SARS-CoV-2 Infection and Lung Cancer: Potential Therapeutic Modalities

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. SARS-CoV-2 and Its Key Entry Genes
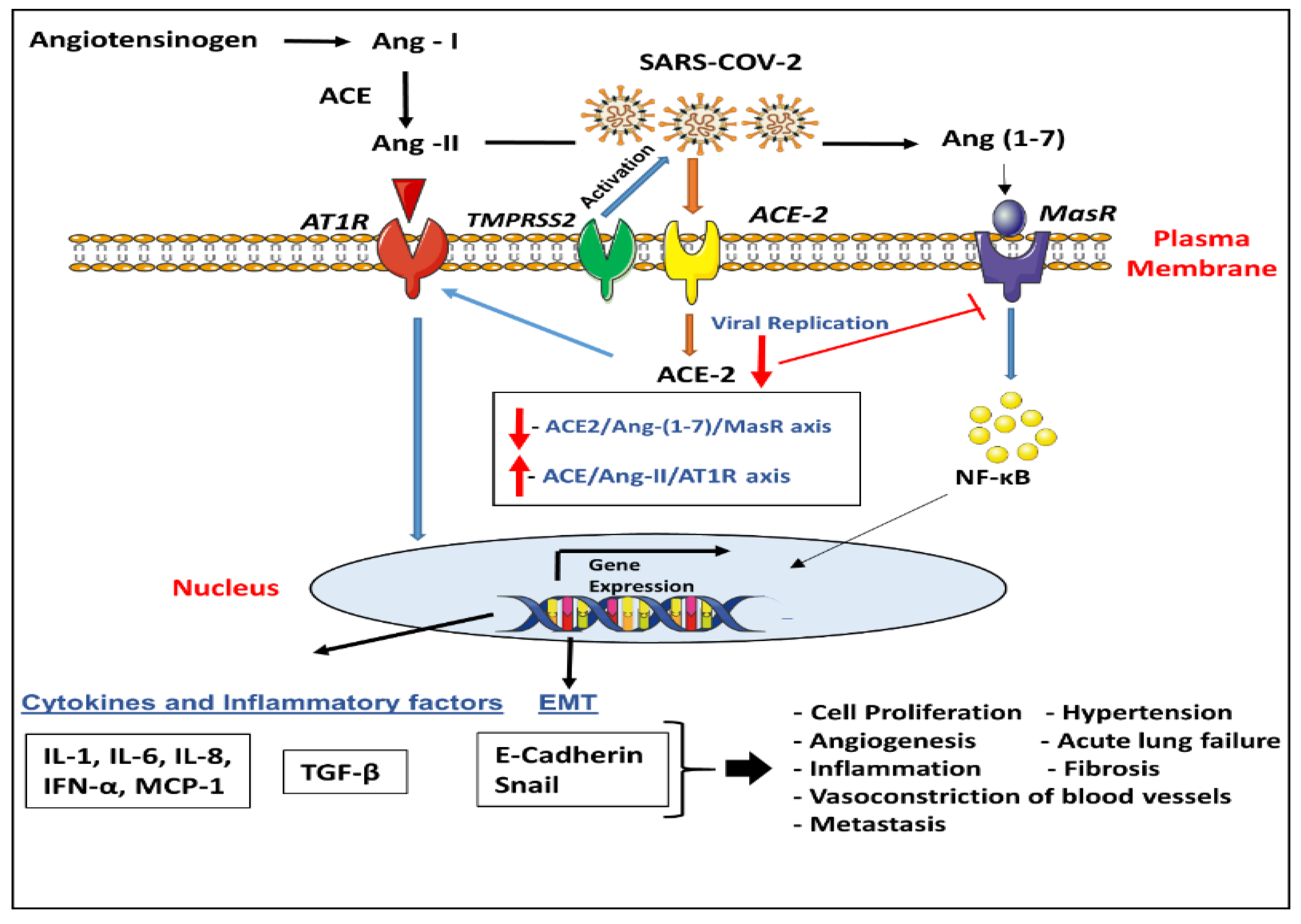
3. Role of ACE-2 and TMPRSS2 in Lung Disease/COVID-19
4. Key Entry Genes of SARS-CoV-2 and Human Cancers Including Lung Cancer
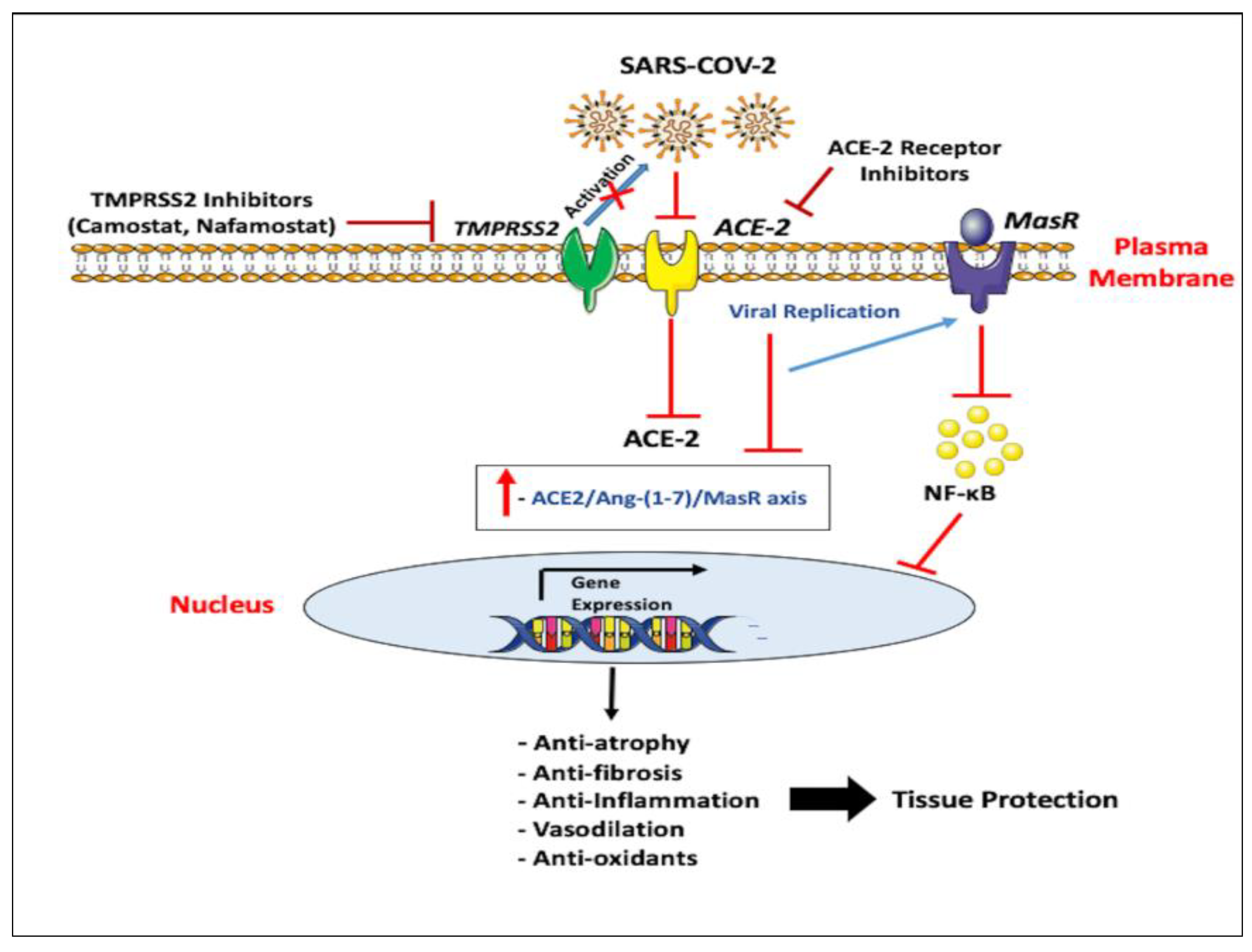
5. Emerging Therapeutic Approaches for ACE-2
6. Emerging Therapeutic Approaches for TMPRSS2
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Lemjabbar-Alaoui, H.; Hassan, O.U.; Yang, Y.-W.; Buchanan, P. Lung cancer: Biology and treatment options. Biochim. Biophys. Acta 2015, 1856, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Youlden, D.R.; Cramb, S.M.; Baade, P.D. The International Epidemiology of Lung Cancer: Geographical distribution and secular trends. J. Thorac. Oncol. 2008, 3, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Sasco, A.J.; Secretan, M.B.; Straif, K. Tobacco smoking and cancer: A brief review of recent epidemiological evidence. Lung Cancer 2004, 45 (Suppl. 2), S3–S9. [Google Scholar] [CrossRef]
- Littman, A.J.; Jackson, L.A.; Vaughan, T.L. Chlamydia pneumoniae and lung cancer: Epidemiologic evidence. Cancer Epidemiol. Biomark. Prev. 2005, 14, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Lanoix, J.-P.; Pluquet, E.; Lescure, F.X.; Bentayeb, H.; Lecuyer, E.; Boutemy, M.; Dumont, P.; Jounieaux, V.; Schmit, J.L.; Dayen, C.; et al. Bacterial infection profiles in lung cancer patients with febrile neutropenia. BMC Infect. Dis. 2011, 11, 183. [Google Scholar] [CrossRef]
- Greathouse, K.L.; White, J.R.; Vargas, A.J.; Bliskovsky, V.V.; Beck, J.A.; von Muhlinen, N.; Polley, E.C.; Bowman, E.D.; Khan, M.A.; Robles, A.I.; et al. Interaction between the microbiome and TP53 in human lung cancer. Genome Biol. 2018, 19, 123. [Google Scholar] [CrossRef]
- Robinson, L.A.; Jaing, C.J.; Pierce Campbell, C.; Magliocco, A.; Xiong, Y.; Magliocco, G.; Thissen, J.B.; Antonia, S. Molecular evidence of viral DNA in non-small cell lung cancer and non-neoplastic lung. Br. J. Cancer 2016, 115, 497–504. [Google Scholar] [CrossRef]
- Cardona, A.; Ruiz-Patiño, A.; Ricaurte, L.; Rojas, L.; Zatarain-Barrón, Z.L.; Arrieta, O.; Rosell, R. Human Papillomavirus Infection and Lung Cancer. In Current Perspectives in Human Papillomavirus; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Syrjänen, K.J. HPV infections and lung cancer. J. Clin. Pathol. 2002, 55, 885–891. [Google Scholar] [CrossRef]
- Kheir, F.; Zhao, M.; Strong, M.J.; Yu, Y.; Nanbo, A.; Flemington, E.K.; Morris, G.F.; Reiss, K.; Li, L.; Lin, Z. Detection of Epstein-Barr Virus Infection in Non-Small Cell Lung Cancer. Cancers 2019, 11, 759. [Google Scholar] [CrossRef]
- Zur Hausen, H. The search for infectious causes of human cancers: Where and why. Virology 2009, 392, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Z.; Zheng, B.; He, Z.; Winberg, G.; Ernberg, I. An update on viral association of human cancers. Arch. Virol. 2013, 158, 1433–1443. [Google Scholar] [CrossRef] [PubMed]
- Vijayanand, P.; Wilkins, E.; Woodhead, M. Severe acute respiratory syndrome (SARS): A review. Clin. Med. 2004, 4, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, N.; Shaib, H. Middle East respiratory syndrome coronavirus (MERS-CoV): A review. Germs 2019, 9, 35–42. [Google Scholar] [CrossRef]
- Kumar, D.; Malviya, R.; Kumar Sharma, P. Corona Virus: A Review of COVID-19. EJMO 2020, 4, 8–25. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Yuan, S.; Kok, K.-H.; To, K.K.-W.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.-Y.; Poon, R.W.-S.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, Q.; Zhang, Z. Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Curr. Biol. 2020, 30, 1346–1351.e2. [Google Scholar] [CrossRef]
- Shen, L.W.; Mao, H.J.; Wu, Y.L.; Tanaka, Y.; Zhang, W. TMPRSS2: A potential target for treatment of influenza virus and coronavirus infections. Biochimie 2017, 142, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Reguera, J.; Mudgal, G.; Santiago, C.; Casasnovas, J.M. A structural view of coronavirus–receptor interactions. Virus Res. 2014, 194, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.F. Angiotensin-I-converting enzyme and its relatives. Genome Biol. 2003, 4, 225. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient Activation of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein by the Transmembrane Protease TMPRSS2. J. Virol. 2020, 84, 12658–12664. [Google Scholar] [CrossRef]
- Iwata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Burkard, C.; Verheije, M.H.; Wicht, O.; van Kasteren, S.I.; van Kuppeveld, F.J.; Haagmans, B.L.; Pelkmans, L.; Rottier, P.J.; Bosch, B.J.; de Haan, C.A. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog. 2014, 10, e1004502. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA 2014, 111, 15214–15219. [Google Scholar] [CrossRef] [PubMed]
