pairojkul, C.; Yongvanit, P. Quantitative changes in tumor-associated M2 macrophages characterize cholangiocarcinoma and their association with metastasis. Asian Pac. J. Cancer Prev. 2015, 16, 3043–3050. [Google Scholar] [CrossRef]
- Subimerb, C.; Pinlaor, S.; Lulitanond, V.; Khuntikeo, N.; Okada, S.; McGrath, M.S.; Wongkham, S. Circulating CD14(+) CD16(+) monocyte levels predict tissue invasive character of cholangiocarcinoma. Clin. Exp. Immunol. 2010, 161, 471–479. [Google Scholar] [CrossRef]
- Paillet, J.; Kroemer, G.; Pol, J.G. Immune contexture of cholangiocarcinoma. Curr. Opin. Gastroenterol. 2020, 36, 70–76. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef]
- Fabris, L.; Perugorria, M.J.; Mertens, J.; Bjorkstrom, N.K.; Cramer, T.; Lleo, A.; Solinas, A.; Sanger, H.; Lukacs-Kornek, V.; Moncsek, A.; et al. The tumour microenvironment and immune milieu of cholangiocarcinoma. Liver Int. 2019, 39 (Suppl. 1), 63–78. [Google Scholar] [CrossRef] [PubMed]
- Hasita, H.; Komohara, Y.; Okabe, H.; Masuda, T.; Ohnishi, K.; Lei, X.F.; Beppu, T.; Baba, H.; Takeya, M. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci. 2010, 101, 1913–1919. [Google Scholar] [CrossRef] [PubMed]
- Brivio, S.; Cadamuro, M.; Strazzabosco, M.; Fabris, L. Tumor reactive stroma in cholangiocarcinoma: The fuel behind cancer aggressiveness. World J. Hepatol. 2017, 9, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Raggi, C.; Correnti, M.; Sica, A.; Andersen, J.B.; Cardinale, V.; Alvaro, D.; Chiorino, G.; Forti, E.; Glaser, S.; Alpini, G.; et al. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J. Hepatol. 2017, 66, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, G.; Dietel, C.; Feldbrugge, L.; Benzing, C.; Krenzien, F.; Brandl, A.; Mann, E.; Englisch, J.P.; Schierle, K.; Robson, S.C.; et al. Tumor necrosis and infiltrating macrophages predict survival after curative resection for cholangiocarcinoma. Oncoimmunology 2017, 6, e1331806. [Google Scholar] [CrossRef]
- Gu, F.M.; Gao, Q.; Shi, G.M.; Zhang, X.; Wang, J.; Jiang, J.H.; Wang, X.Y.; Shi, Y.H.; Ding, Z.B.; Fan, J.; et al. Intratumoral IL-17(+) cells and neutrophils show strong prognostic significance in intrahepatic cholangiocarcinoma. Ann. Surg. Oncol. 2012, 19, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Kitano, Y.; Okabe, H.; Yamashita, Y.I.; Nakagawa, S.; Saito, Y.; Umezaki, N.; Tsukamoto, M.; Yamao, T.; Yamamura, K.; Arima, K.; et al. Tumour-infiltrating inflammatory and immune cells in patients with extrahepatic cholangiocarcinoma. Br. J. Cancer 2018, 118, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.Y.; Zhu, G.Q.; Xiong, M.; Ren, L.; Bai, L. Prognostic value of neutrophil distribution in cholangiocarcinoma. World J. Gastroenterol. 2015, 21, 4961–4968. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.L.; Dai, Z.; Zhou, Z.J.; Chen, Q.; Wang, Z.; Xiao, Y.S.; Hu, Z.Q.; Huang, X.Y.; Yang, G.H.; Shi, Y.H.; et al. CXCL5 contributes to tumor metastasis and recurrence of intrahepatic cholangiocarcinoma by recruiting infiltrative intratumoral neutrophils. Carcinogenesis 2014, 35, 597–605. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Lin, Y.; Li, B.; Yang, X.; Cai, Q.; Liu, W.; Tian, M.; Luo, H.; Yin, W.; Song, Y.; Shi, Y.; et al. Fibroblastic FAP promotes intrahepatic cholangiocarcinoma growth via MDSCs recruitment. Neoplasia 2019, 21, 1133–1142. [Google Scholar] [CrossRef]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016, 76, 4124–4135. [Google Scholar] [CrossRef]
