Abstract
The incidence of thyroid cancer is rapidly increasing, mostly due to the overdiagnosis and overtreatment of differentiated thyroid cancer (TC). The increasing use of potent preclinical models, high throughput molecular technologies, and gene expression microarrays have provided a deeper understanding of molecular characteristics in cancer. Hence, molecular markers have become a potent tool also in TC management to distinguish benign from malignant lesions, predict aggressive biology, prognosis, recurrence, as well as for identification of novel therapeutic targets. In differentiated TC, molecular markers are mainly used as an adjunct to guide management of indeterminate nodules on fine needle aspiration biopsies. In contrast, in advanced thyroid cancer, molecular markers enable targeted treatments of affected signalling pathways. Identification of the driver mutation of targetable kinases in advanced TC can select treatment with mutation targeted tyrosine kinase inhibitors (TKI) to slow growth and reverse adverse effects of the mutations, when traditional treatments fail. This review will outline the molecular landscape and discuss the impact of molecular markers on diagnosis, surveillance and treatment of differentiated, poorly differentiated and anaplastic follicular TC.
1. Introduction
According to the Global Cancer Observatory GLOBOCAN 2018 database, thyroid cancer (TC) accounts for 3.1% of all new cancer diagnoses worldwide and, therefore, ranks in 9th position with regard to incidence [1,2]. Moreover, with an estimated annual 41,000 deaths, mortality rates (0.4–0.5%) remain comparably stable and low [1,3]. However, it is one of the fastest growing cancer entities worldwide and its incidence in the United States has nearly tripled over the last 30 years from 4.9 to 14.3 per 100,000 people [3,4,5]. Many reasons for this epidemiological development have been discussed [2,6,7,8]. While some authors also suggest a contributing true increase of incidence [9], the main reason for the increase seems to be a consequence of oversurveillance and overdiagnosis of small differentiated tumours [3]. This can be attributed to significant improvements of diagnostic techniques, particularly higher sensitivity of ultrasounds (US), US-guided fine needle aspiration (FNA) and computed tomography [7,10]. The resulting overtreatment or continuous surveillance for clinically insignificant findings not only influences incidence but is also a significant factor in times where modern healthcare systems become under significant cost pressure [11,12].
Therefore, it is crucial to identify adjuncts such as molecular markers, which can distinguish between benign and malignant tumours, predict aggressive biology, prognosis, recurrence and efficacy of treatment as well as represent possible novel therapeutic targets. The development of potent preclinical models, high throughput molecular technologies such as next-generation sequencing (NGS) and gene expression microarrays have provided a deeper understanding of genetic alterations and molecular characteristics in cancer [13]. This offers the potential for development of personalized cancer treatment where patients are treated according to their individual tumour characteristics, with potential to improve patient’s outcome, rather than to a “one-fits-all” strategy [14]. The increasing importance of molecular markers in diagnostic and treatment strategies of TC was also further acknowledged by the latest American Thyroid Association (ATA) guidelines as well as American Association of Endocrine Surgeons guidelines [15,16,17].
TC subtypes derived from follicular cells can be histologically divided into differentiated (DTC), poorly differentiated (PDTC), and anaplastic thyroid cancer (ATC) [18]. However, since the discovery of the oncogenic role of the BRAFV600E mutation in 2003, various molecular and genetic markers in TC have been identified, and added an additional layer to the classification of TC [19,20,21]. Although the total mutational burden (TMB) is lower than in most other cancer entities, the increasing importance of molecular alterations was underlined by The Cancer Genome Atlas (TCGA), which identified genetic alterations in 97% of the studied tumours [20,22]. While the TCGA analysis only included papillary thyroid cancer (PTC), subsequent studies have shown several markers and their combination play a significant role in the development of other TC subtypes as well as the progression and dedifferentiation to more advanced cancer subtypes [23,24,25]. An example of evolving biomarkers for diagnosis and prognosis in TC are MicroRNAs (miRNA). By regulating target genes of various signalling pathways including gene expression of known oncogenes and tumour suppressor genes, they play a crucial role for cell differentiation, migration, invasion and even epithelial-to-mesenchymal transition (EMT) [26,27,28]. Furthermore, different and distinct miRNA profiles were identified in different TC subtypes [28,29,30,31,32,33].
In this review, we will outline the molecular landscape of follicular cell derived TC and discuss the impact of molecular markers on diagnosis, surveillance, and treatment of DTC, as well as PDTC and ATC. Moreover, we will give an outlook on the emergence of novel molecular characteristics and techniques, which have potential to guide decision making in TC management in the future leading to personalisation of TC care and avoiding overtreatment in low risk cancers.