- Tigerstedt, R.; Bergman, P. Niere und Kreislauf 1. Skand. Arch. Für Physiol. 1898, 8, 223–271. [Google Scholar] [CrossRef]
- N Campbell, K.; Raij, L.; Mundel, P. Role of angiotensin II in the development of nephropathy and podocytopathy of diabetes. Curr. Diabetes Rev. 2011, 7, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Peach, M.J. Renin-angiotensin system: Biochemistry and mechanisms of action. Physiol. Rev. 1977, 57, 313–370. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, C.; Zhang, L. Angiotensin II receptors and drug discovery in cardiovascular disease. Drug Discov. Today 2011, 16, 22–34. [Google Scholar] [CrossRef]
- Ager, E.I.; Neo, J.; Christophi, C. The renin–angiotensin system and malignancy. Carcinogenesis 2008, 29, 1675–1684. [Google Scholar] [CrossRef]
- Gul, R.; Ramdas, M.; Mandavia, C.H.; Sowers, J.R.; Pulakat, L. RAS-mediated adaptive mechanisms in cardiovascular tissues: Confounding factors of RAS blockade therapy and alternative approaches. Cardiorenal Med. 2012, 2, 268–280. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R. A novel angiotensin-converting enzyme–related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000, 87, e1–e9. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Penninger, J.M. Recombinant human angiotensin-converting enzyme 2 as a new renin-angiotensin system peptidase for heart failure therapy. Curr. Heart Fail. Rep. 2011, 8, 176–183. [Google Scholar] [CrossRef]
- Skeggs, L.T.; Kahn, J.R.; Shumway, N.P. The preparation and function of the hypertensin-converting enzyme. J. Exp. Med. 1956, 103, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Angus, P.W.; Burrell, L.M.; Herath, C.; Gibson, P.R.; Lubel, J.S. The pathophysiological roles of the renin–angiotensin system in the gastrointestinal tract. Aliment. Pharmacol. Ther. 2012, 35, 414–428. [Google Scholar] [CrossRef]
- Jiang, F.; Yang, J.; Zhang, Y.; Dong, M.; Wang, S.; Zhang, Q.; Liu, F.F.; Zhang, K.; Zhang, C. Angiotensin-converting enzyme 2 and angiotensin 1–7: Novel therapeutic targets. Nat. Rev. Cardiol. 2014, 11, 413. [Google Scholar] [CrossRef] [PubMed]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [PubMed]
- Abuohashish, H.M.; Ahmed, M.M.; Sabry, D.; Khattab, M.M.; Al-Rejaie, S.S. Angiotensin (1-7) ameliorates the structural and biochemical alterations of ovariectomy-induced osteoporosis in rats via activation of ACE-2/Mas receptor axis. Sci. Rep. 2017, 7, 2293. [Google Scholar] [CrossRef]
- Santos, R.A.; e Silva, A.C.S.; Maric, C.; Silva, D.M.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.B.; Lopes, M.T.; Bader, M. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [PubMed]
- Iusuf, D.; Henning, R.H.; van Gilst, W.H.; Roks, A.J. Angiotensin-(1–7): Pharmacological properties and pharmacotherapeutic perspectives. Eur. J. Pharmacol. 2008, 585, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Allred, A.J.; Diz, D.I.; Ferrario, C.M.; Chappell, M.C. Pathways for angiotensin-(1—7) metabolism in pulmonary and renal tissues. Am. J. Physiol. Ren. Physiol. 2000, 279, F841–F850. [Google Scholar] [CrossRef]
- Hooper, J.D.; Clements, J.A.; Quigley, J.P.; Antalis, T.M. Type II transmembrane serine proteases. Insights into an emerging class of cell surface proteolytic enzymes. J. Biol. Chem. 2001, 276, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Bugge, T.H.; Antalis, T.M.; Wu, Q. Type II Transmembrane Serine Proteases. J. Biol. Chem. 2009, 284, 23177–23181. [Google Scholar] [CrossRef] [PubMed]
- Appel, L.F.; Prout, M.; Abu-Shumays, R.; Hammonds, A.; Garbe, J.C.; Fristrom, D.; Fristrom, J. The Drosophila Stubble-stubbloid gene encodes an apparent transmembrane serine protease required for epithelial morphogenesis. Proc. Natl. Acad. Sci. USA 1993, 90, 4937–4941. [Google Scholar] [CrossRef] [PubMed]
- Irving, P.; Troxler, L.; Heuer, T.S.; Belvin, M.; Kopczynski, C.; Reichhart, J.-M.; Hoffmann, J.A.; Hetru, C. A genome-wide analysis of immune responses in Drosophila. Proc. Natl. Acad. Sci. USA 2001, 98, 15119–15124. [Google Scholar] [CrossRef] [PubMed]
- Paoloni-Giacobino, A.; Chen, H.; Peitsch, M.C.; Rossier, C.; Antonarakis, S.E. Cloning of the TMPRSS2 Gene, Which Encodes a Novel Serine Protease with Transmembrane, LDLRA, and SRCR Domains and Maps to 21q22.3. Genomics 1997, 44, 309–320. [Google Scholar] [CrossRef]
- Jacquinet, E.; Rao, N.V.; Rao, G.V.; Zhengming, W.; Albertine, K.H.; Hoidal, J.R. Cloning and characterization of the cDNA and gene for human epitheliasin. Eur. J. Biochem. 2001, 268, 2687–2699. [Google Scholar] [CrossRef] [PubMed]
