Molecular Markers Guiding Thyroid Cancer Management

 , ,
, ,
Abstract
1. Introduction
2. The Molecular Landscape of Follicular Cell Derived Thyroid Cancer and Oncogenesis
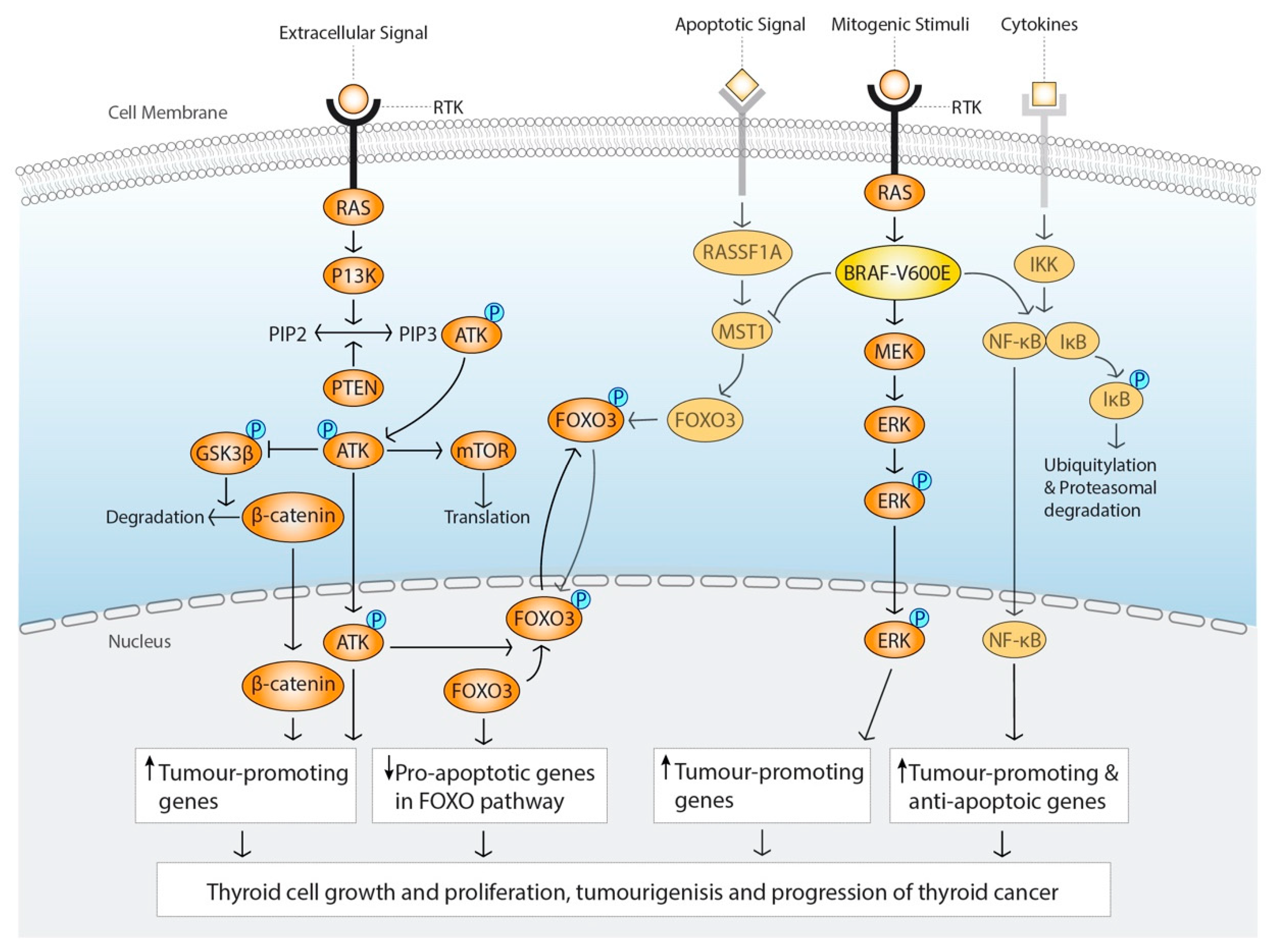
2.1. The MAPK-Signalling Pathway
2.2. PI3K/AKT Signalling Pathway
2.3. Gene Translocations
2.4. Gene Amplification and Copy Number Alterations
2.5. Epigenetic Regulation
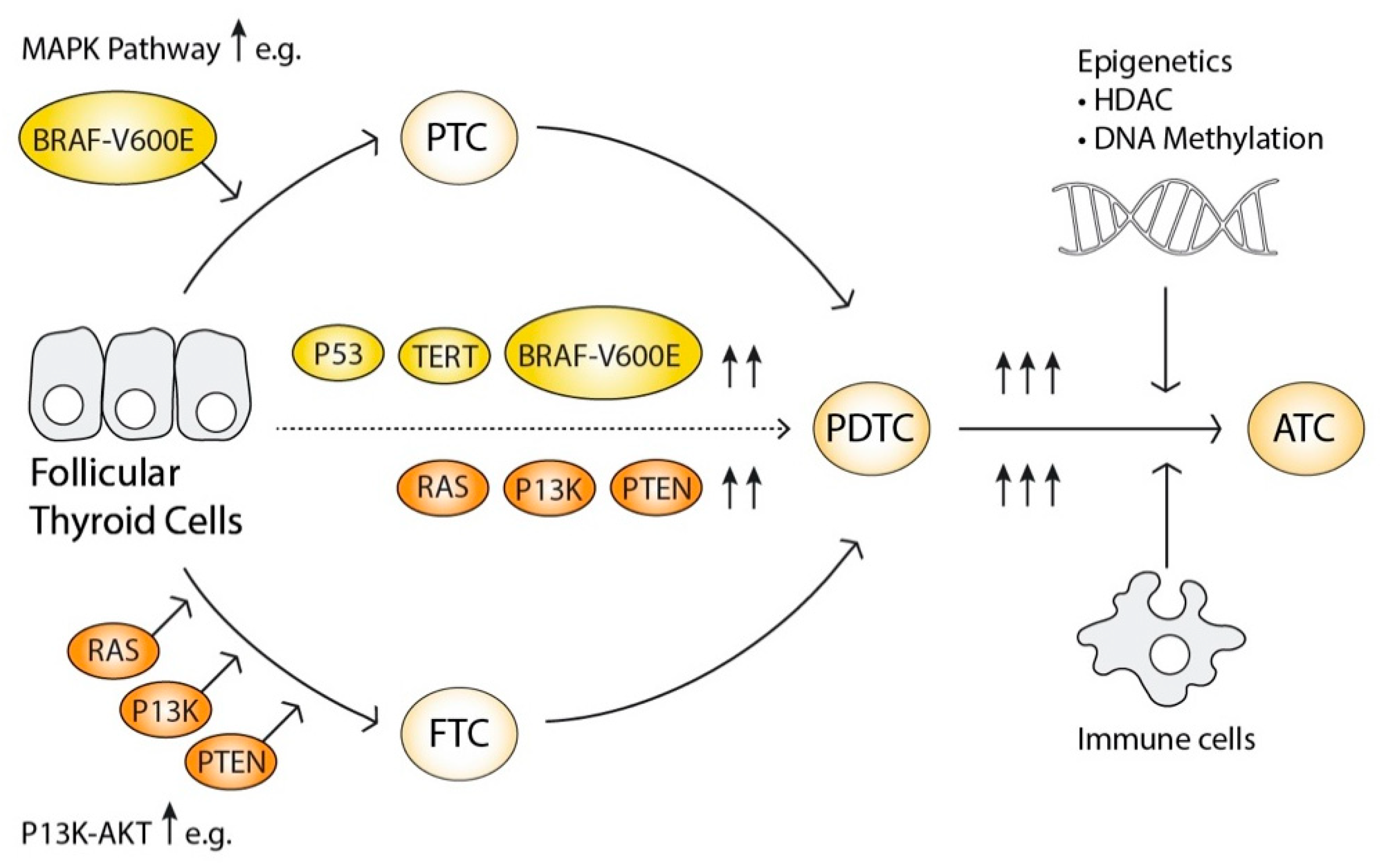
2.6. The Progressive Oncogenesis Model
3. Molecular Marker for Diagnosis and Management of Thyroid Cancer in DTC
3.1. The Diagnostic and Therapeutic Challenge of Indeterminate Nodules

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnostic Category | ROM 1 (NIFTP 2 Considered as Cancer (%)) | ROM 1 (NIFTP 2 NOT Considered as Cancer (%)) | Management Options | |
|---|---|---|---|---|
| I. | Nondiagnostic/unsatisfactory (Cyst fluid, acellular specimen, other (e.g., obscuring blood, artefacts)) | 5–10 | 5–10 | Correlate with clinical/radiological findings Consider repeat FNA 3 |
| II. | Benign (Benign follicular nodule, Chronic lymphocytic (Hashimoto) thyroiditis, Granulomatous (subacute) thyroiditis) | 0–3 | 0–3 | Surveillance and US 4 follow up |
| III. | Atypia of undetermined significance, or follicular lesion of undetermined significance | 6–18 | 10–30 | Correlate with clinical/radiological findings Consider repeat FNA 3 Consider molecular testing |
| IV. | Follicular neoplasm OR Follicular carcinoma (specify if Hürtle cell features) | 10–40 | 25–40 | Consider molecular testing, Lobectomy |
| V. | Suspicious for malignancy | 45–60 | 50–75 | Total thyroidectomy or lobectomy |