2. The Molecular Landscape of Follicular Cell Derived Thyroid Cancer and Oncogenesis
The understanding of the molecular landscape of follicular cell derived TC has improved vastly with the publications of the genomic studies of TC, which has enabled promising new targets of treatment. The mutations identified in the TCGA show distinct patterns mostly affecting two signalling pathways involved in thyroid growth and proliferation, the mitogen-activating protein kinase (MAPK), and the Phosphatidylinositol-3-kinase (PI3K)/AKT-pathways [34] (Figure 1). Genetic and epigenetic dysfunctions of kinases and transcription factors related to these pathways lead to constitutive activation that initiate and aggravate the tumorigenesis and progression of TC. In this section, we will describe the main identified molecular alterations in follicular-derived TC.
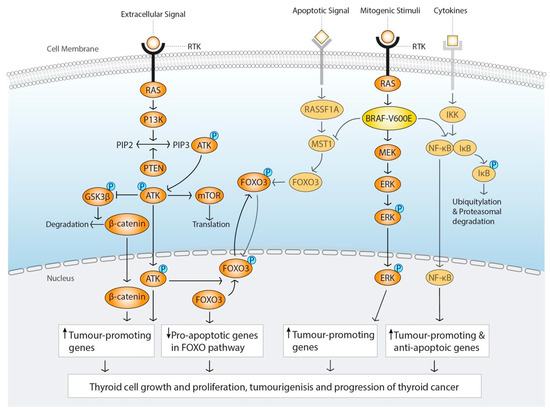
Figure 1.
Molecular landscape in thyroid cancer.
2.1. The MAPK-Signalling Pathway
The MAPK-signalling pathway is crucial for growth and proliferation of the cell (Figure 1). The signalling cascade is initiated on the cell surface by a “mitogen” stimuli, a growth factor, that binds to the tyrosine kinase receptor (RTK) and ultimately results in the phosphorylation and activation of Mitogen-Activated Protein Kinase Kinase (MAP2K), which is also known as MEK, and the downstream extra cellular signal-regulated-kinase (ERK). Phosphorylation of ERK leads to activation of transcription factors targeting genes to induce cell cycle entry and to suppress negative regulators of cell cycle [35]. The MAPK-signalling pathway is delicately regulated in several steps by enzymes and factors, to which genetic and epigenetic dysregulations can lead to a constitutively active pathway, an uncontrolled growth and proliferation of the cell, hallmarks of oncogenesis [36]. Mutations of key regulators of the MAPK-signalling pathway are present in about 70% of all PTC [37] and constitutive MAPK activation leads to dedifferentiation of PTC in preclinical models [38].
This illustration summarizes the two main affected signalling pathways in thyroid cancer (TC); Phosphatidylinositol-3-kinase (PI3K)-AKT and mitogen-activating protein kinase (MAPK) signalling pathway. Mutations or genetic rearrangements of targets in the respective pathway lead to constitutive activation that ultimately leads to translocation of transcription factors to the nucleus upregulating transcription of genes promoting tumourigenesis and progression.
A point mutation at the loci T1799A in codon 600 in the B-Raf proto-oncogene, BRAF, a gene coding for a key serine/threonine kinase in the MAPK signalling cascade, leads to a BRAFV600E mutant protein first described in TC by Cohen et al. in 2003 [19]. BRAFV600E mutation leads to a constitutive active BRAF kinase independent of its upstream target, RAS, ultimately leading to increased ERK1/2 activation [39]. The BRAFV600E kinase domain is about 500 fold more active than the wild type BRAF kinase [40] and its mutation is regarded to be one of the fundamental initiating events in the tumorigenesis and progression of PTC [41].
In addition to activating the downstream targets in the MAPK-signalling cascade, BRAFV600E mutation is also capable of activating the nuclear factor kappa beta (NFκB) pathway [42], which is important for the inflammatory response and activated in oncogenesis. BRAFV600E mutation can also inhibit the class O of forkhead box transcription factors (FOXO)-pathway through macrophage stimulating 1 (MST1), leading to inhibition of proapoptotic genes [43].
The discovery of telomerase reverse transcriptase (TERT) promoter mutations in TC in 2013 [44], represents an important event in the TC field and has gained much attention since in its role in modifying the effect of driver mutations such as BRAF. The proposed mechanism of TERT promoter mutation is to generate de novo binding elements for the ETS-transcription family of transcription factors such as GABPA, which leads to increased telomerase expression and immortalisation [45]. ETS-transcription factors are activated by MAPK signalling, and this process enhances the effect of the MAPK signalling further, and indeed, co-existent mutations of BRAFV600E and TERT are associated with more aggressive forms of PTC [46].