- Afar, D.E.; Vivanco, I.; Hubert, R.S.; Kuo, J.; Chen, E.; Saffran, D.C.; Raitano, A.B.; Jakobovits, A. Catalytic cleavage of the androgen-regulated TMPRSS2 protease results in its secretion by prostate and prostate cancer epithelia. Cancer Res. 2001, 61, 1686–1692. [Google Scholar]
- Chen, Y.-W.; Lee, M.-S.; Lucht, A.; Chou, F.-P.; Huang, W.; Havighurst, T.C.; Kim, K.; Wang, J.-K.; Antalis, T.M.; Johnson, M.D.; et al. TMPRSS2, a Serine Protease Expressed in the Prostate on the Apical Surface of Luminal Epithelial Cells and Released into Semen in Prostasomes, Is Misregulated in Prostate Cancer Cells. Am. J. Pathol. 2010, 176, 2986–2996. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Hirsh, A.; Li, D.C.; Holloway, G.; Chao, J.; Boucher, R.C.; Gabriel, S.E. Regulation of the epithelial sodium channel by serine proteases in human airways. J. Biol. Chem. 2002, 277, 8338–8345. [Google Scholar] [CrossRef]
- Rey-Parra, G.; Vadivel, A.; Coltan, L.; Hall, A.; Eaton, F.; Schuster, M.; Loibner, H.; Penninger, J.; Kassiri, Z.; Oudit, G. Angiotensin converting enzyme 2 abrogates bleomycin-induced lung injury. J. Mol. Med. 2012, 90, 637–647. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.-C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef]
- Zheng, Y.-Y.; Ma, Y.-T.; Zhang, J.-Y.; Xie, X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 2020, 17, 259–260. [Google Scholar] [CrossRef]
- Barnard, D.L.; Kumaki, Y. Recent developments in anti-severe acute respiratory syndrome coronavirus chemotherapy. Future Virol. 2011, 6, 615–631. [Google Scholar] [CrossRef] [PubMed]
- Yilin, Z.; Yandong, N.; Faguang, J. Role of angiotensin-converting enzyme (ACE) and ACE2 in a rat model of smoke inhalation induced acute respiratory distress syndrome. Burns 2015, 41, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Tolouian, R.; Vahed, S.Z.; Ghiyasvand, S.; Tolouian, A.; Ardalan, M. COVID-19 interactions with angiotensin-converting enzyme 2 (ACE2) and the kinin system; looking at a potential treatment. J. Ren. Inj. Prev. 2020, 9, 19. [Google Scholar] [CrossRef]
- Stewart, C.A.; Gay, C.M.; Ramkumar, K.; Cargill, K.R.; Cardnell, R.J.; Nilsson, M.B.; Heeke, S.; Park, E.M.; Kundu, S.T.; Diao, L.; et al. SARS-CoV-2 infection induces EMT-like molecular changes, including ZEB1-mediated repression of the viral receptor ACE2, in lung cancer models. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lin, B.; Ferguson, C.; White, J.T.; Wang, S.; Vessella, R.; True, L.D.; Hood, L.; Nelson, P.S. Prostate-localized and androgen-regulated expression of the membrane-bound serine protease TMPRSS2. Cancer Res. 1999, 59, 4180–4184. [Google Scholar]
- Lucas, J.; True, L.; Hawley, S.; Matsumura, M.; Morrissey, C.; Vessella, R.; Nelson, P. The androgen-regulated type II serine protease TMPRSS2 is differentially expressed and mislocalized in prostate adenocarcinoma. J. Pathol. 2008, 215, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Vaarala, M.H.; Porvari, K.S.; Kellokumpu, S.; Kyllönen, A.P.; Vihko, P.T. Expression of transmembrane serine protease TMPRSS2 in mouse and human tissues. J. Pathol 2001, 193, 134–140. [Google Scholar] [CrossRef]
- Böttcher-Friebertshäuser, E. Membrane-Anchored Serine Proteases: Host Cell Factors in Proteolytic Activation of Viral Glycoproteins. In Activation of Viruses by Host Proteases; Springer: Cham, Switzerland, 2018. [Google Scholar] [CrossRef]
- Böttcher, E.; Matrosovich, T.; Beyerle, M.; Klenk, H.-D.; Garten, W.; Matrosovich, M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 2006, 80, 9896–9898. [Google Scholar] [CrossRef]
- Böttcher-Friebertshäuser, E.; Freuer, C.; Sielaff, F.; Schmidt, S.; Eickmann, M.; Uhlendorff, J.; Steinmetzer, T.; Klenk, H.D.; Garten, W. Cleavage of influenza virus hemagglutinin by airway proteases TMPRSS2 and HAT differs in subcellular localization and susceptibility to protease inhibitors. J. Virol. 2010, 84, 5605–5614. [Google Scholar] [CrossRef]
- Chaipan, C.; Kobasa, D.; Bertram, S.; Glowacka, I.; Steffen, I.; Tsegaye, T.S.; Takeda, M.; Bugge, T.H.; Kim, S.; Park, Y.; et al. Proteolytic activation of the 1918 influenza virus hemagglutinin. J. Virol. 2009, 83, 3200–3211. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Heinlein, C.; Hackman, R.C.; Nelson, P.S. Phenotypic analysis of mice lacking the Tmprss2-encoded protease. Mol. Cell. Biol. 2006, 26, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Jaspers, I. Respiratory protease/antiprotease balance determines susceptibility to viral infection and can be modified by nutritional antioxidants. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1189–L1201. [Google Scholar] [CrossRef] [PubMed]
- Abboud, R.T.; Vimalanathan, S. Pathogenesis of COPD. Part I. The role of protease-antiprotease imbalance in emphysema. Int. J. Tuberc. Lung Dis. 2008, 12, 361–367. [Google Scholar]