- Dunne, R.F.; Figueroa, N.; Belt, B.; Findeis-Hosey, J.; Lunt, M.; McMahon, J.Y.; Baran, A.; Shubin, A.; Smith Noel, M.; Abdulaziz Tejani, M.; et al. The role of myeloid derived suppressor cells in cholangiocarcinoma: A potential target for therapy. J. Clin. Oncol. 2016, 34, 273. [Google Scholar] [CrossRef]
- Thepmalee, C.; Panya, A.; Sujjitjoon, J.; Sawasdee, N.; Poungvarin, N.; Junking, M.; Yenchitsomanus, P.T. Suppression of TGF-beta and IL-10 receptors on self-differentiated dendritic cells by short-hairpin RNAs enhanced activation of effector T-cells against cholangiocarcinoma cells. Hum. Vaccin. Immunother. 2020, 1–10. [Google Scholar] [CrossRef]
- Panya, A.; Thepmalee, C.; Sawasdee, N.; Sujjitjoon, J.; Phanthaphol, N.; Junking, M.; Wongkham, S.; Yenchitsomanus, P.T. Cytotoxic activity of effector T cells against cholangiocarcinoma is enhanced by self-differentiated monocyte-derived dendritic cells. Cancer Immunol. Immunother. 2018, 67, 1579–1588. [Google Scholar] [CrossRef]
- Martin-Sierra, C.; Martins, R.; Laranjeira, P.; Abrantes, A.M.; Oliveira, R.C.; Tralhao, J.G.; Botelho, M.F.; Furtado, E.; Domingues, R.; Paiva, A. Functional Impairment of Circulating FcepsilonRI(+) Monocytes and Myeloid Dendritic Cells in Hepatocellular Carcinoma and Cholangiocarcinoma Patients. Cytom. B Clin. Cytom. 2019, 96, 490–495. [Google Scholar] [CrossRef]
- Jung, I.H.; Kim, D.H.; Yoo, D.K.; Baek, S.Y.; Jeong, S.H.; Jung, D.E.; Park, S.W.; Chung, Y.Y. In Vivo Study of Natural Killer (NK) Cell Cytotoxicity Against Cholangiocarcinoma in a Nude Mouse Model. In Vivo 2018, 32, 771–781. [Google Scholar] [CrossRef]
- Fukuda, Y.; Asaoka, T.; Eguchi, H.; Yokota, Y.; Kubo, M.; Kinoshita, M.; Urakawa, S.; Iwagami, Y.; Tomimaru, Y.; Akita, H.; et al. Endogenous CXCL9 affects prognosis by regulating tumor-infiltrating natural killer cells in intrahepatic cholangiocarcinoma. Cancer Sci. 2020, 111, 323–333. [Google Scholar] [CrossRef]
- Schrumpf, E.; Tan, C.; Karlsen, T.H.; Sponheim, J.; Bjorkstrom, N.K.; Sundnes, O.; Alfsnes, K.; Kaser, A.; Jefferson, D.M.; Ueno, Y.; et al. The biliary epithelium presents antigens to and activates natural killer T cells. Hepatology 2015, 62, 1249–1259. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Okada, K.; Shimizu, Y.; Nambu, S.; Higuchi, K.; Watanabe, A. Interleukin-6 functions as an autocrine growth factor in a cholangiocarcinoma cell line. J. Gastroenterol. Hepatol. 1994, 9, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan-Kynurenine-Aryl Hydrocarbon Axis. Clin. Cancer Res. 2019, 25, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Gunther, J.; Dabritz, J.; Wirthgen, E. Limitations and Off-Target Effects of Tryptophan-Related IDO Inhibitors in Cancer Treatment. Front. Immunol. 2019, 10, 1801. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, J.W.; Iversen, T.Z.; Engell-Noerregaard, L.; Mellemgaard, A.; Andersen, M.H.; Svane, I.M. Durable Clinical Responses and Long-Term Follow-Up of Stage III-IV Non-Small-Cell Lung Cancer (NSCLC) Patients Treated With IDO Peptide Vaccine in a Phase I Study-A Brief Research Report. Front. Immunol. 2018, 9, 2145. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- David, P.; Megger, D.A.; Kaiser, T.; Werner, T.; Liu, J.; Chen, L.; Sitek, B.; Dittmer, U.; Zelinskyy, G. The PD-1/PD-L1 Pathway Affects the Expansion and Function of Cytotoxic CD8(+) T Cells During an Acute Retroviral Infection. Front. Immunol. 2019, 10, 54. [Google Scholar] [CrossRef]