2.2. PI3K/AKT Signalling Pathway
The PI3K/AKT signalling pathway is involved in the regulation of apoptosis, proliferation, cell cycle progression, angiogenesis, cytoskeleton integrity, and energy metabolism, and ultimately leads to AKT and mTOR activation [47] (Figure 1). One of the most commonly mutated targets in the PI3K/AKT cascade is RAS. RAS, a small G protein anchored to the inner membrane of the cell, is the upstream kinase of BRAF kinase and active when bound to GTP. RAS mutation downregulates the activity of GTPase, the enzyme that hydrolyses GTP to GDP, and the mutation leaves RAS in a constitutively active GTP-bound state. Due to its location in the cell, adjacent to the membrane bound tyrosine kinase receptor, RAS mutations can activate both MAPK- and PI3K/AKT pathways. However, it seems like RAS mutations preferable activate the PI3K-AKT pathway in thyroid oncogenesis [48]. There are three isoforms of RAS-NRAS, HRAS and KRAS, where NRAS is the most commonly mutated in human TC [49]. RAS mutation is thought to be a premalignant mutation, and that additional mutations are needed to set off carcinogenesis [49]. Indeed, in a KRAS transgenic mouse model, deletion of a tumour suppressor gene, PTEN, was needed for development of follicular thyroid cancer (FTC) [50]. Mutations or deletions of PTEN activates PI3K-AKT pathway, which is the genetic basis for follicular thyroid cell tumorigenesis in Cowden’s disease [51]. Mutations in PI3K3CA gene which encodes for the p110alpha catalytic subunit of PI3K is also common in follicular thyroid cancer (FTC), but also poorly differentiated thyroid cancer (PDTC) and anaplastic thyroid cancer (ATC) [48].
2.3. Gene Translocations
Gene translocations can lead to oncogenetic chromosomal rearrangements. The most common group of translocation in TC is known as RET/PTC and was first described in PTC by Fusco et al. 1987 [52]. It is important to note that RET/PTC does not refer to a single translocation, but to a group of different translocations involving RET and several other genes. For example, the most common translocation known as RET/PTC1 and RET/PTC3 actually represent translocations between the RET gene and CCDC6 (RET-CCDC6) and the RET gene and NCOA4 (RET-NCOA4), respectively [53]. RET/PTC rearrangements occurs as a consequence of genetic recombination between the 3´tyrosine kinase portion of RET and the 5´portion of a partner gene due to spatial proximity in the nucleus [54]. RET is a classical proto-onco gene, a gene that when activated by mutation on rearrangement can contribute to cancer. The RET/PTC rearrangement encodes for a fusion oncogene containing a receptor tyrosine kinase (RTK) that is ligand independent, enabling constitutive activation of both MAPK and the PI3K-AKT pathways. The PAX8/PPARG, NTRK1, and -3 and ALK are other prominent recombinant oncogenes [55], where the fusion of the oncogenes alters the transcription of the target genes, predisposing for oncogenesis in TC.
2.4. Gene Amplification and Copy Number Alterations
Oncogenic gene amplifications are genetic mechanisms in thyroid oncogenesis, commonly occurring with RTKs (receptor tyrosine kinases) and genes encoding targets in the PI3K-AKT pathways. Genetic copy number alterations (CNA) are more common in ATC suggesting mechanism of progression and aggressiveness [25]. CNA through genetic amplification or chromosomal instability and aneuploidy (extra chromosome) leads to increased protein expression and hence activation of downstream signalling pathways. Gains of genes encoding RTKs ultimately increase phosphorylation and hence activation of both AKT and ERK [48].
2.5. Epigenetic Regulation
“Epi” comes from the Greek prefix meaning “on top of”, which refers to a regulative mechanism “on top of the DNA sequence”. Epigenetic modifications of the genome and gene expression include histone modifications, DNA methylation and microRNAs (miRNA). These epigenetic marks influence the central dogma of DNA transcription, RNA (mRNA) translation and protein synthesis in the cell. Epigenetic modifications are common in human cancer and are also present in TC [56]. DNA methylation of promotor regions often lead to suppression of gene transcription. In array-based genome-wide DNA methylation studies of PTC, the first performed by Hou et al. [57] shows that, in similarity to sequencing studies revealing low mutation frequency in PTC, there is also low frequency of DNA methylation alteration. In contrast, ATC exhibits a high frequency of DNA methylation alterations, 10-fold higher than PTC [58]. One confounding factor in performing DNA methylation studies on ATC is however the infiltration of immune cells making it possible that part of the hypermethylation may come from these cells and not the TC itself [56]. Some specific targets that often have been shown to be hypermethylated in TCs is PTEN, RASSF1A, CDKN2A or P16INK4A and DAPK [56].