- Kersul, A.L.; Iglesias, A.; Ríos, Á.; Noguera, A.; Forteza, A.; Serra, E.; Agustí, A.; Cosío, B.G. Molecular mechanisms of inflammation during exacerbations of chronic obstructive pulmonary disease. Arch. Bronconeumol. 2011, 47, 176–183. [Google Scholar] [CrossRef]
- Sakai, K.; Ami, Y.; Tahara, M.; Kubota, T.; Anraku, M.; Abe, M.; Nakajima, N.; Sekizuka, T.; Shirato, K.; Suzaki, Y.; et al. The Host Protease TMPRSS2 Plays a Major Role in In Vivo Replication of Emerging H7N9 and Seasonal Influenza Viruses. J. Virol. 2014, 88, 5608–5616. [Google Scholar] [CrossRef]
- Shirato, K.; Kawase, M.; Matsuyama, S. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J. Virol. 2013, 87, 12552–12561. [Google Scholar] [CrossRef]
- Cheng, Z.; Zhou, J.; To, K.K.; Chu, H.; Li, C.; Wang, D.; Yang, D.; Zheng, S.; Hao, K.; Bossé, Y.; et al. Identification of TMPRSS2 as a Susceptibility Gene for Severe 2009 Pandemic A(H1N1) Influenza and A(H7N9) Influenza. J. Infect. Dis. 2015, 212, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.V.; Harris, P.A.; Tollefson, S.J.; Halburnt-Rush, L.L.; Pingsterhaus, J.M.; Edwards, K.M.; Wright, P.F.; Crowe, J.E., Jr. Human metapneumovirus and lower respiratory tract disease in otherwise healthy infants and children. N. Engl. J. Med. 2004, 350, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Shirogane, Y.; Takeda, M.; Iwasaki, M.; Ishiguro, N.; Takeuchi, H.; Nakatsu, Y.; Tahara, M.; Kikuta, H.; Yanagi, Y. Efficient Multiplication of Human Metapneumovirus in Vero Cells Expressing the Transmembrane Serine Protease TMPRSS2. J. Virol. 2008, 82, 8942–8946. [Google Scholar] [CrossRef] [PubMed]
- Hatesuer, B.; Bertram, S.; Mehnert, N.; Bahgat, M.M.; Nelson, P.S.; Pöhlmann, S.; Schughart, K. Tmprss2 is essential for influenza H1N1 virus pathogenesis in mice. PLoS Pathog. 2013, 9, e1003774. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhu, F.; Xie, L.; Wang, C.; Wang, J.; Chen, R.; Jia, P.; Guan, H.; Peng, L.; Chen, Y. Clinical characteristics of COVID-19-infected cancer patients: A retrospective case study in three hospitals within Wuhan, China. Ann. Oncol. 2020, 31, 894–901. [Google Scholar]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, P.; Wei, Y.; Yue, H.; Wang, Y.; Hu, M.; Zhang, S.; Cao, T.; Yang, C.; Li, M.; et al. Histopathologic Changes and SARS-CoV-2 Immunostaining in the Lung of a Patient With COVID-19. Ann. Intern. Med. 2020, 172, 629–632. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Tian, S.; Xiao, S.Y. Pathology of 2019 Novel Coronavirus Pneumonia: A Dynamic Disease Process. J. Thorac. Oncol. 2020, 15, e67–e68. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Correction to: Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020, 46, 1294–1297. [Google Scholar] [CrossRef]
- Zhou, Y.; Vedantham, P.; Lu, K.; Agudelo, J.; Carrion, R., Jr.; Nunneley, J.W.; Barnard, D.; Pöhlmann, S.; McKerrow, J.H.; Renslo, A.R.; et al. Protease inhibitors targeting coronavirus and filovirus entry. Antivir. Res. 2015, 116, 76–84. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F.; et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7001–7003. [Google Scholar] [CrossRef]
- Meng, T.; Cao, H.; Zhang, H.; Kang, Z.; Xu, D.; Gong, H.; Wang, J.; Li, Z.; Cui, X.; Xu, H.; et al. The transmembrane serine protease inhibitors are potential antiviral drugs for 2019-nCoV targeting the insertion sequence-induced viral infectivity enhancement. bioRxiv 2020. [Google Scholar] [CrossRef]
- Li, R.; Qiao, S.; Zhang, G. Analysis of angiotensin-converting enzyme 2 (ACE2) from different species sheds some light on cross-species receptor usage of a novel coronavirus 2019-nCoV. J. Infect. 2020, 80, 469–496. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef] [PubMed]
- Bertram, S.; Glowacka, I.; Müller, M.A.; Lavender, H.; Gnirss, K.; Nehlmeier, I.; Niemeyer, D.; He, Y.; Simmons, G.; Drosten, C.; et al. Cleavage and Activation of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein by Human Airway Trypsin-Like Protease. J. Virol. 2011, 85, 13363–13372. [Google Scholar] [CrossRef] [PubMed]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, 39, e105114. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q. Analysis the susceptibility of lung cancer patients to SARS-CoV-2 infection. Mol. Cancer 2020, 19, 1–5. [Google Scholar] [CrossRef]
- Pinto, B.G.; Oliveira, A.E.; Singh, Y.; Jimenez, L.; Goncalves, A.N.; Ogava, R.L.; Creighton, R.; Peron, J.P.; Nakaya, H.I. ACE2 Expression is Increased in the Lungs of Patients with Comorbidities Associated with Severe COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Kim, W.J.; Lim, J.H.; Lee, J.S.; Lee, S.-D.; Kim, J.H.; Oh, Y.-M. Comprehensive Analysis of Transcriptome Sequencing Data in the Lung Tissues of COPD Subjects. Int. J. Genom. 2015, 2015, 206937. [Google Scholar] [CrossRef]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B., Jr. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L84–L96. [Google Scholar] [CrossRef]