| VI. | Malignant (Papillary thyroid carcinoma, Poorly differentiated thyroid carcinoma, Medullary thyroid carcinoma, Anaplastic thyroid carcinoma, Squamous cell carcinoma, Carcinoma with mixed features, Metastatic malignancy, Non-Hodgkin lymphoma, Other) | 94–96 | 97–99 | Total thyroidectomy or lobectomy |
3.2. Molecular Markers and Clinical Profiles in DTC
3.2.1. Genetic Markers
3.2.2. MiRNA Markers
3.2.3. Long Noncoding RNA Markers
3.2.4. Proteome Based Markers
3.3. Molecular Testing (Table 2)
3.4. Current Challenges and Limitations of Molecular Testing
4. Molecular Marker for Surveillance in DTC
5. Molecular Markers to Guide Targeted Treatment for Advanced TC
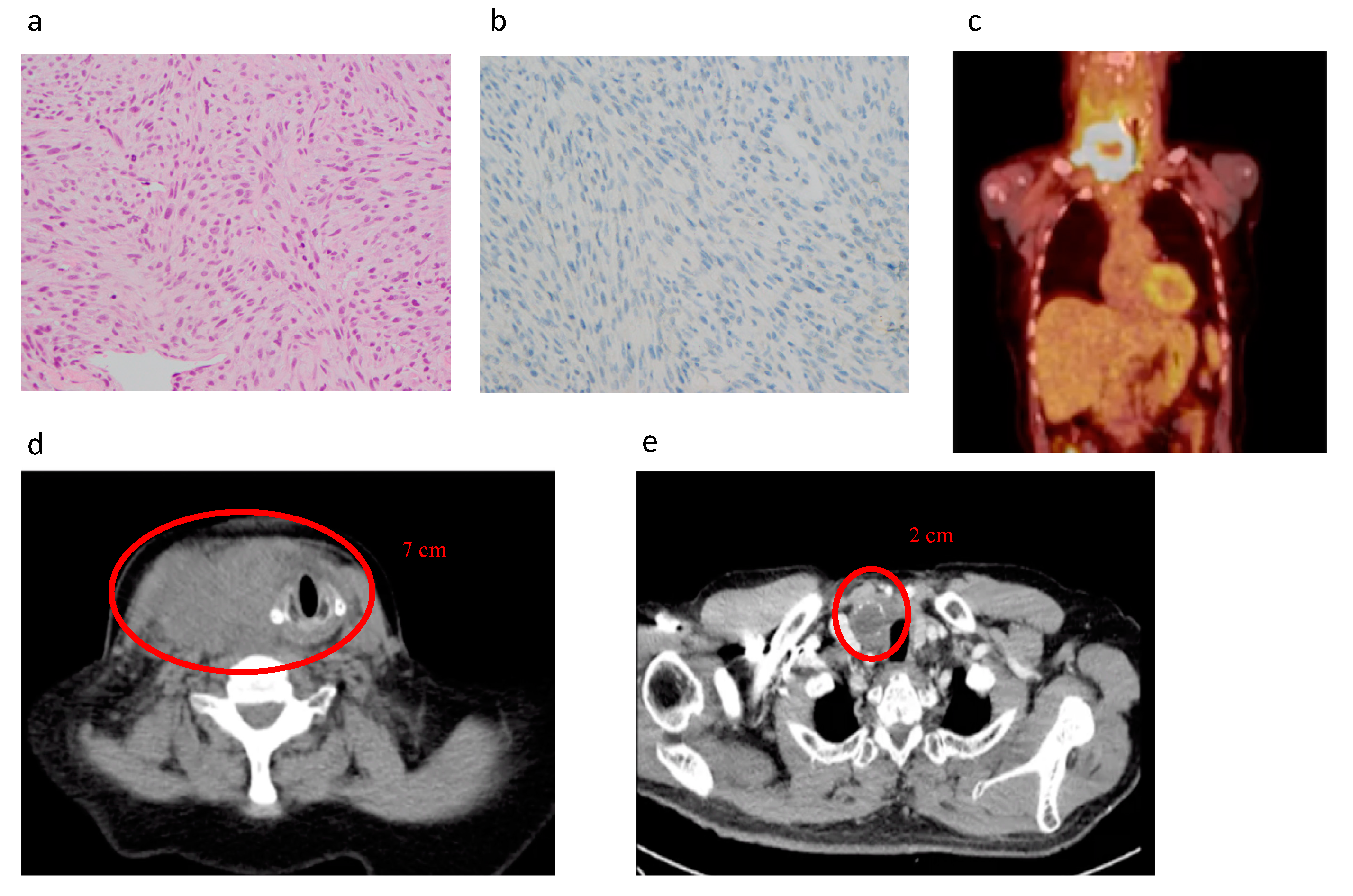
5.1. Diagnosis of Advanced TC, PDTC, and ATC
5.2. Impairment of Iodine-Handling Machinery in Advanced Thyroid Cancer
5.3. Tyrosine Kinase Inhibitors
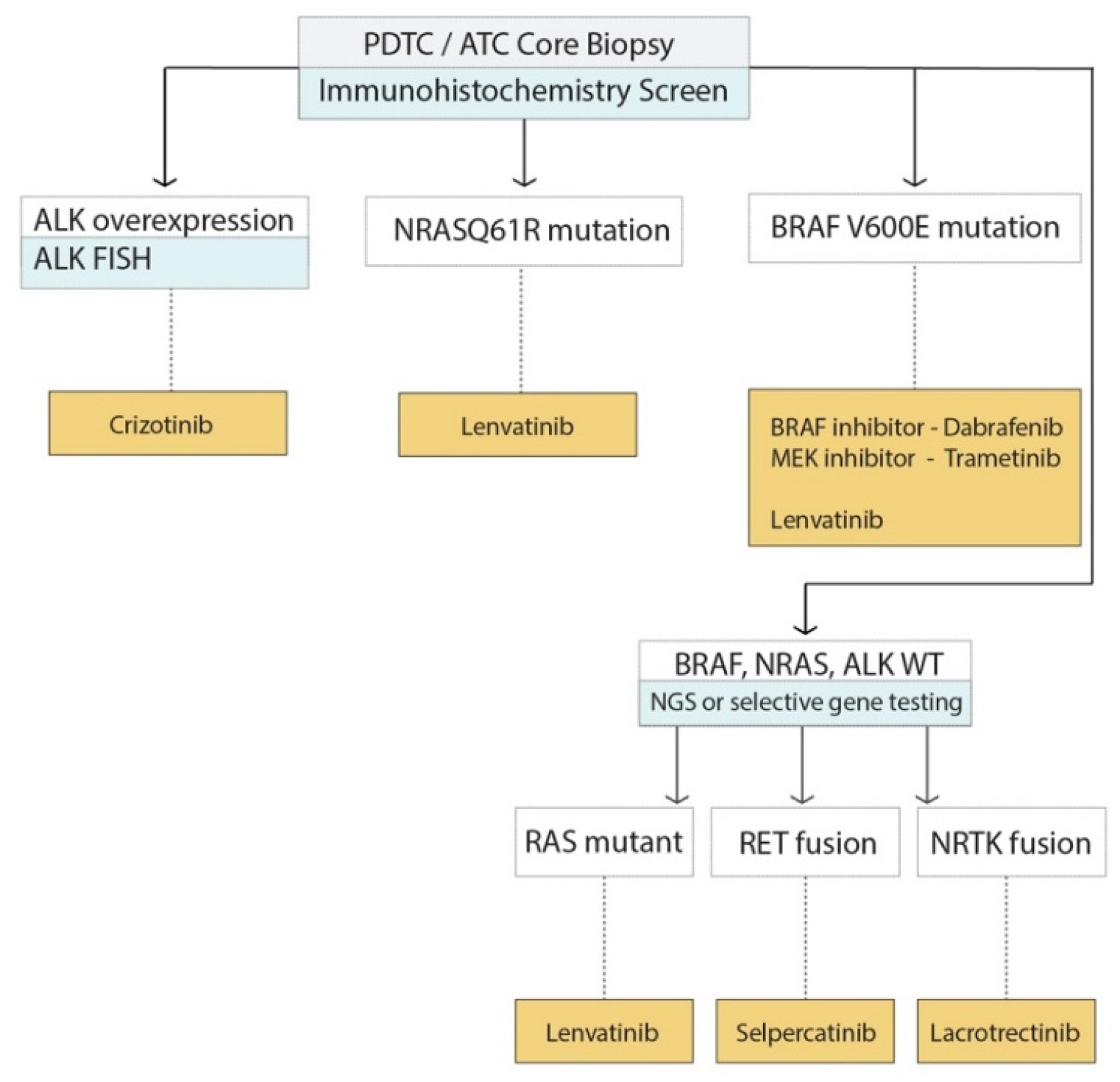
5.4. Application of Molecular Markers to Decision Making for Advanced TC
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Seib, C.D.; Sosa, J.A. Evolving understanding of the epidemiology of thyroid cancer. Endocrinol. Metab. Clin. N. Am. 2019, 48, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.; Welch, H.G. Current thyroid cancer trends in the united states. JAMA Otolaryngol. Head. Neck. Surg. 2014, 140, 317–322. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Howlader, N.; Noone, A.; Krapcho, M.; Miller, D.; Bishop, K.; Altekruse, S.F.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; et al. (Eds.) Seer Cancer Statistics Review, 1975–2013; National Cancer Institute: Bethesda, MD, USA, 2015. Available online: https://seer.cancer.gov/archive/csr/1975_2013/ (accessed on 14 May 2020).