Histones are the scaffold proteins that DNA is tightly winded around and function to condense and package DNA in the nucleus. Histones have molecules attached to them that when modified changes the chromatin structure and the availability of the DNA sequence to transcription [59]. There are several different known histone modifications and enzymes responsible for these modifications. Most of the histone modifications identified are also in the more advanced TCs, and the most studied is the histone deacetylase (HDAC) [60].
He et al. [61] were the first to acknowledge the involvement of miRNA in PTC. MiRNA are noncoding small RNAs that in simplified terms recognise a target messenger RNA (mRNA) and repress translation of the mRNA into protein, and will be discussed further below. In addition to miRNA, the involvement of multiple long noncoding RNAs (lncRNA) in TC have recently been established by several researchers [62,63]. Dysregulated lncRNA are involved in the epigenetic regulation of gene transcription and implicated in tumour suppression and oncogenic functions [64]
2.6. The Progressive Oncogenesis Model
The currently accepted model of TC oncogenesis is the multistep oncogenesis model, that suggests that mature follicular cells can transform into well differentiated TC cells and then progress into undifferentiated TC cell [65] (Figure 2). The clinical findings that well differentiated TC can harbour foci of PDTC and APC, strengthen this hypothesis that the progress is possible due to stepwise additions of molecular pathogenesis with increased mutational, copy number, and epigenetic events [66]. Mutations including TERT promoter, TP53, EIF1AX, and genes involved in the PIK3CA-AKT-mTOR pathway, SW1/SNF complex and mismatch repair genes are particularly involved in the more advanced TCs [67].
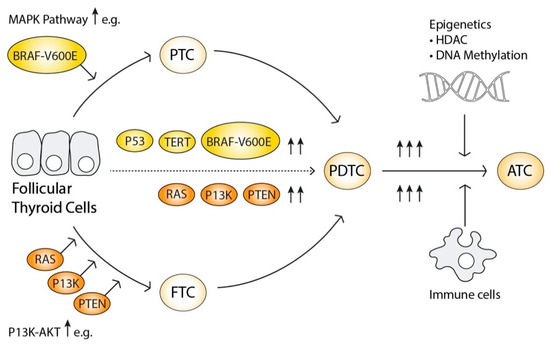
Figure 2.
Illustration of the oncogenesis mode.
Progression from Follicular thyroid cells to papillary thyroid cancer (PTC) or follicular thyroid cancer (FTC) is marked by mutations activating the MAPK- or PI3K-AKT signalling pathways, illustrated by the arrows. Further progression to poorly differentiated thyroid cancer (PDTC) is characterised by additional mutations and rearrangements, augmenting both signalling pathways simultaneously. Anaplastic thyroid cancer (ATC) is characterised by further genetic events, especially in oncogenes like P53, as well as epigenetic alterations and infiltration of immune cells, creating an intracellular microclimate that favours genetic instability and oncogenesis.
Interestingly, simultaneous activation of MAPK and PI3K-AKT signalling pathways becomes more frequent as the grade of the tumour progresses [49]. Overactive PI3K-AKT and MAPK signalling pathways can activate the Wnt/β-catenin pathway known to regulate growth, proliferation as well as stem cell differentiation, as well as the HIF1α-pathway involved in angiogenesis and altered cell metabolism. Catenin Beta 1 (CTNNB1) is involved in cell adhesion and invasiveness, and HIF1, is a strong activator of vascular endothelial growth factor A, VEGFA. Both MAPK and PI3K-AKT signalling pathways can also stimulate FOXO- and NFκB pathways as previously described. Specific genetic alterations also present in the more advanced cancers includes TP53 that induces genomic instability, ALK and epidermal growth factor receptor (EGFR) [49,67].
ATC harbours more copy number variations and epigenetic alterations than PDTC and PDTC more than DTC, which also supports the model of progressive oncogenesis. In conclusion, the progression from DTC to PDTC and further to ATC is hence an accumulation of genetic and epigenetic events that synergistically amplify their tumorigenicity [68] (Figure 2).
3. Molecular Marker for Diagnosis and Management of Thyroid Cancer in DTC
3.1. The Diagnostic and Therapeutic Challenge of Indeterminate Nodules
One major way this knowledge of the molecular biology of thyroid cancer is being applied to management is in the diagnosis of thyroid nodules. The introduction of the Bethesda System for Reporting Thyroid Cytopathology (BSRTC) in 2009 established a standardized reporting system of cytology results of thyroid fine needle aspiration (FNA) [69] (Table 1). It enabled users to reliably estimate the occurrence of malignancy in thyroid nodules for benign (BII) and malignant (BVI) Bethesda categories in 70 to 80% [70]. However, 20–30% remain indeterminate (BIII-BV) and risk of malignancy (ROM) demonstrate high variability ranging from 5–75%, the reasons for this are a matter of debate [69]. While intra- and interobserver variability is certainly one explanation, selection and publication bias need to be considered as well and result in a general overestimation of the ROM [69,71,72,73]. Moreover, the reclassification of the Encapsulated Follicular Variant of Papillary Thyroid Carcinoma (EFVPTC) to Non-invasive Follicular Thyroid Neoplasm with papillary-like nuclear Features (NIFTP) [74] not only established a new tumour classification [75] but as a consequence significantly lowered the rates of carcinomas in the indeterminate categories of the BSRTC [76]. Hence, predictions of ROM in the latest BSRTC classification differ from the previous version and, therefore impact decision making for the management of indeterminate nodules even more (Table 1).