- Totura, A.L.; Whitmore, A.; Agnihothram, S.; Schäfer, A.; Katze, M.G.; Heise, M.T.; Baric, R.S. Toll-Like Receptor 3 Signaling via TRIF Contributes to a Protective Innate Immune Response to Severe Acute Respiratory Syndrome Coronavirus Infection. mBio 2015, 6, e00638-15. [Google Scholar] [CrossRef]
- Liang, W.; Guan, W.; Chen, R.; Wang, W.; Li, J.; Xu, K.; Li, C.; Ai, Q.; Lu, W.; Liang, H. Cancer patients in SARS-CoV-2 infection: A nationwide analysis in China. Lancet Oncol. 2020, 21, 335–337. [Google Scholar] [CrossRef]
- Hsueh, W.A.; Wyne, K. Renin-angiotensin-aldosterone system in diabetes and hypertension. J. Clin. Hypertens. 2011, 13, 224–237. [Google Scholar] [CrossRef]
- Lau, S.T.; Leung, P.S. Role of the RAS in pancreatic cancer. Curr. Cancer Drug Targets 2011, 11, 412–420. [Google Scholar] [CrossRef]
- Geuna, E.; Lombardi, P.; Martinello, R.; Garino, D.; Bonzano, A.; Galizia, D.; Nuzzo, A.; Berchialla, P.; Becco, P.; Mangioni, M. Treatment with Beta-Blockers and ACE-Inhibitors in Breast Cancer Patients Receiving Adjuvant Trastuzumab-Based Therapy and Developing Mild Cardiac Toxicity: A Prospective Study. Cancers 2020, 12, 327. [Google Scholar] [CrossRef]
- Babacan, T.; Balakan, O.; Kuzan, T.Y.; Sarici, F.; Koca, E.; Kertmen, N.; Petekkaya, I.; Altundag, K. The effect of renin-angiotensin-system inhibition on survival and recurrence of N3+ breast cancer patients. J. Buon 2015, 20, 50–56. [Google Scholar]
- Ino, K.; Shibata, K.; Yamamoto, E.; Kajiyama, H.; Nawa, A.; Mabuchi, Y.; Yagi, S.; Minami, S.; Tanizaki, Y.; Kobayashi, A. Role of the renin-angiotensin system in gynecologic cancers. Curr. Cancer Drug Targets 2011, 11, 405–411. [Google Scholar] [CrossRef]
- Kuniyasu, H. Multiple roles of angiotensin in colorectal cancer. World J. Clin. Oncol. 2012, 3, 150. [Google Scholar] [CrossRef]
- De Paula, G.A.; Palmeira, V.; Ribeiro, T.; Costa, L.; de Sá, R.K. ACE2/Angiotensin-(1-7)/Mas receptor axis in human cancer: Potential role for pediatric tumors. Curr. Drug Targets 2020. [Google Scholar] [CrossRef]
- Ramírez-Expósito, M.J.; Martínez-Martos, J.M. Anti-Inflammatory and antitumor effects of hydroxytyrosol but not oleuropein on experimental glioma in vivo. A putative role for the renin-angiotensin system. Biomedicines 2018, 6, 11. [Google Scholar]
- Tikellis, C.; Thomas, M. Angiotensin-converting enzyme 2 (ACE2) is a key modulator of the renin angiotensin system in health and disease. Int. J. Pept. 2012, 2012, 256294. [Google Scholar] [CrossRef]
- Passos-Silva, D.G.; Brandan, E.; Santos, R.A.S. Angiotensins as therapeutic targets beyond heart disease. Trends Pharmacol. Sci. 2015, 36, 310–320. [Google Scholar] [CrossRef]
- Rasha, F.; Ramalingam, L.; Gollahon, L.; Rahman, R.; Rahman, S.M.; Menikdiwela, K.; Moustaid-Moussa, N. Mechanisms linking the renin-angiotensin system, obesity, and breast cancer. Endocr. Relat. Cancer 2019, 26, 653–672. [Google Scholar] [CrossRef]
- Qian, Y.-R.; Guo, Y.; Wan, H.-Y.; Fan, L.; Feng, Y.; Ni, L.; Xiang, Y.; Li, Q.-Y. Angiotensin-converting enzyme 2 attenuates the metastasis of non-small cell lung cancer through inhibition of epithelial-mesenchymal transition. Oncol. Rep. 2013, 29, 2408–2414. [Google Scholar] [CrossRef]
- Ni, L.; Feng, Y.; Wan, H.; Ma, Q.; Fan, L.; Qian, Y.; Li, Q.; Xiang, Y.; Gao, B. Angiotensin-(1-7) inhibits the migration and invasion of A549 human lung adenocarcinoma cells through inactivation of the PI3K/Akt and MAPK signaling pathways. Oncol. Rep. 2012, 27, 783–790. [Google Scholar]
- Zhou, L.; Zhang, R.; Zhang, L.; Yao, W.; Li, J.; Yuan, Y. Angiotensin-converting enzyme 2 acts as a potential molecular target for pancreatic cancer therapy. Cancer Lett. 2011, 307, 18–25. [Google Scholar] [CrossRef]
- Yu, C.; Tang, W.; Wang, Y.; Shen, Q.; Wang, B.; Cai, C.; Meng, X.; Zou, F. Downregulation of ACE2/Ang-(1–7)/Mas axis promotes breast cancer metastasis by enhancing store-operated calcium entry. Cancer Lett. 2016, 376, 268–277. [Google Scholar] [CrossRef]
- e Silva, A.C.S.; Sampaio, W.O. The Role of Angiotensin–(1-7) in Cancer. In Angiotensin-(1-7); Springer: Berlin/Heidelberg, Germany, 2019; pp. 219–229. [Google Scholar]
- e Silva, A.C.S.; Teixeira, M.M. ACE inhibition, ACE2 and angiotensin-(1–7) axis in kidney and cardiac inflammation and fibrosis. Pharmacol. Res. 2016, 107, 154–162. [Google Scholar] [CrossRef]
- Gallagher, P.; Arter, A.; Deng, G.; Tallant, E. Angiotensin-(1-7): A peptide hormone with anti-cancer activity. Curr. Med. Chem. 2014, 21, 2417–2423. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Jiang, C.; Penninger, J.M. Lessons from SARS: Control of acute lung failure by the SARS receptor ACE2. J. Mol. Med. 2006, 84, 814–820. [Google Scholar] [CrossRef]