- Noone, A.M.; Cronin, K.A.; Altekruse, S.F.; Howlader, N.; Lewis, D.R.; Petkov, V.I.; Penberthy, L. Cancer incidence and survival trends by subtype using data from the surveillance epidemiology and end results program, 1992–2013. Cancer Epidemiol. Biomark. Prev. 2017, 26, 632–641. [Google Scholar] [CrossRef]
- La Vecchia, C.; Malvezzi, M.; Bosetti, C.; Garavello, W.; Bertuccio, P.; Levi, F.; Negri, E. Thyroid cancer mortality and incidence: A global overview. Int. J. Cancer 2015, 136, 2187–2195. [Google Scholar] [CrossRef]
- Kim, J.; Gosnell, J.E.; Roman, S.A. Geographic influences in the global rise of thyroid cancer. Nat. Rev. Endocrinol. 2020, 16, 17–29. [Google Scholar] [CrossRef]
- Lim, H.; Devesa, S.S.; Sosa, J.A.; Check, D.; Kitahara, C.M. Trends in thyroid cancer incidence and mortality in the united states, 1974–2013. JAMA 2017, 317, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.S.; Kim, H.J.; Welch, H.G. Korea’s thyroid-cancer “epidemic”—Screening and overdiagnosis. N. Engl. J. Med. 2014, 371, 1765–1767. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.F.; Jonker, P.K.C.; Cunich, M.; Sidhu, S.B.; Delbridge, L.W.; Glover, A.R.; Learoyd, D.L.; Aniss, A.; Kruijff, S.; Sywak, M.S. Surgery alone for papillary thyroid microcarcinoma is less costly and more effective than long term active surveillance. Surgery 2020, 167, 110–116. [Google Scholar] [CrossRef]
- Aschebrook-Kilfoy, B.; Schechter, R.B.; Shih, Y.C.; Kaplan, E.L.; Chiu, B.C.; Angelos, P.; Grogan, R.H. The clinical and economic burden of a sustained increase in thyroid cancer incidence. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The i-predict study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 american thyroid association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: The american thyroid association guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Baloch, Z.; Bernet, V.; Chen, A.; Fahey, T.J., 3rd; Ganly, I.; Hodak, S.P.; Kebebew, E.; Patel, K.N.; Shaha, A.; et al. American thyroid association statement on surgical application of molecular profiling for thyroid nodules: Current impact on perioperative decision making. Thyroid 2015, 25, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.N.; Yip, L.; Lubitz, C.C.; Grubbs, E.G.; Miller, B.S.; Shen, W.; Angelos, P.; Chen, H.; Doherty, G.M.; Fahey, T.J., 3rd; et al. The american association of endocrine surgeons guidelines for the definitive surgical management of thyroid disease in adults. Ann. Surg. 2020, 271, e21–e93. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas, M.E.; McFadden, D.G.; Durante, C. Thyroid cancer. Lancet 2016, 388, 2783–2795. [Google Scholar] [CrossRef]
- Cohen, Y.; Xing, M.; Mambo, E.; Guo, Z.; Wu, G.; Trink, B.; Beller, U.; Westra, W.H.; Ladenson, P.W.; Sidransky, D. Braf mutation in papillary thyroid carcinoma. J. Natl. Cancer Inst. 2003, 95, 625–627. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef]
- Prete, A.; Borges de Souza, P.; Censi, S.; Muzza, M.; Nucci, N.; Sponziello, M. Update on fundamental mechanisms of thyroid cancer. Front Endocrinol. (Lausanne) 2020, 11, 102. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Bishop, J.; Zhu, G.; Zhang, T.; Ladenson, P.W.; Xing, M. Mortality risk stratification by combining braf v600e and tert promoter mutations in papillary thyroid cancer: Genetic duet of braf and tert promoter mutations in thyroid cancer mortality. JAMA Oncol. 2017, 3, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Acquaviva, G.; Visani, M.; Repaci, A.; Rhoden, K.J.; de Biase, D.; Pession, A.; Giovanni, T. Molecular pathology of thyroid tumours of follicular cells: A review of genetic alterations and their clinicopathological relevance. Histopathology 2018, 72, 6–31. [Google Scholar] [CrossRef] [PubMed]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef]
- Calabrese, G.; Dolcimascolo, A.; Caruso, G.; Forte, S. Mir-19a is involved in progression and malignancy of anaplastic thyroid cancer cells. Onco. Targets Ther. 2019, 12, 9571–9583. [Google Scholar] [CrossRef]
- Celano, M.; Rosignolo, F.; Maggisano, V.; Pecce, V.; Iannone, M.; Russo, D.; Bulotta, S. Micrornas as biomarkers in thyroid carcinoma. Int. J. Genom. 2017, 2017, 6496570. [Google Scholar] [CrossRef]
- Santiago, K.; Chen Wongworawat, Y.; Khan, S. Differential microrna-signatures in thyroid cancer subtypes. J. Oncol. 2020, 2020, 2052396. [Google Scholar] [CrossRef]
- Marini, F.; Luzi, E.; Brandi, M.L. Microrna role in thyroid cancer development. J. Thyroid Res. 2011, 2011, 407123. [Google Scholar] [CrossRef]
- Calabrese, G.; Dolcimascolo, A.; Torrisi, F.; Zappala, A.; Gulino, R.; Parenti, R. Mir-19a overexpression in ftc-133 cell line induces a more de-differentiated and aggressive phenotype. Int. J. Mol. Sci. 2018, 19, 3944. [Google Scholar] [CrossRef]
- Lee, J.C.; Gundara, J.S.; Glover, A.; Serpell, J.; Sidhu, S.B. Microrna expression profiles in the management of papillary thyroid cancer. Oncologist 2014, 19, 1141–1147. [Google Scholar] [CrossRef]
- Lee, J.C.; Zhao, J.T.; Clifton-Bligh, R.J.; Gill, A.; Gundara, J.S.; Ip, J.C.; Glover, A.; Sywak, M.S.; Delbridge, L.W.; Robinson, B.G.; et al. Microrna-222 and microrna-146b are tissue and circulating biomarkers of recurrent papillary thyroid cancer. Cancer 2013, 119, 4358–4365. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Li, C.; Liu, C.; Zhao, S.; Wang, Y.; Fu, Z. Expressions of mirnas in papillary thyroid carcinoma and their associations with the clinical characteristics of ptc. Cancer Biomark. 2017, 18, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Giordano, T.J. Follicular cell thyroid neoplasia: Insights from genomics and the cancer genome atlas research network. Curr. Opin. Oncol. 2016, 28, 1–4. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A comprehensive review on mapk: A promising therapeutic target in cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, A.R. Signal integration by jnk and p38 mapk pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Adeniran, A.J.; Zhu, Z.; Gandhi, M.; Steward, D.L.; Fidler, J.P.; Giordano, T.J.; Biddinger, P.W.; Nikiforov, Y.E. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am. J. Surg. Pathol. 2006, 30, 216–222. [Google Scholar] [CrossRef]
- Chakravarty, D.; Santos, E.; Ryder, M.; Knauf, J.A.; Liao, X.H.; West, B.L.; Bollag, G.; Kolesnick, R.; Thin, T.H.; Rosen, N.; et al. Small-molecule mapk inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional braf activation. J. Clin. Investig. 2011, 121, 4700–4711. [Google Scholar] [CrossRef]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the raf-erk signaling pathway by oncogenic mutations of b-raf. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Rusinek, D.; Swierniak, M.; Chmielik, E.; Kowal, M.; Kowalska, M.; Cyplinska, R.; Czarniecka, A.; Piglowski, W.; Korfanty, J.; Chekan, M.; et al. Brafv600e-associated gene expression profile: Early changes in the transcriptome, based on a transgenic mouse model of papillary thyroid carcinoma. PLoS ONE 2015, 10, e0143688. [Google Scholar] [CrossRef]
- Fagin, J.A. Challenging dogma in thyroid cancer molecular genetics--role of ret/ptc and braf in tumor initiation. J. Clin. Endocrinol. Metab. 2004, 89, 4264–4266. [Google Scholar] [CrossRef]
- Ikenoue, T.; Hikiba, Y.; Kanai, F.; Tanaka, Y.; Imamura, J.; Imamura, T.; Ohta, M.; Ijichi, H.; Tateishi, K.; Kawakami, T.; et al. Functional analysis of mutations within the kinase activation segment of b-raf in human colorectal tumors. Cancer Res. 2003, 63, 8132–8137. [Google Scholar] [PubMed]
- Lee, S.J.; Lee, M.H.; Kim, D.W.; Lee, S.; Huang, S.; Ryu, M.J.; Kim, Y.K.; Kim, S.J.; Kim, S.J.; Hwang, J.H.; et al. Cross-regulation between oncogenic braf(v600e) kinase and the mst1 pathway in papillary thyroid carcinoma. PLoS ONE 2011, 6, e16180. [Google Scholar]
- Liu, X.; Bishop, J.; Shan, Y.; Pai, S.; Liu, D.; Murugan, A.K.; Sun, H.; El-Naggar, A.K.; Xing, M. Highly prevalent tert promoter mutations in aggressive thyroid cancers. Endocr. Relat. Cancer 2013, 20, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent tert promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Qu, S.; Liu, R.; Sheng, C.; Shi, X.; Zhu, G.; Murugan, A.K.; Guan, H.; Yu, H.; Wang, Y.; et al. Tert promoter mutations and their association with braf v600e mutation and aggressive clinicopathological characteristics of thyroid cancer. J. Clin. Endocrinol. Metab. 2014, 99, E1130–E1136. [Google Scholar] [CrossRef]