Of note, malignancy of cytologically indeterminate nodules cannot be reliably diagnosed intraoperatively on fresh frozen sections due to low sensitivity and high false negative rates and increases cost and duration of operation [77,78]. In line with the 2015 ATA and the 2020 American Association of Endocrine Surgeons (AAES) guidelines, possible management strategies range from repeat FNA or surveillance to diagnostic surgery [15,17]. However, only 10–40% of indeterminate nodules submitted to surgery will be malignant on histopathology [79]. Consequently, molecular testing (MT) is considered as an adjunct to refine the cancer risk and to reduce the rate of diagnostic surgery. Furthermore, it may minimize the extent of the surgical procedure if no other indications for surgery are present (size, compressive symptoms, personal preference, high-risk features on imaging) [15,16,17,80]. However, in many health settings, commercially available MT is not easily available, and its cost can be prohibitive to use as will be discussed further (Table 2).

Table 2.
Summary of recent commercially available molecular tests [17,82,83].
Table 1.
The revised 2017 Bethesda System for Reporting Thyroid Cytopathology (TBSRTC) [81].
Table 1.
The revised 2017 Bethesda System for Reporting Thyroid Cytopathology (TBSRTC) [81].
| Diagnostic Category | ROM 1 (NIFTP 2 Considered as Cancer (%)) | ROM 1 (NIFTP 2 NOT Considered as Cancer (%)) | Management Options | |
|---|---|---|---|---|
| I. | Nondiagnostic/unsatisfactory (Cyst fluid, acellular specimen, other (e.g., obscuring blood, artefacts)) | 5–10 | 5–10 | Correlate with clinical/radiological findings Consider repeat FNA 3 |
| II. | Benign (Benign follicular nodule, Chronic lymphocytic (Hashimoto) thyroiditis, Granulomatous (subacute) thyroiditis) | 0–3 | 0–3 | Surveillance and US 4 follow up |
| III. | Atypia of undetermined significance, or follicular lesion of undetermined significance | 6–18 | 10–30 | Correlate with clinical/radiological findings Consider repeat FNA 3 Consider molecular testing |
| IV. | Follicular neoplasm OR Follicular carcinoma (specify if Hürtle cell features) | 10–40 | 25–40 | Consider molecular testing, Lobectomy |
| V. | Suspicious for malignancy | 45–60 | 50–75 | Total thyroidectomy or lobectomy |
| VI. | Malignant (Papillary thyroid carcinoma, Poorly differentiated thyroid carcinoma, Medullary thyroid carcinoma, Anaplastic thyroid carcinoma, Squamous cell carcinoma, Carcinoma with mixed features, Metastatic malignancy, Non-Hodgkin lymphoma, Other) | 94–96 | 97–99 | Total thyroidectomy or lobectomy |
1 Risk of malignancy; 2 Noninvasive Follicular Thyroid Neoplasm with papillary-like nuclear features; 3 Fine needle aspiration; 4 Ultrasound.
3.2. Molecular Markers and Clinical Profiles in DTC
To enhance the diagnostic sensitivity of FNA results, enable molecular testing and to help guiding management in indeterminate nodules where no other indication for surgery (size, compressive symptoms, personal preference, high risk features on imaging) are present, molecular profiles and markers need to be identified and their diagnostic and prognostic role defined [87]. Molecular markers currently in use include immunohistochemistry, classical genomic alteration (point mutations, deletions, insertion), gene fusions causing rearrangements and translocation, copy number variations, microRNA (miRNA), and circulating markers of disease [17,88]. The most relevant alterations and markers will be discussed below.
3.2.1. Genetic Markers
With around 80%, PTC is the most common entity of TC [89]. Despite excellent long-term survival, recurrence can cause significant morbidity and molecular markers can help to distinguish an aggressive from an indolent course [90]. Various driver mutations have been identified and associated with different histological subtypes [20]. However, PTCs can be classified by gene expression profiles to either BRAF-like (BRAFV600E, RET/PTC fusion, NTRK fusions) or RAS-like molecular subtypes (RAS, BRAFK601E, PAX8/PPARG, PTEN, EIF1AX) [20].