- Delforce, S.J.; Lumbers, E.R.; de Meaultsart, C.C.; Wang, Y.; Proietto, A.; Otton, G.; Scurry, J.; Verrills, N.M.; Scott, R.J.; Pringle, K.G. Expression of renin–angiotensin system (RAS) components in endometrial cancer. Endocr. Connect. 2017, 6, 9. [Google Scholar] [CrossRef]
- Das, U.N. Reply to: Bioactive Lipids and Coronavirus (COVID-19)-further Discussion. Arch. Med. Res. 2020. [Google Scholar]
- Antalis, T.M.; Bugge, T.H.; Wu, Q. Chapter 1-Membrane-Anchored Serine Proteases in Health and Disease. In Progress in Molecular Biology and Translational Science; Di Cera, E., Ed.; Academic Press: Cambridge, MA, USA, 2011; Volume 99, pp. 1–50. [Google Scholar]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef]
- Yu, J.; Yu, J.; Mani, R.-S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; et al. An Integrated Network of Androgen Receptor, Polycomb, and TMPRSS2-ERG Gene Fusions in Prostate Cancer Progression. Cancer Cell 2010, 17, 443–454. [Google Scholar] [CrossRef]
- Li, Y.; Sarkar, F.H. Down-regulation of invasion and angiogenesis-related genes identified by cDNA microarray analysis of PC3 prostate cancer cells treated with genistein. Cancer Lett. 2002, 186, 157–164. [Google Scholar] [CrossRef]
- Greenberg, D.L.; Mize, G.J.; Takayama, T.K. Protease-activated receptor mediated RhoA signaling and cytoskeletal reorganization in LNCaP cells. Biochemistry 2003, 42, 702–709. [Google Scholar] [CrossRef]
- Cooper, C.R.; Chay, C.H.; Gendernalik, J.D.; Lee, H.L.; Bhatia, J.; Taichman, R.S.; McCauley, L.K.; Keller, E.T.; Pienta, K.J. Stromal factors involved in prostate carcinoma metastasis to bone. Cancer 2003, 97, 739–747. [Google Scholar] [CrossRef]
- Vaarala, M.H.; Porvari, K.; Kyllönen, A.; Vihko, P. Differentially expressed genes in two LNCaP prostate cancer cell lines reflecting changes during prostate cancer progression. Lab. Investig. 2000, 80, 1259–1268. [Google Scholar] [CrossRef]
- Macfarlane, S.R.; Seatter, M.J.; Kanke, T.; Hunter, G.D.; Plevin, R. Proteinase-activated receptors. Pharm. Rev. 2001, 53, 245–282. [Google Scholar]
- Bertram, S.; Glowacka, I.; Blazejewska, P.; Soilleux, E.; Allen, P.; Danisch, S.; Steffen, I.; Choi, S.Y.; Park, Y.; Schneider, H.; et al. TMPRSS2 and TMPRSS4 facilitate trypsin-independent spread of influenza virus in Caco-2 cells. J. Virol. 2010, 84, 10016–10025. [Google Scholar] [CrossRef]
- Yamaya, M.; Shimotai, Y.; Hatachi, Y.; Lusamba Kalonji, N.; Tando, Y.; Kitajima, Y.; Matsuo, K.; Kubo, H.; Nagatomi, R.; Hongo, S.; et al. The serine protease inhibitor camostat inhibits influenza virus replication and cytokine production in primary cultures of human tracheal epithelial cells. Pulm. Pharm. Ther. 2015, 33, 66–74. [Google Scholar] [CrossRef]
- Jung, H.; Lee, K.P.; Park, S.J.; Park, J.H.; Jang, Y.S.; Choi, S.Y.; Jung, J.G.; Jo, K.; Park, D.Y.; Yoon, J.H.; et al. TMPRSS4 promotes invasion, migration and metastasis of human tumor cells by facilitating an epithelial-mesenchymal transition. Oncogene 2008, 27, 2635–2647. [Google Scholar] [CrossRef]
- Yasuoka, S.; Ohnishi, T.; Kawano, S.; Tsuchihashi, S.; Ogawara, M.; Masuda, K.; Yamaoka, K.; Takahashi, M.; Sano, T. Purification, characterization, and localization of a novel trypsin-like protease found in the human airway. Am. J. Respir. Cell Mol. Biol. 1997, 16, 300–308. [Google Scholar] [CrossRef]
- Yamaoka, K.; Masuda, K.; Ogawa, H.; Takagi, K.; Umemoto, N.; Yasuoka, S. Cloning and characterization of the cDNA for human airway trypsin-like protease. J. Biol. Chem. 1998, 273, 11895–11901. [Google Scholar] [CrossRef]
- Guo, C.C.; Dancer, J.Y.; Wang, Y.; Aparicio, A.; Navone, N.M.; Troncoso, P.; Czerniak, B.A. TMPRSS2-ERG gene fusion in small cell carcinoma of the prostate. Hum. Pathol. 2011, 42, 11–17. [Google Scholar] [CrossRef]
- Passaro, A.; Peters, S.; Mok, T.S.K.; Attili, I.; Mitsudomi, T.; de Marinis, F. Testing for COVID-19 in lung cancer patients. Ann. Oncol. 2020, 31, 832–834. [Google Scholar] [CrossRef]
- Zhao, Z.; Bai, H.; Duan, J.; Wang, J. Recommendations of individualized medical treatment and common adverse events management for lung cancer patients during the outbreak of COVID-19 epidemic. Thorac. Cancer 2020. [Google Scholar] [CrossRef]
- Chinese, T.S.; Group, L.C.S.; Association, C.M.; Collaboration, C.R.O. Expert recommendations on the management of patients with advanced non-small cell lung cancer during epidemic of COVID-19 (Trial version). Zhonghua Jie He He Hu Xi Za Zhi = Zhonghua Jiehe He Huxi Zazhi = Chin. J. Tuberc. Respir. Dis. 2020, 43, E031. [Google Scholar]
- Hung, Y.-H.; Hsieh, W.-Y.; Hsieh, J.-S.; Liu, C.; Tsai, C.-H.; Lu, L.-C.; Huang, C.-Y.; Wu, C.-L.; Lin, C.-S. Alternative roles of STAT3 and MAPK signaling pathways in the MMPs activation and progression of lung injury induced by cigarette smoke exposure in ACE2 knockout mice. Int. J. Biol. Sci. 2016, 12, 454. [Google Scholar] [CrossRef]