- Saha, A.; O’Connor, R.S.; Thangavelu, G.; Lovitch, S.B.; Dandamudi, D.B.; Wilson, C.B.; Vincent, B.G.; Tkachev, V.; Pawlicki, J.M.; Furlan, S.N.; et al. Programmed death ligand-1 expression on donor T cells drives graft-versus-host disease lethality. J. Clin. Investig. 2016, 126, 2642–2660. [Google Scholar] [CrossRef]
- Chen, W.X.; Li, G.X.; Hu, Z.N.; Zhu, P.; Zhang, B.X.; Ding, Z.Y. Significant response to anti-PD-1 based immunotherapy plus lenvatinib for recurrent intrahepatic cholangiocarcinoma with bone metastasis: A case report and literature review. Medicine (Baltimore) 2019, 98, e17832. [Google Scholar] [CrossRef] [PubMed]
- Fingas, C.D.; Katsounas, A.; Kahraman, A.; Siffert, W.; Jochum, C.; Gerken, G.; Nuckel, H.; Canbay, A. Prognostic assessment of three single-nucleotide polymorphisms (GNB3 825C > T, BCL2-938C > A, MCL1-386C > G) in extrahepatic cholangiocarcinoma. Cancer Investig. 2010, 28, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Braconi, C.; Roessler, S.; Kruk, B.; Lammert, F.; Krawczyk, M.; Andersen, J.B. Molecular perturbations in cholangiocarcinoma: Is it time for precision medicine? Liver Int. 2019, 39 (Suppl. 1), 32–42. [Google Scholar] [CrossRef] [PubMed]
- Paolicchi, E.; Pacetti, P.; Giovannetti, E.; Mambrini, A.; Orlandi, M.; Crea, F.; Romani, A.A.; Tartarini, R.; Danesi, R.; Peters, G.J.; et al. A single nucleotide polymorphism in EZH2 predicts overall survival rate in patients with cholangiocarcinoma. Oncol. Lett. 2013, 6, 1487–1491. [Google Scholar] [CrossRef]
- Khunluck, T.; Kukongviriyapan, V.; Puapairoj, A.; Khuntikeo, N.; Senggunprai, L.; Zeekpudsa, P.; Prawan, A. Association of NRF2 polymorphism with cholangiocarcinoma prognosis in Thai patients. Asian Pac. J. Cancer Prev. 2014, 15, 299–304. [Google Scholar] [CrossRef]
- Huang, W.Y.; Gao, Y.T.; Rashid, A.; Sakoda, L.C.; Deng, J.; Shen, M.C.; Wang, B.S.; Han, T.Q.; Zhang, B.H.; Chen, B.E.; et al. Selected base excision repair gene polymorphisms and susceptibility to biliary tract cancer and biliary stones: A population-based case-control study in China. Carcinogenesis 2008, 29, 100–105. [Google Scholar] [CrossRef]
- Rankin, C.H. Interactions between two antagonistic reflexes in the nematode Caenorhabditis elegans. J. Comp. Physiol. A 1991, 169, 59–67. [Google Scholar] [CrossRef]
- Narang, N.; Garg, L.C.; Crews, F.T. Adenosine and its analogs stimulate phosphoinositide hydrolysis in the kidney. Pharmacology 1990, 40, 90–95. [Google Scholar] [CrossRef]
- Melum, E.; Karlsen, T.H.; Schrumpf, E.; Bergquist, A.; Thorsby, E.; Boberg, K.M.; Lie, B.A. Cholangiocarcinoma in primary sclerosing cholangitis is associated with NKG2D polymorphisms. Hepatology 2008, 47, 90–96. [Google Scholar] [CrossRef]
- Wu, Y.M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.F.; et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Liau, J.Y.; Tsai, J.H.; Yuan, R.H.; Chang, C.N.; Lee, H.J.; Jeng, Y.M. Morphological subclassification of intrahepatic cholangiocarcinoma: Etiological, clinicopathological, and molecular features. Mod. Pathol. 2014, 27, 1163–1173. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.; Phelps, M.A.; Li, X.; Saji, M.; Goff, L.; Kauh, J.S.; O’Neil, B.H.; Balsom, S.; Balint, C.; Liersemann, R.; et al. Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J. Clin. Oncol. 2011, 29, 2357–2363. [Google Scholar] [CrossRef] [PubMed]