The most common mutation in 40–80% of PTC’s is BRAFV600E [91]. Mainly seen in classic and tall cell PTC and less often in benign nodules, BRAFV600E mutations are associated with a higher frequency of lymph node metastasis, extrathyroidal extension, recurrence, advanced clinical stage as well as poor response to radioiodine [92,93,94,95]. However, despite high specificity and positive predictive value for PTC, the absence of BRAFV600E alterations alone harbour a low negative predictive value mainly due to the involvement of other driver mutations in the development of TC [96,97,98]. There have been conflicting findings on the prognostic value of BRAFV600E [20,99,100] and only in combination with a TERT promotor mutation, it seems to be truly linked to aggressive PTC [101]. Due to this range of heterogenous phenotypes, BRAFV600E is not an ideal exclusive marker to guide surgical management [102].
The rarer BRAFK601E mutation is mainly associated with follicular variant PTC (FVPTC) but also benign adenoma. They demonstrated to show improved long-term outcome and only rarely cause disease related deaths [94,103]. However, BRAFK601E mutation is not diagnostic for cancer due to a rather low positive predictive value (PPV) [104].
Despite being specific for PTC, RET rearrangements/fusion are identified only in about 7% of PTCs but frequencies vary deeply [20]. Almost exclusively found in PTC, the most common are the RET/PTC1 and RET/PTC3 accounting for about 90% of rearrangement in PTCs [20,105]. Some authors suggest that RET/PTC1 relates to prognostically advantageous subtypes (more common in classic or diffuse sclerosing PTC), while RET/PTC3 appears to occur in more aggressive phenotypes (solid variant PTC) [37,106,107]. Although, RET/PTC rearrangements seem to be associated with lymph node metastasis [94] and even multifocality [108] patients respond well to post-operative radioiodine (RAI) ablation and, therefore, follow a less aggressive course [109].
While the three RAS mutations (HRAS, NRAS and KRAS) play a smaller role in PTC (6–20%) [110], they are with 90% most common in FVPTC [111]. This subtype has been associated with infrequent spread to regional lymph nodes and good RAI avidity [20,112]. Interestingly, RAS mutations are, beside PAX8/PPARG and THADA/IGF2BP3 gene fusions (22% of NIFTP) the predominant mutation in 67% of NIFTP [113,114] but also in a large number of benign follicular adenoma (20–25%) [21,115]. Therefore, RAS mutations alone cannot sufficiently predict malignancy but RAS mutated benign or borderline lesions represent precursor of malignancies [116] and may therefore necessitate surgical treatment such as lobectomy and close follow-up [74,117,118,119].
While the molecular landscape of PTC has been well described with current sequencing technologies, the molecular characterization of FTC still requires further clarification. The most common mutation in FTC are RAS and PAX8/PPARG fusion oncogenes, found in 27–50% [110] and 12–53% [21] respectively. However, additional genetic mutations, such as PTEN, PIK3CA, EIF1AX, DICER1, TSH Receptor, but also TERT promotor mutations are thought to be required to promote carcinogenesis and transformation of a follicular adenoma to FTC [21,120,121]. Although, some authors suggest an improved prognosis with PAX8/PPARG [122] rearrangement and worsened outcome with RAS mutation, only the total mutational burden seems to be an independent predictor of worse outcome, rather than single mutation alone [120,123,124]. Hürtle cell carcinomas (HCC), a variant of FTC, are genetically distinct with mainly CNAs and mutations in mitochondrial DNA or non-mitochondrial genes that interact with mitochondrial function. To date, none have been proven reliable [125]. Hence, proof of vascular invasion is still necessary to distinguish HCC from Hürtle cell adenoma (HCA).
3.2.2. MiRNA Markers
Specific patterns of miRNA have been identified in large number of studies and are one of the novel methods to improve diagnosis in TC on FNA and also in “liquid biopsies” such as plasma samples due to their high stability in fluids and tissue specificity [31,126,127,128,129,130].
MiRNAs have been classified into a group of oncogenic and tumour suppressor miRNAs, which are based on their expression profile and their effect on cancer related signalling pathways [28,131,132]. The upregulation of oncogenic MiRNAs inhibits apoptosis and induce cell proliferation and growth, invasion and metastases by modulating target genes of several signalling pathways such as the PI3K/AKT/mTOR-, adipocytokine-, Hippo-, Wnt/β-catenin-signalling pathways [28]. In contrast, downregulation of tumour suppressor miRNAs results in loss of inhibition of cell proliferation and migration, and EMT in the MAPK, PI3K/AKT, NFκB or GSK-3β/β-catenin pathways [27,131,132,133,134]. Although miRNA have extensively been studied in PTC, distinct miRNA patterns were identified in different TC subtypes targeting different target genes [28]. Some examples of target genes in PTC are Zinc Ring Finger 3 (ZNRF3)-, cytokine receptor KIT- or the CXCL12 gene [27,134,135].