- Rubinfeld, H.; Seger, R. The ERK cascade: A prototype of MAPK signaling. Mol. Biotechnol. 2005, 31, 151–174. [Google Scholar] [CrossRef]
- Bang, Y.J.; Kwon, J.H.; Kang, S.H.; Kim, J.W.; Yang, Y.C. Increased MAPK activity and MKP-1 overexpression in human gastric adenocarcinoma. Biochem. Biophys. Res. Commun. 1998, 250, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Lefloch, R.; Pouysségur, J.; Lenormand, P. Total ERK1/2 activity regulates cell proliferation. Cell Cycle 2009, 8, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Khatlani, T.S.; Wislez, M.; Sun, M.; Srinivas, H.; Iwanaga, K.; Ma, L.; Hanna, A.E.; Liu, D.; Girard, L.; Kim, Y.H.; et al. c-Jun N-terminal kinase is activated in non-small-cell lung cancer and promotes neoplastic transformation in human bronchial epithelial cells. Oncogene 2007, 26, 2658–2666. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Fukushi, S.; Murakami, M.; Hirano, T.; Saijo, M.; Kurane, I.; Morikawa, S. Tyrosine dephosphorylation of STAT3 in SARS coronavirus-infected Vero E6 cells. FEBS Lett. 2004, 577, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Fukushi, S.; Saijo, M.; Kurane, I.; Morikawa, S. Phosphorylation of p38 MAPK and its downstream targets in SARS coronavirus-infected cells. Biochem. Biophys. Res. Commun. 2004, 319, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Fukushi, S.; Saijo, M.; Kurane, I.; Morikawa, S. JNK and PI3k/Akt signaling pathways are required for establishing persistent SARS-CoV infection in Vero E6 cells. Biochim. Biophys. Acta 2005, 1741, 4–10. [Google Scholar] [CrossRef]
- Greenberg, A.K.; Basu, S.; Hu, J.; Yie, T.-a.; Tchou-Wong, K.M.; Rom, W.N.; Lee, T.C. Selective p38 Activation in Human Non–Small Cell Lung Cancer. Am. J. Respir. Cell Mol. Biol. 2002, 26, 558–564. [Google Scholar] [CrossRef]
- Chen, I.Y.; Chang, S.C.; Wu, H.Y.; Yu, T.C.; Wei, W.C.; Lin, S.; Chien, C.L.; Chang, M.F. Upregulation of the chemokine (C-C motif) ligand 2 via a severe acute respiratory syndrome coronavirus spike-ACE2 signaling pathway. J. Virol. 2010, 84, 7703–7712. [Google Scholar] [CrossRef]
- Lim, Y.X.; Ng, Y.L.; Tam, J.P.; Liu, D.X. Human Coronaviruses: A Review of Virus-Host Interactions. Diseases 2016, 4, 26. [Google Scholar] [CrossRef]
- Liu, M.; Yang, Y.; Gu, C.; Yue, Y.; Wu, K.K.; Wu, J.; Zhu, Y. Spike protein of SARS-CoV stimulates cyclooxygenase-2 expression via both calcium-dependent and calciumin-dependent protein kinase C pathways. FASEB J. 2007, 21, 1586–1596. [Google Scholar] [CrossRef]
- Fung, T.S.; Liao, Y.; Liu, D.X. The endoplasmic reticulum stress sensor IRE1α protects cells from apoptosis induced by the coronavirus infectious bronchitis virus. J. Virol. 2014, 88, 12752–12764. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Liu, D.X. Activation of the c-Jun NH(2)-terminal kinase pathway by coronavirus infectious bronchitis virus promotes apoptosis independently of c-Jun. Cell Death Dis. 2017, 8, 3215. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033. [Google Scholar] [CrossRef]
- Kawase, M.; Shirato, K.; van der Hoek, L.; Taguchi, F.; Matsuyama, S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef]
- Gurwitz, D. Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug Dev. Res. 2020. [Google Scholar] [CrossRef]
- Touyz, R.M.; Li, H.; Delles, C. ACE2 the Janus-faced protein–from cardiovascular protection to severe acute respiratory syndrome-coronavirus and COVID-19. Clin. Sci. 2020, 134, 747–750. [Google Scholar] [CrossRef]
- Kuster, G.M.; Pfister, O.; Burkard, T.; Zhou, Q.; Twerenbold, R.; Haaf, P.; Widmer, A.F.; Osswald, S. SARS-CoV2: Should inhibitors of the renin–angiotensin system be withdrawn in patients with COVID-19? Eur. Heart J. 2020, 41, 1801–1803. [Google Scholar] [CrossRef]
- McKay, R.R.; Rodriguez, G.E.; Lin, X.; Kaymakcalan, M.D.; Hamnvik, O.-P.R.; Sabbisetti, V.S.; Bhatt, R.S.; Simantov, R.; Choueiri, T.K. Angiotensin system inhibitors and survival outcomes in patients with metastatic renal cell carcinoma. Clin. Cancer Res. 2015, 21, 2471–2479. [Google Scholar] [CrossRef]
- Neo, J.H.; Ager, E.I.; Angus, P.W.; Zhu, J.; Herath, C.B.; Christophi, C. Changes in the renin angiotensin system during the development of colorectal cancer liver metastases. BMC Cancer 2010, 10, 134. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, X.; Cai, L.; Yang, Y.; Sui, G.; Fu, S. Angiotensin II/angiotensin II type I receptor (AT1R) signaling promotes MCF-7 breast cancer cells survival via PI3-kinase/Akt pathway. J. Cell. Physiol. 2010, 225, 168–173. [Google Scholar] [CrossRef]