- Goeppert, B.; Frauenschuh, L.; Renner, M.; Roessler, S.; Stenzinger, A.; Klauschen, F.; Warth, A.; Vogel, M.N.; Mehrabi, A.; Hafezi, M.; et al. BRAF V600E-specific immunohistochemistry reveals low mutation rates in biliary tract cancer and restriction to intrahepatic cholangiocarcinoma. Mod. Pathol. 2014, 27, 1028–1034. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef]
- Hoy, S.M. Pemigatinib: First Approval. Drugs 2020, 80, 923–929. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.A.; Marcano-Bonilla, L.; Roberts, L.R. Gallbladder cancer: Epidemiology and genetic risk associations. Chin. Clin. Oncol. 2019, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Baiu, I.; Visser, B. Gallbladder Cancer. JAMA 2018, 320, 1294. [Google Scholar] [CrossRef]
- Valle, J.W.; Borbath, I.; Khan, S.A.; Huguet, F.; Gruenberger, T.; Arnold, D.; Committee, E.G. Biliary cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v28–v37. [Google Scholar] [CrossRef] [PubMed]
- Koshiol, J.; Castro, F.; Kemp, T.J.; Gao, Y.T.; Roa, J.C.; Wang, B.; Nogueira, L.; Araya, J.C.; Shen, M.C.; Rashid, A.; et al. Association of inflammatory and other immune markers with gallbladder cancer: Results from two independent case-control studies. Cytokine 2016, 83, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.A.; Bizama, C.; Garcia, P.; Ferreccio, C.; Javle, M.; Miquel, J.F.; Koshiol, J.; Roa, J.C. The inflammatory inception of gallbladder cancer. Biochim. Biophys. Acta 2016, 1865, 245–254. [Google Scholar] [CrossRef]
- Gu, J.; Yan, S.; Wang, B.; Shen, F.; Cao, H.; Fan, J.; Wang, Y. Type 2 diabetes mellitus and risk of gallbladder cancer: A systematic review and meta-analysis of observational studies. Diabetes Metab. Res. Rev. 2016, 32, 63–72. [Google Scholar] [CrossRef]
- Wernberg, J.A.; Lucarelli, D.D. Gallbladder cancer. Surg. Clin. N. Am. 2014, 94, 343–360. [Google Scholar] [CrossRef]
- Kim, T.G. Patterns of initial failure after resection for gallbladder cancer: Implications for adjuvant radiotherapy. Radiat. Oncol. J. 2017, 35, 359–367. [Google Scholar] [CrossRef][Green Version]
- Jarnagin, W.R.; Ruo, L.; Little, S.A.; Klimstra, D.; D’Angelica, M.; DeMatteo, R.P.; Wagman, R.; Blumgart, L.H.; Fong, Y. Patterns of initial disease recurrence after resection of gallbladder carcinoma and hilar cholangiocarcinoma: Implications for adjuvant therapeutic strategies. Cancer 2003, 98, 1689–1700. [Google Scholar] [CrossRef]
- Javle, M.; Zhao, H.; Abou-Alfa, G.K. Systemic therapy for gallbladder cancer. Chin. Clin. Oncol. 2019, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Dodagoudar, C.; Doval, D.C.; Mahanta, A.; Goel, V.; Upadhyay, A.; Goyal, P.; Talwar, V.; Singh, S.; John, M.C.; Tiwari, S.; et al. FOLFOX-4 as second-line therapy after failure of gemcitabine and platinum combination in advanced gall bladder cancer patients. Jpn. J. Clin. Oncol. 2016, 46, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Belkouz, A.; de Vos-Geelen, J.; Mathot, R.A.A.; Eskens, F.; van Gulik, T.M.; van Oijen, M.G.H.; Punt, C.J.A.; Wilmink, J.W.; Klumpen, H.J. Efficacy and safety of FOLFIRINOX as salvage treatment in advanced biliary tract cancer: An open-label, single arm, phase 2 trial. Br. J. Cancer 2020, 122, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Z.; Li, X.; Ye, J.; Wu, X.; Tan, Z.; Liu, C.; Shen, B.; Wang, X.A.; Wu, W.; et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat. Genet. 2014, 46, 872–876. [Google Scholar] [CrossRef]
- Bridgewater, J.A.; Goodman, K.A.; Kalyan, A.; Mulcahy, M.F. Biliary Tract Cancer: Epidemiology, Radiotherapy, and Molecular Profiling. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e194–e203. [Google Scholar] [CrossRef]