- Shinohara, M.; Chung, Y.J.; Saji, M.; Ringel, M.D. Akt in thyroid tumorigenesis and progression. Endocrinology 2007, 148, 942–947. [Google Scholar] [CrossRef]
- Liu, Z.; Hou, P.; Ji, M.; Guan, H.; Studeman, K.; Jensen, K.; Vasko, V.; El-Naggar, A.K.; Xing, M. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J. Clin. Endocrinol. Metab. 2008, 93, 3106–3116. [Google Scholar] [CrossRef]
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef]
- Miller, K.A.; Yeager, N.; Baker, K.; Liao, X.H.; Refetoff, S.; Di Cristofano, A. Oncogenic kras requires simultaneous pi3k signaling to induce erk activation and transform thyroid epithelial cells in vivo. Cancer Res. 2009, 69, 3689–3694. [Google Scholar] [CrossRef]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacocke, M.; et al. Germline mutations of the pten gene in cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 1997, 16, 64–67. [Google Scholar] [CrossRef]
- Fusco, A.; Grieco, M.; Santoro, M.; Berlingieri, M.T.; Pilotti, S.; Pierotti, M.A.; Della Porta, G.; Vecchio, G. A new oncogene in human thyroid papillary carcinomas and their lymph-nodal metastases. Nature 1987, 328, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, M.I.; Junit, S.M.; Ng, K.L.; Jayapalan, J.J.; Karikalan, B.; Hashim, O.H. Papillary thyroid cancer: Genetic alterations and molecular biomarker investigations. Int. J. Med. Sci. 2019, 16, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Fagin, J.A.; Mitsiades, N. Molecular pathology of thyroid cancer: Diagnostic and clinical implications. Best Pract. Res. Clin. Endocrinol. Metab. 2008, 22, 955–969. [Google Scholar] [CrossRef] [PubMed]
- Nikiforova, M.N.; Lynch, R.A.; Biddinger, P.W.; Alexander, E.K.; Dorn, G.W., 2nd; Tallini, G.; Kroll, T.G.; Nikiforov, Y.E. Ras point mutations and pax8-ppar gamma rearrangement in thyroid tumors: Evidence for distinct molecular pathways in thyroid follicular carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 2318–2326. [Google Scholar] [CrossRef] [PubMed]
- Zafon, C.; Gil, J.; Pérez-González, B.; Jordà, M. DNA methylation in thyroid cancer. Endocr. Relat. Cancer 2019, 26, R415–R439. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Liu, D.; Xing, M. Genome-wide alterations in gene methylation by the braf v600e mutation in papillary thyroid cancer cells. Endocr. Relat. Cancer 2011, 18, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Bisarro Dos Reis, M.; Barros-Filho, M.C.; Marchi, F.A.; Beltrami, C.M.; Kuasne, H.; Pinto, C.A.L.; Ambatipudi, S.; Herceg, Z.; Kowalski, L.P.; Rogatto, S.R. Prognostic classifier based on genome-wide DNA methylation profiling in well-differentiated thyroid tumors. J. Clin. Endocrinol. Metab. 2017, 102, 4089–4099. [Google Scholar] [CrossRef]
- Audia, J.E.; Campbell, R.M. Histone modifications and cancer. Cold Spring Harb. Perspect Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Marks, P.; Rifkind, R.A.; Richon, V.M.; Breslow, R.; Miller, T.; Kelly, W.K. Histone deacetylases and cancer: Causes and therapies. Nat. Rev. Cancer 2001, 1, 194–202. [Google Scholar] [CrossRef]
- He, H.; Jazdzewski, K.; Li, W.; Liyanarachchi, S.; Nagy, R.; Volinia, S.; Calin, G.A.; Liu, C.G.; Franssila, K.; Suster, S.; et al. The role of microrna genes in papillary thyroid carcinoma. Proc. Natl. Acad. Sci. USA 2005, 102, 19075–19080. [Google Scholar] [CrossRef]
- Sui, F.; Ji, M.; Hou, P. Long non-coding rnas in thyroid cancer: Biological functions and clinical significance. Mol. Cell Endocrinol. 2018, 469, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Sedaghati, M.; Kebebew, E. Long noncoding rnas in thyroid cancer. Curr. Opin. Endocrinol. Diabetes Obes. 2019, 26, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Hauptman, N.; Glavac, D. Long non-coding rna in cancer. Int. J. Mol. Sci. 2013, 14, 4655–4669. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.Y. Thyroid cancer stem cells. Nat. Rev. Endocrinol. 2011, 7, 609–616. [Google Scholar] [CrossRef]
- Hunt, J.L.; Tometsko, M.; LiVolsi, V.A.; Swalsky, P.; Finkelstein, S.D.; Barnes, E.L. Molecular evidence of anaplastic transformation in coexisting well-differentiated and anaplastic carcinomas of the thyroid. Am. J. Surg. Pathol. 2003, 27, 1559–1564. [Google Scholar] [CrossRef]
- Molinaro, E.; Romei, C.; Biagini, A.; Sabini, E.; Agate, L.; Mazzeo, S.; Materazzi, G.; Sellari-Franceschini, S.; Ribechini, A.; Torregrossa, L.; et al. Anaplastic thyroid carcinoma: From clinicopathology to genetics and advanced therapies. Nat. Rev. Endocrinol. 2017, 13, 644–660. [Google Scholar] [CrossRef]
- Kondo, T.; Ezzat, S.; Asa, S.L. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat. Rev. Cancer 2006, 6, 292–306. [Google Scholar] [CrossRef]
- Cibas, E.S.; Ali, S.Z. The bethesda system for reporting thyroid cytopathology. Thyroid 2009, 19, 1159–1165. [Google Scholar] [CrossRef]
- Bongiovanni, M.; Spitale, A.; Faquin, W.C.; Mazzucchelli, L.; Baloch, Z.W. The bethesda system for reporting thyroid cytopathology: A meta-analysis. Acta Cytol. 2012, 56, 333–339. [Google Scholar] [CrossRef]
- Bongiovanni, M.; De Saussure, B.; Kumar, N.; Pache, J.C.; Cibas, E.S. A quality control study on cytotechnologist-cytopathologist concordance and its relationship to the number of dots on the slide. Acta Cytol. 2009, 53, 653–658. [Google Scholar] [CrossRef]
- Cibas, E.S.; Baloch, Z.W.; Fellegara, G.; LiVolsi, V.A.; Raab, S.S.; Rosai, J.; Diggans, J.; Friedman, L.; Kennedy, G.C.; Kloos, R.T.; et al. A prospective assessment defining the limitations of thyroid nodule pathologic evaluation. Ann. Intern. Med. 2013, 159, 325–332. [Google Scholar] [CrossRef]
- Iskandar, M.E.; Bonomo, G.; Avadhani, V.; Persky, M.; Lucido, D.; Wang, B.; Marti, J.L. Evidence for overestimation of the prevalence of malignancy in indeterminate thyroid nodules classified as bethesda category iii. Surgery 2015, 157, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Seethala, R.R.; Tallini, G.; Baloch, Z.W.; Basolo, F.; Thompson, L.D.; Barletta, J.A.; Wenig, B.M.; Al Ghuzlan, A.; Kakudo, K.; et al. Nomenclature revision for encapsulated follicular variant of papillary thyroid carcinoma: A paradigm shift to reduce overtreatment of indolent tumors. JAMA Oncol. 2016, 2, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.V.; Osamura, R.; Klöppel, G.; Rosai, J. WHO Classification of Tumours of Endocrine Organs from the World Health Organization; IARC WHO Classification of Tumours; WHO: Geneva, Switzerland, 2017; Volume 10. [Google Scholar]
- Cibas, E.S.; Ali, S.Z. The 2017 bethesda system for reporting thyroid cytopathology. Thyroid 2017, 27, 1341–1346. [Google Scholar] [CrossRef] [PubMed]
- Mallick, R.; Stevens, T.M.; Winokur, T.S.; Asban, A.; Wang, T.N.; Lindeman, B.M.; Porterfield, J.R.; Chen, H. Is frozen-section analysis during thyroid operation useful in the era of molecular testing? J. Am. Coll Surg. 2019, 228, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Guevara, N.; Lassalle, S.; Benaim, G.; Sadoul, J.L.; Santini, J.; Hofman, P. Role of frozen section analysis in nodular thyroid pathology. Eur. Ann. Otorhinolaryngol. Head. Neck. Dis. 2015, 132, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.S.; Sarti, E.E.; Jain, K.S.; Wang, H.; Nixon, I.J.; Shaha, A.R.; Shah, J.P.; Kraus, D.H.; Ghossein, R.; Fish, S.A.; et al. Malignancy rate in thyroid nodules classified as bethesda category iii (aus/flus). Thyroid 2014, 24, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Nikiforova, M. Update on molecular testing for cytologically indeterminate thyroid nodules. Arch Pathol. Lab Med. 2018, 142, 446–457. [Google Scholar] [CrossRef]
- Ali, S.; Cibas, E.S. The Bethesda System for Reporting Thyroid Cytopathology: Definitions, Criteria, and Explanatory Notes, 2nd ed.; Springer: New York, NY, USA, 2018. [Google Scholar]