Moreover, miRNAs have been successfully demonstrated to be able to discriminate benign from malignant lesions in PTC (e.g., miRNA-221, miRNA-222, miRNA-181b, miRNA-146b) and FTC/HCC (e.g., miRNA 148b–3p, miRNA-484) and could be a guide to avoid unnecessary surgery [27,136,137]. Moreover, certain miRNAs were also identified in APC, such as miRNA-19a [26]. However, panels of miRNAs may have a higher sensitivity and specificity than single miRNA in distinguishing thyroid nodules [127,138]. MiRNA profiling also showed that certain expression levels correlate with clinicopathological features (subtype, tumour size, multifocality, extrathyroidal extension and metastasis) as well as aggressiveness [27,32,139] and offer many opportunities for future research.
3.2.3. Long Noncoding RNA Markers
Several specific lncRNA are dysregulated in TC in comparison to benign thyroid lesions and the presence of certain lncRNA have been associated with poorer TC prognosis [140,141,142,143]. LncRNA panels represent a promising novel adjunct method to distinguish malignant from benign thyroid lesions [144], however, not yet clinically available. An advantage with lncRNA markers is that they are tissue specific and present in circulating blood, enabling the possibility of a blood sample analysis to detect the presence of thyroid cancer in the future [145].
3.2.4. Proteome Based Markers
Protein expression profiles (Proteomics) have been increasingly applied to search for novel diagnostic and prognostic marker [146]. They offer an unbiased platform to further investigate the whole proteome [147]. While several proteins have been identified (e.g., Prohibitin, ATP Synthase D chain, HSP70, PRDX, A100A6, TGFBI, E-Cadherin, A1AT) to be altered in malignant lesions, further preclinical and clinical study are required to assess their clinical applicability [53,146].
3.3. Molecular Testing (Table 2)
As demonstrated, mutational analysis for single genes has not adequately provided enough accuracy to properly guide decision making of thyroid nodules. The choice of MT is based on performance factors such as specificity, sensitivity, PPV, Negative predictive value (NPV), and prevalence. It should either provide the information if a test can “rule out” cancer (likelihood that a nodule is benign) and reduce diagnostic surgery for benign nodules or “rule in” cancer (likelihood that the nodule is malignant) to optimize extent of surgical treatment [82,117]. Therefore, high sensitivity and NPV would be ideal to “rule out” malignancy, while high specificity and high PPV would help to “rule in” malignancies [148].
Two MT techniques have been developed that avoid the use of single marker testing by using multiple markers to improve their performance, utilise somatic mutation and rearrangement testing and gene expression or sequencing classifier (GEC, GSC) [149]. Somatic and mutation and rearrangement testing initially started with the development of multigene panels such as the 7-gene panel (7GP) which included common genetic alterations (BRAFV600E/K601E, N-, H-, K-RAS, PAX8/PPARG, RET/PTC 1/3) [150]. The 7GP has been further extended and incorporated into 2 commercially available tests: The ThyGenX® (7GP) + ThyraMIR® (10 miRNA panel) [85] and ThyroSeq® test. The ThyraGeNEXT/ThyraMIR® is a further extension of the ThyGenX® + ThyraMIR® test and includes additional DNA and RNA, such as the prognostically relevant TERT mutation as well as specific alterations for Hürtle cell lesions. The first study of clinical performance reports that patients with Bethesda III and IV nodules with low risk mutations had high probability (94%) of remaining cancer-free (2 year follow-up), whereas moderate- and high-risk results demonstrated a probability of malignancy of 53% and 70% respectively [151]. This test might help to guide surgical planning for initial resection, rather than a 2-stage diagnostic lobectomy followed by definitive surgery as well as avoiding surgery for a benign disease [151]. The extent of surgery has not been addressed [85,151].
Following the initial ThyroSeq v0 (7GP) [150], the application of next generation sequencing (NGS) allowed to progress to the current version of the ThyroSeq v3, which tests for 112 genes, including copy number alterations (CNAs) [152,153]. As a “rule out” test with a benign call rate (proportion of FNA with negative MT as predictor of avoidable diagnostic surgery) of 61% of all Bethesda III and IV nodules and 82% of indeterminate nodules the ThyroSeq v3 could help to prevent diagnostic lobectomy [86,152]. However, further studies are needed to evaluate the clinical relevant performance [154].