- Valle, J.W.; Lamarca, A.; Goyal, L.; Barriuso, J.; Zhu, A.X. New Horizons for Precision Medicine in Biliary Tract Cancers. Cancer Discov. 2017, 7, 943–962. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Lassen, U.N.; Elez, E.; Italiano, A.; Curigliano, G.; De Braud, F.G.; Prager, G.; Greil, R.; Stein, A.; Fasolo, A.; et al. Efficacy and safety of dabrafenib (D) and trametinib (T) in patients (pts) with BRAF V600E–mutated biliary tract cancer (BTC): A cohort of the ROAR basket trial. J. Clin. Oncol. 2019, 37, 187. [Google Scholar] [CrossRef]
- Deshpande, V.; Nduaguba, A.; Zimmerman, S.M.; Kehoe, S.M.; Macconaill, L.E.; Lauwers, G.Y.; Ferrone, C.; Bardeesy, N.; Zhu, A.X.; Hezel, A.F. Mutational profiling reveals PIK3CA mutations in gallbladder carcinoma. BMC Cancer 2011, 11, 60. [Google Scholar] [CrossRef]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Sanchez-Vega, F.; Jonsson, P.; Chatila, W.K.; Hechtman, J.F.; Ku, G.Y.; Riches, J.C.; Tuvy, Y.; Kundra, R.; Bouvier, N.; et al. Genetic Predictors of Response to Systemic Therapy in Esophagogastric Cancer. Cancer Discov. 2018, 8, 49–58. [Google Scholar] [CrossRef]
- Hempelmann, J.A.; Lockwood, C.M.; Konnick, E.Q.; Schweizer, M.T.; Antonarakis, E.S.; Lotan, T.L.; Montgomery, B.; Nelson, P.S.; Klemfuss, N.; Salipante, S.J.; et al. Microsatellite instability in prostate cancer by PCR or next-generation sequencing. J. Immunother. Cancer 2018, 6, 29. [Google Scholar] [CrossRef]
- Weinberg, B.A.; Xiu, J.; Lindberg, M.R.; Shields, A.F.; Hwang, J.J.; Poorman, K.; Salem, M.E.; Pishvaian, M.J.; Holcombe, R.F.; Marshall, J.L.; et al. Molecular profiling of biliary cancers reveals distinct molecular alterations and potential therapeutic targets. J. Gastrointest. Oncol. 2019, 10, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Sugai, T.; Habano, W.; Nakamura, S.; Uesugi, N.; Funato, O.; Saito, K. Microsatellite instability in gallbladder carcinoma: Two independent genetic pathways of gallbladder carcinogenesis. J. Gastroenterol. 2000, 35, 768–774. [Google Scholar] [CrossRef]
- Rojas-Sepulveda, D.; Tittarelli, A.; Gleisner, M.A.; Avalos, I.; Pereda, C.; Gallegos, I.; Gonzalez, F.E.; Lopez, M.N.; Butte, J.M.; Roa, J.C.; et al. Tumor lysate-based vaccines: On the road to immunotherapy for gallbladder cancer. Cancer Immunol. Immunother. 2018, 67, 1897–1910. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Rankin, C.J.; Ben-Josef, E.; Lenz, H.J.; Gold, P.J.; Hamilton, R.D.; Govindarajan, R.; Eng, C.; Blanke, C.D. SWOG 0514: A phase II study of sorafenib in patients with unresectable or metastatic gallbladder carcinoma and cholangiocarcinoma. Investig. New Drugs 2012, 30, 1646–1651. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Rankin, C.; Siegel, A.B.; Iqbal, S.; Gong, I.Y.; Micetich, K.C.; Kayaleh, O.R.; Lenz, H.J.; Blanke, C.D. S0941: A phase 2 SWOG study of sorafenib and erlotinib in patients with advanced gallbladder carcinoma or cholangiocarcinoma. Br. J. Cancer 2014, 110, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Xiao, Y.P.; Tan, L.H.; Wang, L.T.; Cao, Q.; Qu, G.F.; Xiao, S.; Duan, H.X. Efficacy and safety of chemotherapy with or without targeted therapy in biliary tract cancer: A meta-analysis of 7 randomized controlled trials. J. Huazhong Univ. Sci. Technol. Med. Sci. 2017, 37, 172–178. [Google Scholar] [CrossRef]