- Muzza, M.; Colombo, C.; Pogliaghi, G.; Karapanou, O.; Fugazzola, L. Molecular markers for the classification of cytologically indeterminate thyroid nodules. J. Endocrinol. Investig. 2020, 43, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.; Yip, L. Decision making in indeterminate thyroid nodules and the role of molecular testing. Surg. Clin. N. Am. 2019, 99, 587–598. [Google Scholar] [CrossRef]
- Patel, K.N.; Angell, T.E.; Babiarz, J.; Barth, N.M.; Blevins, T.; Duh, Q.Y.; Ghossein, R.A.; Harrell, R.M.; Huang, J.; Kennedy, G.C.; et al. Performance of a genomic sequencing classifier for the preoperative diagnosis of cytologically indeterminate thyroid nodules. JAMA Surg. 2018, 153, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Labourier, E.; Shifrin, A.; Busseniers, A.E.; Lupo, M.A.; Manganelli, M.L.; Andruss, B.; Wylie, D.; Beaudenon-Huibregtse, S. Molecular testing for mirna, mrna, and DNA on fine-needle aspiration improves the preoperative diagnosis of thyroid nodules with indeterminate cytology. J. Clin. Endocrinol. Metab. 2015, 100, 2743–2750. [Google Scholar] [CrossRef] [PubMed]
- Steward, D.L.; Carty, S.E.; Sippel, R.S.; Yang, S.P.; Sosa, J.A.; Sipos, J.A.; Figge, J.J.; Mandel, S.; Haugen, B.R.; Burman, K.D.; et al. Performance of a multigene genomic classifier in thyroid nodules with indeterminate cytology: A prospective blinded multicenter study. JAMA Oncol. 2019, 5, 204–212. [Google Scholar] [CrossRef]
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Khatami, F.; Larijani, B.; Nikfar, S.; Hasanzad, M.; Fendereski, K.; Tavangar, S.M. Personalized treatment options for thyroid cancer: Current perspectives. Pharmgenom. Pers. Med. 2019, 12, 235–245. [Google Scholar] [CrossRef]
- Zhu, X.; Yao, J.; Tian, W. Microarray technology to investigate genes associated with papillary thyroid carcinoma. Mol. Med. Rep. 2015, 11, 3729–3733. [Google Scholar] [CrossRef] [PubMed]
- Grogan, R.H.; Kaplan, S.P.; Cao, H.; Weiss, R.E.; Degroot, L.J.; Simon, C.A.; Embia, O.M.; Angelos, P.; Kaplan, E.L.; Schechter, R.B. A study of recurrence and death from papillary thyroid cancer with 27 years of median follow-up. Surgery 2013, 154, 1436–1446. [Google Scholar] [CrossRef]
- Vuong, H.G.; Duong, U.N.; Altibi, A.M.; Ngo, H.T.; Pham, T.Q.; Tran, H.M.; Gandolfi, G.; Hassell, L. A meta-analysis of prognostic roles of molecular markers in papillary thyroid carcinoma. Endocr. Connect 2017, 6, R8–R17. [Google Scholar] [CrossRef]
- Xing, M.; Alzahrani, A.S.; Carson, K.A.; Viola, D.; Elisei, R.; Bendlova, B.; Yip, L.; Mian, C.; Vianello, F.; Tuttle, R.M.; et al. Association between braf v600e mutation and mortality in patients with papillary thyroid cancer. JAMA 2013, 309, 1493–1501. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, E.S.; Kim, Y.S. Clinicopathologic significance of braf v600e mutation in papillary carcinomas of the thyroid: A meta-analysis. Cancer 2007, 110, 38–46. [Google Scholar] [CrossRef]
- Yip, L.; Nikiforova, M.N.; Yoo, J.Y.; McCoy, K.L.; Stang, M.T.; Armstrong, M.J.; Nicholson, K.J.; Ohori, N.P.; Coyne, C.; Hodak, S.P.; et al. Tumor genotype determines phenotype and disease-related outcomes in thyroid cancer: A study of 1510 patients. Ann. Surg. 2015, 262, 519–525. [Google Scholar] [CrossRef]
- Zatelli, M.C.; Trasforini, G.; Leoni, S.; Frigato, G.; Buratto, M.; Tagliati, F.; Rossi, R.; Cavazzini, L.; Roti, E.; degli Uberti, E.C. Braf v600e mutation analysis increases diagnostic accuracy for papillary thyroid carcinoma in fine-needle aspiration biopsies. Eur. J. Endocrinol. 2009, 161, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Park, Y.J.; Lim, J.A.; Ahn, H.Y.; Lee, E.K.; Lee, Y.J.; Kim, K.W.; Hahn, S.K.; Youn, Y.K.; Kim, K.H.; et al. The association of the braf(v600e) mutation with prognostic factors and poor clinical outcome in papillary thyroid cancer: A meta-analysis. Cancer 2012, 118, 1764–1773. [Google Scholar] [CrossRef]
- Fnais, N.; Soobiah, C.; Al-Qahtani, K.; Hamid, J.S.; Perrier, L.; Straus, S.E.; Tricco, A.C. Diagnostic value of fine needle aspiration braf(v600e) mutation analysis in papillary thyroid cancer: A systematic review and meta-analysis. Hum. Pathol. 2015, 46, 1443–1454. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, E.K.; Kwak, J.Y.; Moon, H.J.; Yoon, J.H. What to do with thyroid nodules showing benign cytology and braf(v600e) mutation? A study based on clinical and radiologic features using a highly sensitive analytic method. Surgery 2015, 157, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Antonello, Z.A.; Hsu, N.; Bhasin, M.; Roti, G.; Joshi, M.; Van Hummelen, P.; Ye, E.; Lo, A.S.; Karumanchi, S.A.; Bryke, C.R.; et al. Vemurafenib-resistance via de novo rbm genes mutations and chromosome 5 aberrations is overcome by combined therapy with palbociclib in thyroid carcinoma with braf(v600e). Oncotarget 2017, 8, 84743–84760. [Google Scholar] [CrossRef]
- Finkel, A.; Liba, L.; Simon, E.; Bick, T.; Prinz, E.; Sabo, E.; Ben-Izhak, O.; Hershkovitz, D. Subclonality for braf mutation in papillary thyroid carcinoma is associated with earlier disease stage. J. Clin. Endocrinol. Metab. 2016, 101, 1407–1413. [Google Scholar] [CrossRef]
- Vuong, H.G.; Altibi, A.M.A.; Duong, U.N.P.; Hassell, L. Prognostic implication of braf and tert promoter mutation combination in papillary thyroid carcinoma-a meta-analysis. Clin. Endocrinol. (Oxf.) 2017, 87, 411–417. [Google Scholar] [CrossRef]
- Howell, G.M.; Nikiforova, M.N.; Carty, S.E.; Armstrong, M.J.; Hodak, S.P.; Stang, M.T.; McCoy, K.L.; Nikiforov, Y.E.; Yip, L. Braf v600e mutation independently predicts central compartment lymph node metastasis in patients with papillary thyroid cancer. Ann. Surg. Oncol. 2013, 20, 47–52. [Google Scholar] [CrossRef]
- Afkhami, M.; Karunamurthy, A.; Chiosea, S.; Nikiforova, M.N.; Seethala, R.; Nikiforov, Y.E.; Coyne, C. Histopathologic and clinical characterization of thyroid tumors carrying the braf(k601e) mutation. Thyroid 2016, 26, 242–247. [Google Scholar] [CrossRef]
- Ohori, N.P.; Singhal, R.; Nikiforova, M.N.; Yip, L.; Schoedel, K.E.; Coyne, C.; McCoy, K.L.; LeBeau, S.O.; Hodak, S.P.; Carty, S.E.; et al. Braf mutation detection in indeterminate thyroid cytology specimens: Underlying cytologic, molecular, and pathologic characteristics of papillary thyroid carcinoma. Cancer Cytopathol. 2013, 121, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Grogan, R.H.; Mitmaker, E.J.; Clark, O.H. The evolution of biomarkers in thyroid cancer-from mass screening to a personalized biosignature. Cancers 2010, 2, 885–912. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Qadri, Q.; Makhdoomi, M.J.; Wani, M.A.; Malik, A.A.; Niyaz, M.; Masoodi, S.R.; Andrabi, K.I.; Ahmad, R.; Mudassar, S. Ret/ptc gene rearrangements in thyroid carcinogenesis: Assessment and clinico-pathological correlations. Pathol. Oncol. Res. 2020, 26, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Rowland, J.M.; Bove, K.E.; Monforte-Munoz, H.; Fagin, J.A. Distinct pattern of ret oncogene rearrangements in morphological variants of radiation-induced and sporadic thyroid papillary carcinomas in children. Cancer Res. 1997, 57, 1690–1694. [Google Scholar]
- Su, X.; He, C.; Ma, J.; Tang, T.; Zhang, X.; Ye, Z.; Long, Y.; Shao, Q.; Shao, J.; Yang, A. Ret/ptc rearrangements are associated with elevated postoperative tsh levels and multifocal lesions in papillary thyroid cancer without concomitant thyroid benign disease. PLoS ONE 2016, 11, e0165596. [Google Scholar] [CrossRef]
- Paulson, V.A.; Rudzinski, E.R.; Hawkins, D.S. Thyroid cancer in the pediatric population. Genes 2019, 10, 723. [Google Scholar] [CrossRef]
- Howell, G.M.; Hodak, S.P.; Yip, L. Ras mutations in thyroid cancer. Oncologist 2013, 18, 926–932. [Google Scholar] [CrossRef]
- Gupta, N.; Dasyam, A.K.; Carty, S.E.; Nikiforova, M.N.; Ohori, N.P.; Armstrong, M.; Yip, L.; LeBeau, S.O.; McCoy, K.L.; Coyne, C.; et al. Ras mutations in thyroid fna specimens are highly predictive of predominantly low-risk follicular-pattern cancers. J. Clin. Endocrinol. Metab. 2013, 98, E914–E922. [Google Scholar] [CrossRef]
- Sabra, M.M.; Dominguez, J.M.; Grewal, R.K.; Larson, S.M.; Ghossein, R.A.; Tuttle, R.M.; Fagin, J.A. Clinical outcomes and molecular profile of differentiated thyroid cancers with radioiodine-avid distant metastases. J. Clin. Endocrinol. Metab. 2013, 98, E829–E836. [Google Scholar] [CrossRef]