- Aydiner, A.; Ciftci, R.; Sen, F. Renin-Angiotensin system blockers may prolong survival of metastatic non-small cell lung cancer patients receiving erlotinib. Medicine 2015, 94, e887. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Torres, E.; Oyarzún, A.; Mondaca-Ruff, D.; Azocar, A.; Castro, P.F.; Jalil, J.E.; Chiong, M.; Lavandero, S.; Ocaranza, M.P. ACE2 and vasoactive peptides: Novel players in cardiovascular/renal remodeling and hypertension. Ther. Adv. Cardiovasc. Dis. 2015, 9, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.K.; Li, W.; Moore, M.J.; Choe, H.; Farzan, M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 2004, 279, 3197–3201. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mathieu, M.; Brezski, R.J. IgG Fc engineering to modulate antibody effector functions. Protein Cell 2018, 9, 63–73. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Kruse, R.L. Therapeutic strategies in an outbreak scenario to treat the novel coronavirus originating in Wuhan, China. F1000Research 2020, 9, 72. [Google Scholar] [CrossRef]
- Moore, M.J.; Dorfman, T.; Li, W.; Wong, S.K.; Li, Y.; Kuhn, J.H.; Coderre, J.; Vasilieva, N.; Han, Z.; Greenough, T.C. Retroviruses pseudotyped with the severe acute respiratory syndrome coronavirus spike protein efficiently infect cells expressing angiotensin-converting enzyme 2. J. Virol. 2004, 78, 10628–10635. [Google Scholar] [CrossRef]
- Abe, M.; Tahara, M.; Sakai, K.; Yamaguchi, H.; Kanou, K.; Shirato, K.; Kawase, M.; Noda, M.; Kimura, H.; Matsuyama, S.; et al. TMPRSS2 is an activating protease for respiratory parainfluenza viruses. J. Virol. 2013, 87, 11930–11935. [Google Scholar] [CrossRef]
- Shin, W.J.; Seong, B.L. Type II transmembrane serine proteases as potential target for anti-influenza drug discovery. Expert Opin. Drug Discov. 2017, 12, 1139–1152. [Google Scholar] [CrossRef]
- Yamamoto, M.; Matsuyama, S.; Li, X.; Takeda, M.; Kawaguchi, Y.; Inoue, J.-i.; Matsuda, Z. Identification of Nafamostat as a Potent Inhibitor of Middle East Respiratory Syndrome Coronavirus S Protein-Mediated Membrane Fusion Using the Split-Protein-Based Cell-Cell Fusion Assay. Antimicrob. Agents Chemother. 2016, 60, 6532–6539. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Böttcher-Friebertshäuser, E.; Lu, Y.; Meyer, D.; Sielaff, F.; Steinmetzer, T.; Klenk, H.D.; Garten, W. Hemagglutinin activating host cell proteases provide promising drug targets for the treatment of influenza A and B virus infections. Vaccine 2012, 30, 7374–7380. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Datta, A.; Talwar, S.; Saleem, S.N.; Mondal, D.; Abdel-Mageed, A.B. Estradiol-ERβ2 signaling axis confers growth and migration of CRPC cells through TMPRSS2-ETV5 gene fusion. Oncotarget 2016, 8, 62820–62833. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kong, D.; Wang, Z.; Ahmad, A.; Bao, B.; Padhye, S.; Sarkar, F.H. Inactivation of AR/TMPRSS2-ERG/Wnt signaling networks attenuates the aggressive behavior of prostate cancer cells. Cancer Prev. Res. 2011, 4, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Semaan, L.; Mander, N.; Cher, M.L.; Chinni, S.R. TMPRSS2-ERG fusions confer efficacy of enzalutamide in an in vivo bone tumor growth model. BMC Cancer 2019, 19, 972. [Google Scholar] [CrossRef] [PubMed]
- Ateeq, B.; Vellaichamy, A.; Tomlins, S.A.; Wang, R.; Cao, Q.; Lonigro, R.J.; Pienta, K.J.; Varambally, S. Role of dutasteride in pre-clinical ETS fusion-positive prostate cancer models. Prostate 2012, 72, 1542–1549. [Google Scholar] [CrossRef]
- Knuuttila, M.; Mehmood, A.; Mäki-Jouppila, J.; Ryberg, H.; Taimen, P.; Knaapila, J.; Ettala, O.; Boström, P.J.; Ohlsson, C.; Venäläinen, M.S.; et al. Intratumoral androgen levels are linked to TMPRSS2-ERG fusion in prostate cancer. Endocr. Relat. Cancer 2018, 25, 807–819. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fett, C.; Mack, M.; Ten Eyck, P.P.; Meyerholz, D.K.; Perlman, S. Sex-Based Differences in Susceptibility to Severe Acute Respiratory Syndrome Coronavirus Infection. J. Immunol. 2017, 198, 4046–4053. [Google Scholar] [CrossRef]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, I.; Rizeq, B.; Elkord, E.; Vranic, S.; Al Moustafa, A.-E. SARS-CoV-2 Infection and Lung Cancer: Potential Therapeutic Modalities. Cancers 2020, 12, 2186. https://doi.org/10.3390/cancers12082186
Gupta I, Rizeq B, Elkord E, Vranic S, Al Moustafa A-E. SARS-CoV-2 Infection and Lung Cancer: Potential Therapeutic Modalities. Cancers. 2020; 12(8):2186. https://doi.org/10.3390/cancers12082186
Chicago/Turabian StyleGupta, Ishita, Balsam Rizeq, Eyad Elkord, Semir Vranic, and Ala-Eddin Al Moustafa. 2020. "SARS-CoV-2 Infection and Lung Cancer: Potential Therapeutic Modalities" Cancers 12, no. 8: 2186. https://doi.org/10.3390/cancers12082186
APA StyleGupta, I., Rizeq, B., Elkord, E., Vranic, S., & Al Moustafa, A.-E. (2020). SARS-CoV-2 Infection and Lung Cancer: Potential Therapeutic Modalities. Cancers, 12(8), 2186. https://doi.org/10.3390/cancers12082186