- Ueno, M.; Ikeda, M.; Morizane, C.; Kobayashi, S.; Ohno, I.; Kondo, S.; Okano, N.; Kimura, K.; Asada, S.; Namba, Y.; et al. Nivolumab alone or in combination with cisplatin plus gemcitabine in Japanese patients with unresectable or recurrent biliary tract cancer: A non-randomised, multicentre, open-label, phase 1 study. Lancet Gastroenterol. Hepatol. 2019, 4, 611–621. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Treatment | Target | Pathway | Trial Type (Number of Patients) | Agent | Clinicaltrails.Gov Reference |
|---|---|---|---|---|---|
| Molecular targets | Respiratory electron transport | Phase 2 (36) | Olaparib | NCT04042831 | |
| ARID1A | ATP synthesis by chemiosmosis coupling | ||||
| Heat production by uncoupling proteins | |||||
| Oxytocin signaling pathway | Phase 2 (68) | Varlitinib | NCT03231176 | ||
| Direct p53 effector | Phase 2/3 (490) | Varlitinib | NCT03093870 | ||
| EGFR | Phase 1/1b (48) | Afatinib | NCT02451553 | ||
| Phase 2 (6452) | Afatinib | NCT02465060 | |||
| Phase 1/2 (25) | Erlotinib | NCT02273362 | |||
| PTEN | Metabolism of proteins | Phase 2 (6452) | Taselisib | NCT02465060 | |
| Direct p53 effector | |||||
| BRCA1 | Metabolism of proteins | Phase 1/2 (110) | Rucaparib | NCT03337087 | |
| ERK signaling | |||||
| Angiogenesis | Phase 2 (143) | Derazantinib | NCT03230318 | ||
| Wound healing | |||||
| FGFR2 | Cell migration | ||||
| Neural outgrowth | |||||
| Embryonic development | |||||
| RET signaling | Phase 2 (57) | RC48-ADC | NCT04329429 | ||
| ERBB2/HER2 | Phase 2 (15) | Trastuzumab | NCT03613168 | ||
| Phase 2 (100) | Trastuzumab | NCT03185988 | |||
| Phase 1/2 (82) | A166 | NCT03602079 | |||
| PARP | Post-translational modification | Phase 2 (35) | Rucaparib | NCT03639935 | |
| Phase 1/2 (110) | Rucaparib | NCT03337087 | |||
| MEK | Oxytocin signaling pathway | Phase 2 (57) | Selumetinib | NCT02151084 | |
| Phase 3 (50) | Sorafenib | NCT04163237 | |||
| Phase 1 (17) | Sorafenib | NCT02292173 | |||
| Checkpoint inhibitors | PD-1 | T-Cell receptor and co-stimulatory signaling | Observational study (100) | Nivolumab/ pembrolizumab | NCT03695952 |
| Class I MHC mediated antigen processing and presentation | Phase 2 (30) | Nivolumab | NCT04057365 | ||
| Phase 1/2 (40) | Nivolumab | NCT03785873 | |||
| Phase 3 (200) | Toripalimab | NCT03949231 | |||
| PD-L1 | Class I MHC mediated antigen processing and presentation | Phase 2 (90) | Durvalumab | NCT02821754 | |
| Cell adhesion molecules | Phase 2 (50) | Durvalumab | NCT04238637 | ||
| CTLA-4 | T-Cell receptor and co-stimulatory signaling | Phase 2 (45) | Ipilimumab | NCT03222076 | |
| Class I MHC mediated antigen processing and presentation |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malenica, I.; Donadon, M.; Lleo, A. Molecular and Immunological Characterization of Biliary Tract Cancers: A Paradigm Shift Towards a Personalized Medicine. Cancers 2020, 12, 2190. https://doi.org/10.3390/cancers12082190
Malenica I, Donadon M, Lleo A. Molecular and Immunological Characterization of Biliary Tract Cancers: A Paradigm Shift Towards a Personalized Medicine. Cancers. 2020; 12(8):2190. https://doi.org/10.3390/cancers12082190
Chicago/Turabian StyleMalenica, Ines, Matteo Donadon, and Ana Lleo. 2020. "Molecular and Immunological Characterization of Biliary Tract Cancers: A Paradigm Shift Towards a Personalized Medicine" Cancers 12, no. 8: 2190. https://doi.org/10.3390/cancers12082190
APA StyleMalenica, I., Donadon, M., & Lleo, A. (2020). Molecular and Immunological Characterization of Biliary Tract Cancers: A Paradigm Shift Towards a Personalized Medicine. Cancers, 12(8), 2190. https://doi.org/10.3390/cancers12082190