- Nikiforov, Y.E. Molecular diagnostics of thyroid tumors. Arch. Pathol. Lab. Med. 2011, 135, 569–577. [Google Scholar]
- Brandler, T.C.; Liu, C.Z.; Cho, M.; Zhou, F.; Cangiarella, J.; Yee-Chang, M.; Shi, Y.; Simsir, A.; Sun, W. Does noninvasive follicular thyroid neoplasm with papillary-like nuclear features (niftp) have a unique molecular profile? Am. J. Clin. Pathol. 2018, 150, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Xing, M. Clinical utility of ras mutations in thyroid cancer: A blurred picture now emerging clearer. BMC Med. 2016, 14, 12. [Google Scholar] [CrossRef] [PubMed]
- Giannini, R.; Ugolini, C.; Poma, A.M.; Urpi, M.; Niccoli, C.; Elisei, R.; Chiarugi, M.; Vitti, P.; Miccoli, P.; Basolo, F. Identification of two distinct molecular subtypes of non-invasive follicular neoplasm with papillary-like nuclear features by digital rna counting. Thyroid 2017, 27, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- D’Cruz, A.K.; Vaish, R.; Vaidya, A.; Nixon, I.J.; Williams, M.D.; Vander Poorten, V.; Lopez, F.; Angelos, P.; Shaha, A.R.; Khafif, A.; et al. Molecular markers in well-differentiated thyroid cancer. Eur. Arch. Otorhinolaryngol. 2018, 275, 1375–1384. [Google Scholar] [CrossRef]
- Hodak, S.; Tuttle, R.M.; Maytal, G.; Nikiforov, Y.E.; Randolph, G. Changing the cancer diagnosis: The case of follicular variant of papillary thyroid cancer-primum non nocere and niftp. Thyroid 2016, 26, 869–871. [Google Scholar] [CrossRef]
- Patel, S.G.; Carty, S.E.; McCoy, K.L.; Ohori, N.P.; LeBeau, S.O.; Seethala, R.R.; Nikiforova, M.N.; Nikiforov, Y.E.; Yip, L. Preoperative detection of ras mutation may guide extent of thyroidectomy. Surgery 2017, 161, 168–175. [Google Scholar] [CrossRef]
- Yang, J.; Gong, Y.; Yan, S.; Chen, H.; Qin, S.; Gong, R. Association between tert promoter mutations and clinical behaviors in differentiated thyroid carcinoma: A systematic review and meta-analysis. Endocrine 2020, 67, 44–57. [Google Scholar] [CrossRef]
- Grani, G.; Lamartina, L.; Durante, C.; Filetti, S.; Cooper, D.S. Follicular thyroid cancer and hurthle cell carcinoma: Challenges in diagnosis, treatment, and clinical management. Lancet Diabetes Endocrinol. 2018, 6, 500–514. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Yang, H.; Yip, L.; Ohori, N.P.; McCoy, K.L.; Stang, M.T.; Hodak, S.P.; Nikiforova, M.N.; Carty, S.E.; Nikiforov, Y.E. Pax8/ppargamma rearrangement in thyroid nodules predicts follicular-pattern carcinomas, in particular the encapsulated follicular variant of papillary carcinoma. Thyroid 2014, 24, 1369–1374. [Google Scholar] [CrossRef]
- Boos, L.A.; Dettmer, M.; Schmitt, A.; Rudolph, T.; Steinert, H.; Moch, H.; Sobrinho-Simoes, M.; Komminoth, P.; Perren, A. Diagnostic and prognostic implications of the pax8-ppargamma translocation in thyroid carcinomas-a tma-based study of 226 cases. Histopathology 2013, 63, 234–241. [Google Scholar] [CrossRef]
- Nicolson, N.G.; Murtha, T.D.; Dong, W.; Paulsson, J.O.; Choi, J.; Barbieri, A.L.; Brown, T.C.; Kunstman, J.W.; Larsson, C.; Prasad, M.L.; et al. Comprehensive genetic analysis of follicular thyroid carcinoma predicts prognosis independent of histology. J. Clin. Endocrinol. Metab. 2018, 103, 2640–2650. [Google Scholar] [CrossRef] [PubMed]
- Ganly, I.; Makarov, V.; Deraje, S.; Dong, Y.; Reznik, E.; Seshan, V.; Nanjangud, G.; Eng, S.; Bose, P.; Kuo, F.; et al. Integrated genomic analysis of hurthle cell cancer reveals oncogenic drivers, recurrent mitochondrial mutations, and unique chromosomal landscapes. Cancer Cell 2018, 34, 256–270.e255. [Google Scholar] [CrossRef] [PubMed]
- Stokowy, T.; Gawel, D.; Wojtas, B. Differences in mirna and mrna profile of papillary thyroid cancer variants. Int. J. Endocrinol. 2016, 2016, 1427042. [Google Scholar] [CrossRef] [PubMed]
- Stokowy, T.; Wojtas, B.; Jarzab, B.; Krohn, K.; Fredman, D.; Dralle, H.; Musholt, T.; Hauptmann, S.; Lange, D.; Hegedus, L.; et al. Two-mirna classifiers differentiate mutation-negative follicular thyroid carcinomas and follicular thyroid adenomas in fine needle aspirations with high specificity. Endocrine 2016, 54, 440–447. [Google Scholar] [CrossRef]
- Kwok, G.T.; Zhao, J.T.; Weiss, J.; Mugridge, N.; Brahmbhatt, H.; MacDiarmid, J.A.; Robinson, B.G.; Sidhu, S.B. Translational applications of micrornas in cancer, and therapeutic implications. Noncoding RNA Res. 2017, 2, 143–150. [Google Scholar] [CrossRef]
- Larrea, E.; Sole, C.; Manterola, L.; Goicoechea, I.; Armesto, M.; Arestin, M.; Caffarel, M.M.; Araujo, A.M.; Araiz, M.; Fernandez-Mercado, M.; et al. New concepts in cancer biomarkers: Circulating mirnas in liquid biopsies. Int. J. Mol. Sci. 2016, 17, 627. [Google Scholar] [CrossRef]
- Rosignolo, F.; Memeo, L.; Monzani, F.; Colarossi, C.; Pecce, V.; Verrienti, A.; Durante, C.; Grani, G.; Lamartina, L.; Forte, S.; et al. Microrna-based molecular classification of papillary thyroid carcinoma. Int. J. Oncol. 2017, 50, 1767–1777. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Shirvani-Farsani, Z.; Taheri, M. The role of micrornas in the pathogenesis of thyroid cancer. Noncoding RNA Res. 2020, 5, 88–98. [Google Scholar] [CrossRef]
- Ramirez-Moya, J.; Wert-Lamas, L.; Santisteban, P. Microrna-146b promotes pi3k/akt pathway hyperactivation and thyroid cancer progression by targeting pten. Oncogene 2018, 37, 3369–3383. [Google Scholar] [CrossRef]
- Riesco-Eizaguirre, G.; Wert-Lamas, L.; Perales-Paton, J.; Sastre-Perona, A.; Fernandez, L.P.; Santisteban, P. The mir-146b-3p/pax8/nis regulatory circuit modulates the differentiation phenotype and function of thyroid cells during carcinogenesis. Cancer Res. 2015, 75, 4119–4130. [Google Scholar] [CrossRef]
- Deng, X.; Wu, B.; Xiao, K.; Kang, J.; Xie, J.; Zhang, X.; Fan, Y. Mir-146b-5p promotes metastasis and induces epithelial-mesenchymal transition in thyroid cancer by targeting znrf3. Cell Physiol. Biochem. 2015, 35, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Jin, M.; Li, Y.; Ren, P.; Liu, J. Mir-137 acts as a tumor suppressor in papillary thyroid carcinoma by targeting cxcl12. Oncol. Rep. 2016, 35, 2151–2158. [Google Scholar] [CrossRef] [PubMed]
- Paskas, S.; Jankovic, J.; Zivaljevic, V.; Tatic, S.; Bozic, V.; Nikolic, A.; Radojkovic, D.; Savin, S.; Cvejic, D. Malignant risk stratification of thyroid fna specimens with indeterminate cytology based on molecular testing. Cancer Cytopathol. 2015, 123, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Zheng, X.; Hu, M.; Cui, Y.; Zhong, Q.; Wang, S.; Huang, F. Mirna-221/222 in thyroid cancer: A meta-analysis. Clin. Chim. Acta 2018, 484, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.J.; Shen, C.T.; Song, H.J.; Qiu, Z.L.; Luo, Q.Y. Micrornas as a potential tool in the differential diagnosis of thyroid cancer: A systematic review and meta-analysis. Clin. Endocrinol. (Oxf.) 2016, 84, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Wang, P.; Hurst, Z.A.; Chang, Y.S.; Chen, G. Active surveillance for papillary thyroid microcarcinoma: Challenges and prospects. Front. Endocrinol. (Lausanne) 2018, 9, 736. [Google Scholar] [CrossRef]
- Hardin, H.; Helein, H.; Meyer, K.; Robertson, S.; Zhang, R.; Zhong, W.; Lloyd, R.V. Thyroid cancer stem-like cell exosomes: Regulation of emt via transfer of lncrnas. Lab Invest. 2018, 98, 1133–1142. [Google Scholar] [CrossRef]
- Zhang, R.; Hardin, H.; Huang, W.; Chen, J.; Asioli, S.; Righi, A.; Maletta, F.; Sapino, A.; Lloyd, R.V. Malat1 long non-coding rna expression in thyroid tissues: Analysis by in situ hybridization and real-time pcr. Endocr. Pathol. 2017, 28, 7–12. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, X.; Fu, X.; Yan, W.; Lin, F.; Kuang, P.; Luo, Y.; Lin, E.; Hong, X.; Wu, G. Long non-coding rna bancr regulates cancer stem cell markers in papillary thyroid cancer via the raf/mek/erk signaling pathway. Oncol. Rep. 2018, 40, 859–866. [Google Scholar] [CrossRef]
- Lan, X.; Sun, W.; Dong, W.; Wang, Z.; Zhang, T.; He, L.; Zhang, H. Downregulation of long noncoding rna h19 contributes to the proliferation and migration of papillary thyroid carcinoma. Gene 2018, 646, 98–105. [Google Scholar] [CrossRef]
- Zhang, R.; Hardin, H.; Chen, J.; Guo, Z.; Lloyd, R.V. Non-coding rnas in thyroid cancer. Endocr. Pathol. 2016, 27, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Arita, T.; Ichikawa, D.; Konishi, H.; Komatsu, S.; Shiozaki, A.; Shoda, K.; Kawaguchi, T.; Hirajima, S.; Nagata, H.; Kubota, T.; et al. Circulating long non-coding rnas in plasma of patients with gastric cancer. Anticancer Res. 2013, 33, 3185–3193. [Google Scholar] [PubMed]
- Martinez-Aguilar, J.; Clifton-Bligh, R.; Molloy, M.P. Proteomics of thyroid tumours provides new insights into their molecular composition and changes associated with malignancy. Sci. Rep. 2016, 6, 23660. [Google Scholar] [CrossRef] [PubMed]
- Navas-Carrillo, D.; Rodriguez, J.M.; Montoro-Garcia, S.; Orenes-Pinero, E. High-resolution proteomics and metabolomics in thyroid cancer: Deciphering novel biomarkers. Crit. Rev. Clin. Lab. Sci. 2017, 54, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E. Role of molecular markers in thyroid nodule management: Then and now. Endocr. Pract. 2017, 23, 979–988. [Google Scholar] [CrossRef]
- Yip, L.; Sosa, J.A. Molecular-directed treatment of differentiated thyroid cancer: Advances in diagnosis and treatment. JAMA Surg. 2016, 151, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Steward, D.L.; Robinson-Smith, T.M.; Haugen, B.R.; Klopper, J.P.; Zhu, Z.; Fagin, J.A.; Falciglia, M.; Weber, K.; Nikiforova, M.N. Molecular testing for mutations in improving the fine-needle aspiration diagnosis of thyroid nodules. J. Clin. Endocrinol. Metab. 2009, 94, 2092–2098. [Google Scholar] [CrossRef]
- Sistrunk, J.W.; Shifrin, A.; Frager, M.; Bardales, R.H.; Thomas, J.; Fishman, N.; Goldberg, P.; Guttler, R.; Grant, E. Clinical performance of multiplatform mutation panel and microrna risk classifier in indeterminate thyroid nodules. J. Am. Soc. Cytopathol. 2020. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Mercurio, S.; Wald, A.I.; Barbi de Moura, M.; Callenberg, K.; Santana-Santos, L.; Gooding, W.E.; Yip, L.; Ferris, R.L.; Nikiforov, Y.E. Analytical performance of the thyroseq v3 genomic classifier for cancer diagnosis in thyroid nodules. Cancer 2018, 124, 1682–1690. [Google Scholar] [CrossRef]
- Ohori, N.P.; Landau, M.S.; Carty, S.E.; Yip, L.; LeBeau, S.O.; Manroa, P.; Seethala, R.R.; Schoedel, K.E.; Nikiforova, M.N.; Nikiforov, Y.E. Benign call rate and molecular test result distribution of thyroseq v3. Cancer Cytopathol. 2019, 127, 161–168. [Google Scholar] [CrossRef]
- Nikiforov, Y.E.; Baloch, Z.W. Clinical validation of the thyroseq v3 genomic classifier in thyroid nodules with indeterminate fna cytology. Cancer Cytopathol. 2019, 127, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Chudova, D.; Wilde, J.I.; Wang, E.T.; Wang, H.; Rabbee, N.; Egidio, C.M.; Reynolds, J.; Tom, E.; Pagan, M.; Rigl, C.T.; et al. Molecular classification of thyroid nodules using high-dimensionality genomic data. J. Clin. Endocrinol. Metab. 2010, 95, 5296–5304. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Song, Y.S.; Kim, Y.A.; Lim, J.A.; Cho, S.W.; Moon, J.H.; Hahn, S.; Park, D.J.; Park, Y.J. Effects of coexistent braf(v600e) and tert promoter mutations on poor clinical outcomes in papillary thyroid cancer: A meta-analysis. Thyroid 2017, 27, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Aragon Han, P.; Olson, M.T.; Fazeli, R.; Prescott, J.D.; Pai, S.I.; Schneider, E.B.; Tufano, R.P.; Zeiger, M.A. The impact of molecular testing on the surgical management of patients with thyroid nodules. Ann. Surg. Oncol. 2014, 21, 1862–1869. [Google Scholar] [CrossRef] [PubMed]
- Noureldine, S.I.; Najafian, A.; Aragon Han, P.; Olson, M.T.; Genther, D.J.; Schneider, E.B.; Prescott, J.D.; Agrawal, N.; Mathur, A.; Zeiger, M.A.; et al. Evaluation of the effect of diagnostic molecular testing on the surgical decision-making process for patients with thyroid nodules. JAMA Otolaryngol. Head. Neck. Surg. 2016, 142, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Noureldine, S.I.; Zeiger, M.A.; Tufano, R.P. Gene expression classifier testing and the surgical decision-making process for patients with thyroid nodules-reply. JAMA Otolaryngol. Head. Neck. Surg. 2016, 142, 807. [Google Scholar] [CrossRef]
- Sahli, Z.T.; Smith, P.W.; Umbricht, C.B.; Zeiger, M.A. Preoperative molecular markers in thyroid nodules. Front. Endocrinol. (Lausanne) 2018, 9, 179. [Google Scholar] [CrossRef]
- Nicholson, K.J.; Roberts, M.S.; McCoy, K.L.; Carty, S.E.; Yip, L. Molecular testing versus diagnostic lobectomy in bethesda iii/iv thyroid nodules: A cost-effectiveness analysis. Thyroid 2019, 29, 1237–1243. [Google Scholar] [CrossRef]
- Wang, L.Y.; Ganly, I. Post-treatment surveillance of thyroid cancer. Eur. J. Surg. Oncol. 2018, 44, 357–366. [Google Scholar] [CrossRef]
- Spencer, C.A.; Takeuchi, M.; Kazarosyan, M.; Wang, C.C.; Guttler, R.B.; Singer, P.A.; Fatemi, S.; LoPresti, J.S.; Nicoloff, J.T. Serum thyroglobulin autoantibodies: Prevalence, influence on serum thyroglobulin measurement, and prognostic significance in patients with differentiated thyroid carcinoma. J. Clin. Endocrinol. Metab. 1998, 83, 1121–1127. [Google Scholar] [CrossRef]
- Lee, Z.J.O.; Eslick, G.D.; Edirimanne, S. Investigating anti-thyroglobulin antibody as a prognostic marker for differentiated thyroid cancer: A meta-analysis and systematic review. Thyroid 2020. [Google Scholar] [CrossRef]
- Durante, C.; Tognini, S.; Montesano, T.; Orlandi, F.; Torlontano, M.; Puxeddu, E.; Attard, M.; Costante, G.; Tumino, S.; Meringolo, D.; et al. Clinical aggressiveness and long-term outcome in patients with papillary thyroid cancer and circulating anti-thyroglobulin autoantibodies. Thyroid 2014, 24, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Rosignolo, F.; Sponziello, M.; Giacomelli, L.; Russo, D.; Pecce, V.; Biffoni, M.; Bellantone, R.; Lombardi, C.P.; Lamartina, L.; Grani, G.; et al. Identification of thyroid-associated serum microrna profiles and their potential use in thyroid cancer follow-up. J. Endocr. Soc. 2017, 1, 3–13. [Google Scholar] [PubMed]
- Nixon, A.M.; Provatopoulou, X.; Kalogera, E.; Zografos, G.N.; Gounaris, A. Circulating thyroid cancer biomarkers: Current limitations and future prospects. Clin. Endocrinol. (Oxf.) 2017, 87, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Volante, M.; Collini, P.; Nikiforov, Y.E.; Sakamoto, A.; Kakudo, K.; Katoh, R.; Lloyd, R.V.; LiVolsi, V.A.; Papotti, M.; Sobrinho-Simoes, M.; et al. Poorly differentiated thyroid carcinoma: The turin proposal for the use of uniform diagnostic criteria and an algorithmic diagnostic approach. Am. J. Surg. Pathol. 2007, 31, 1256–1264. [Google Scholar] [CrossRef]
- Ibrahimpasic, T.; Ghossein, R.; Shah, J.P.; Ganly, I. Poorly differentiated carcinoma of the thyroid gland: Current status and future prospects. Thyroid 2019, 29, 311–321. [Google Scholar] [CrossRef]
- Chintakuntlawar, A.V.; Foote, R.L.; Kasperbauer, J.L.; Bible, K.C. Diagnosis and management of anaplastic thyroid cancer. Endocrinol. Metab. Clin. N. Am. 2019, 48, 269–284. [Google Scholar] [CrossRef]
- Kunstman, J.W.; Juhlin, C.C.; Goh, G.; Brown, T.C.; Stenman, A.; Healy, J.M.; Rubinstein, J.C.; Choi, M.; Kiss, N.; Nelson-Williams, C.; et al. Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum. Mol. Genet 2015, 24, 2318–2329. [Google Scholar] [CrossRef]
- Pozdeyev, N.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Deaver, K.E.; Davis, S.; French, J.D.; Borre, P.V.; LaBarbera, D.V.; Tan, A.C.; et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin. Cancer Res. 2018, 24, 3059–3068. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in multiple nonmelanoma cancers with braf v600 mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Godbert, Y.; Henriques de Figueiredo, B.; Bonichon, F.; Chibon, F.; Hostein, I.; Pérot, G.; Dupin, C.; Daubech, A.; Belleannée, G.; Gros, A.; et al. Remarkable response to crizotinib in woman with anaplastic lymphoma kinase-rearranged anaplastic thyroid carcinoma. J. Clin. Oncol. 2015, 33, e84–e87. [Google Scholar] [CrossRef] [PubMed]
- Bible, K.C. Treating advanced radioresistant differentiated thyroid cancer. Lancet Oncol. 2012, 13, 854–855. [Google Scholar] [CrossRef]
- Fagin, J.A.; Wells, S.A., Jr. Biologic and clinical perspectives on thyroid cancer. N. Engl. J. Med. 2016, 375, 1054–1067. [Google Scholar] [CrossRef]
- Durante, C.; Haddy, N.; Baudin, E.; Leboulleux, S.; Hartl, D.; Travagli, J.P.; Caillou, B.; Ricard, M.; Lumbroso, J.D.; De Vathaire, F.; et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: Benefits and limits of radioiodine therapy. J. Clin. Endocrinol. Metab. 2006, 91, 2892–2899. [Google Scholar] [CrossRef] [PubMed]
- Clark, O.; Duh, Q.-Y.; Kebebew, E. Textbook of Endocrine Surgery, 3rd ed.; JP Medical Ltd.: New Delhi, India, 2016. [Google Scholar]
- Ricarte-Filho, J.C.; Ryder, M.; Chitale, D.A.; Rivera, M.; Heguy, A.; Ladanyi, M.; Janakiraman, M.; Solit, D.; Knauf, J.A.; Tuttle, R.M.; et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for braf, pik3ca, and akt1. Cancer Res. 2009, 69, 4885–4893. [Google Scholar] [CrossRef] [PubMed]
- Costamagna, E.; García, B.; Santisteban, P. The functional interaction between the paired domain transcription factor pax8 and smad3 is involved in transforming growth factor-beta repression of the sodium/iodide symporter gene. J. Biol. Chem. 2004, 279, 3439–3446. [Google Scholar] [CrossRef]
- Gild, M.L.; Topliss, D.J.; Learoyd, D.; Parnis, F.; Tie, J.; Hughes, B.; Walsh, J.P.; McLeod, D.S.A.; Clifton-Bligh, R.J.; Robinson, B.G. Clinical guidance for radioiodine refractory differentiated thyroid cancer. Clin. Endocrinol. (Oxf.) 2018, 88, 529–537. [Google Scholar] [CrossRef]
- Okamoto, K.; Kodama, K.; Takase, K.; Sugi, N.H.; Yamamoto, Y.; Iwata, M.; Tsuruoka, A. Antitumor activities of the targeted multi-tyrosine kinase inhibitor lenvatinib (e7080) against ret gene fusion-driven tumor models. Cancer Lett. 2013, 340, 97–103. [Google Scholar] [CrossRef]
- Tohyama, O.; Matsui, J.; Kodama, K.; Hata-Sugi, N.; Kimura, T.; Okamoto, K.; Minoshima, Y.; Iwata, M.; Funahashi, Y. Antitumor activity of lenvatinib (e7080): An angiogenesis inhibitor that targets multiple receptor tyrosine kinases in preclinical human thyroid cancer models. J. Thyroid Res. 2014, 2014, 638747. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both raf and vegf and pdgf receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef]
- Brose, M.S.; Cabanillas, M.E.; Cohen, E.E.; Wirth, L.J.; Riehl, T.; Yue, H.; Sherman, S.I.; Sherman, E.J. Vemurafenib in patients with braf(v600e)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: A non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1272–1282. [Google Scholar] [CrossRef]
- Liu, D.; Hu, S.; Hou, P.; Jiang, D.; Condouris, S.; Xing, M. Suppression of braf/mek/map kinase pathway restores expression of iodide-metabolizing genes in thyroid cells expressing the v600e braf mutant. Clin. Cancer Res. 2007, 13, 1341–1349. [Google Scholar] [CrossRef]
- Ho, A.L.; Grewal, R.K.; Leboeuf, R.; Sherman, E.J.; Pfister, D.G.; Deandreis, D.; Pentlow, K.S.; Zanzonico, P.B.; Haque, S.; Gavane, S.; et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N. Engl. J. Med. 2013, 368, 623–632. [Google Scholar] [CrossRef]
- Iravani, A.; Solomon, B.; Pattison, D.A.; Jackson, P.; Ravi Kumar, A.; Kong, G.; Hofman, M.S.; Akhurst, T.; Hicks, R.J. Mitogen-activated protein kinase pathway inhibition for redifferentiation of radioiodine refractory differentiated thyroid cancer: An evolving protocol. Thyroid 2019, 29, 1634–1645. [Google Scholar] [CrossRef]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; Cabanillas, M.E.; Urbanowitz, G.; et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic braf v600-mutant anaplastic thyroid cancer. J. Clin. Oncol. 2018, 36, 7–13. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. Ntrk fusion-positive cancers and trk inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Harris, E.J.; Hanna, G.J.; Chau, N.; Rabinowits, G.; Haddad, R.; Margalit, D.N.; Schoenfeld, J.; Tishler, R.B.; Barletta, J.A.; Nehs, M.; et al. Everolimus in anaplastic thyroid cancer: A case series. Front. Oncol. 2019, 9, 106. [Google Scholar] [CrossRef]
- Gambacorti-Passerini, C.; Orlov, S.; Zhang, L.; Braiteh, F.; Huang, H.; Esaki, T.; Horibe, K.; Ahn, J.S.; Beck, J.T.; Edenfield, W.J.; et al. Long-term effects of crizotinib in alk-positive tumors (excluding nsclc): A phase 1b open-label study. Am. J. Hematol. 2018, 93, 607–614. [Google Scholar] [CrossRef]
- Subbiah, V.; Velcheti, V.; Tuch, B.B.; Ebata, K.; Busaidy, N.L.; Cabanillas, M.E.; Wirth, L.J.; Stock, S.; Smith, S.; Lauriault, V.; et al. Selective ret kinase inhibition for patients with ret-altered cancers. Ann. Oncol. 2018, 29, 1869–1876. [Google Scholar] [CrossRef] [PubMed]
- Registrational Results of Loxo-292 in Patients with Ret-altered Thyroid Cancers. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-selpercatinib-lung-and-thyroid-cancers-ret-gene-mutations-or-fusions (accessed on 10 May 2020).
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Sheu, N.W.; Jiang, H.J.; Wu, C.W.; Chiang, F.Y.; Chiou, H.C.; Hsiao, P.J. Lenvatinib complementary with radioiodine therapy for patients with advanced differentiated thyroid carcinoma: Case reports and literature review. World J. Surg. Oncol. 2019, 17, 84. [Google Scholar] [CrossRef]
- Nikiforov, Y.E.; Erickson, L.A.; Nikiforova, M.N.; Caudill, C.M.; Lloyd, R.V. Solid variant of papillary thyroid carcinoma: Incidence, clinical-pathologic characteristics, molecular analysis, and biologic behavior. Am. J. Surg. Pathol. 2001, 25, 1478–1484. [Google Scholar] [CrossRef]
- Tiedje, V.; Stuschke, M.; Weber, F.; Dralle, H.; Moss, L.; Fuhrer, D. Anaplastic thyroid carcinoma: Review of treatment protocols. Endocr. Relat. Cancer 2018, 25, R153–R161. [Google Scholar] [CrossRef]




| Affirma® GSC | ThyGenX/ThyraMIR® ThyraGeNEXT/ThyraMIR®, * | Thyroseq v3® | |
|---|---|---|---|
| Methodology | mRNA gene sequencing and gene expression | NGS and mRNA sequencing, polymerase chain reaction miRNA expression | NGS of DNA and mRNA |
| Tested alterations | 511 mRNA panel, gene expression of 1115 genes, CAN 3 | 7–10 DNA and mRNA panel, 10 miRNA | 112 DNA and mRNA panel (>12,000 variants and 150 gene fusions), CAN 3, gene expression |
| Ideally applicable for | Bethesda III/IV, Hürtle cell lesions | Bethesda III/IV/V | Bethesda III/IV/V, Hürtle Cell lesions |
| Report |
|
|
|
| Interpretation of test results | Benign: consider surveillance Suspicious: consider surgery | ThyGenX/ThyraGeNEXT positive: consider surgery ThyraMIR high risk: consider surgery ThyraMIR low risk: consider surveillance | Negative: consider surveillance Positive: consider surgery |
| Validation Study | Patel et al. [84] | Labourier et al. [85] | Steward et al. [86] |
| Nodules examined | n = 190 | n = 109 | n = 247 |
| Prevalence of cancer | 24% | 32% | 28% |
| Sensitivity | 91% (79–98) | 89% (73–97) | 94% (85–100) |
| Specificity | 68% (60–67) | 85% (75–92) | 82% (63–84) |
| NPV 1 | 96% (90–99) | 94% (85–98) | 97% (93–99) |
| PPV 2 | 47% (36–58) | 74% (58–86) | 66% (56–75) |
| “Rule out” | “Rule in” and “Rule out” | “Rule in” and “Rule out” |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nylén, C.; Mechera, R.; Maréchal-Ross, I.; Tsang, V.; Chou, A.; Gill, A.J.; Clifton-Bligh, R.J.; Robinson, B.G.; Sywak, M.S.; Sidhu, S.B.; et al. Molecular Markers Guiding Thyroid Cancer Management. Cancers 2020, 12, 2164. https://doi.org/10.3390/cancers12082164
Nylén C, Mechera R, Maréchal-Ross I, Tsang V, Chou A, Gill AJ, Clifton-Bligh RJ, Robinson BG, Sywak MS, Sidhu SB, et al. Molecular Markers Guiding Thyroid Cancer Management. Cancers. 2020; 12(8):2164. https://doi.org/10.3390/cancers12082164
Chicago/Turabian StyleNylén, Carolina, Robert Mechera, Isabella Maréchal-Ross, Venessa Tsang, Angela Chou, Anthony J. Gill, Roderick J. Clifton-Bligh, Bruce G. Robinson, Mark S. Sywak, Stan B. Sidhu, and et al. 2020. "Molecular Markers Guiding Thyroid Cancer Management" Cancers 12, no. 8: 2164. https://doi.org/10.3390/cancers12082164
APA StyleNylén, C., Mechera, R., Maréchal-Ross, I., Tsang, V., Chou, A., Gill, A. J., Clifton-Bligh, R. J., Robinson, B. G., Sywak, M. S., Sidhu, S. B., & Glover, A. R. (2020). Molecular Markers Guiding Thyroid Cancer Management. Cancers, 12(8), 2164. https://doi.org/10.3390/cancers12082164