The Affirma Gene Sequencing Classifier (GSC) [84] is the latest version of the mRNA-based Affirma Gene Expression Classifier (GSC), which initially consisted of a 167-gene microarray and 6 gene malignancy classifier panel and was a classical “rule out” test [155]. The Affirma GCS incorporated BRAF and RET/PTC and additional expression marker for Medullary thyroid cancer (MTC) and Hürtle cell lesions and improved specificity, while maintaining high sensitivity and NPV of the classical “rule out” GEC [84]. The GCS therefore remains a good tool but seems to be inferior to other tests in terms of benign call rate of 54% and 68% of histologically benign thyroid nodules classified as negative [84].
To date, no MT has been studied to determine the extent of surgery (lobectomy versus total thyroidectomy) under the 2015 ATA guidelines, which significantly modified the recommendation of surgical management of 1–4cm TCs [15]. Only multiple mutations, such as BRAFV600E with TERT promoter have been demonstrated to significantly impact PTC recurrence and mortality, and might therefore, be included in the decision making of surgical management, and offer the opportunity for further research [156].
3.4. Current Challenges and Limitations of Molecular Testing
There are several concerns regarding a wide implementation of molecular testing. First, validation studies of currently available MT were performed before the introduction of NIFTP and likely classified many NIFTP as malignant, which leads to altered predictive values of molecular tests. Hence, validation studies need refinement to appropriately address the impact of the NIFTP reclassification. Second, the identification of the appropriate clinical scenario, where MT might be useful, as well as the interpretation of results is challenging, and might result in over- or under-treatment [157]. Third, some studies showed, that only 7.9–8.4% of patients had altered surgical decision making as a result of molecular testing [158,159]. Fourth, MT has not been validated in paediatric patients [17]. Fifth, the correlation between presence of mutations and occurrence of malignancy is imprecise, since benign nodules harbour mutations, while some malignancies have none detectable with commonly used platforms [160]. Sixth, molecular tests are expensive, ranging from 1675–3500 USD [80], reports on cost effectiveness are conflicting, since expenses on MT might indeed be lower than diagnostic lobectomy [161] and are dependent on local health care settings. However, the impact of long-term surveillance on cost of conservatively managed patients need further work-up. Seventh, MT is not available worldwide and delays of treatment need to be considered and weighed up against the benefits. Finally, molecular marker negative patients lack histological confirmation on a surgical specimen and final diagnosis cannot be established. While many issues are yet to be resolved, MT is widely used in some health care settings and long-term follow up of these patients and further research will hopefully further clarify the benefit of MT in the management of thyroid nodules and thyroid cancer.
4. Molecular Marker for Surveillance in DTC
One molecular marker that is widely used for surveillance is serum thyroglobulin (Tg) level, which reflects remaining benign or malignant thyroid tissue. Tg achieves the highest sensitivity and specificity when used for surveillance with very low serum levels in patients who underwent total thyroidectomy, removal of involved lymph nodes and radioiodine ablation [162]. While Tg levels should be measured every 6–12 month during initial follow-up for all DTC patients, the timing of further Tg measurements depend on ATA risk stratification and recommendations vary [15]. Of note, serum Tg levels are influenced by the degree of thyroid stimulating hormone (TSH) stimulation [15]. A further limitation of measuring Tg is the presence of Thyroglobulin antibodies (TgAb), which is detected in 20% of TC patients and causes unreliable and false negative Tg results [163]. A recent meta-analysis demonstrated that the risk of developing lymph node metastasis and cancer persistence/recurrence was two times, respectively three times, higher in TgAb positive patients [164,165]. Moreover, TgAb persistence or increase leads to a 10 fold higher risk of cancer persistence/recurrence and 15 fold increase in cancer mortality, especially in patients who showed a post-treatment decrease of <50% [164]. Therefore, TgAbs should be assessed along Tg during follow up [15] and a rise should set off further investigations independently of Tg levels [15].
Another promising approach is the use of miRNA as predictor of early relapse. These patients showed a decline of circulating miRNA 146b-5p, miRNA-221-3p, miRNA-222-3p and miRNA-146a-5p after surgical resection [32,166]. Moreover, serum levels of miRNA-221-3p and 146b-5p increased after 2-year follow-up of PTC with evidence of structural recurrence, even in Thyroglobulin negative assays [166]. The use of circulating miRNA and other circulating biomarkers faces several challenges and further studies are required to address the application in TC diagnosis and surveillance [167].
5. Molecular Markers to Guide Targeted Treatment for Advanced TC
5.1. Diagnosis of Advanced TC, PDTC, and ATC
Patients with rapidly growing lesions in the thyroid require prompt investigation with imaging and biopsy. Molecular markers offer the possibility to further aid management of advanced TC. Advanced TC is defined upon its clinical aggressiveness, while the definition of PDTC and ATC require histological evaluation. PDTC is defined histologically by either the Turin or the MSKCC criteria. The Turin criteria defines a solid, insular or trabecular growth pattern, absence of nuclear features of PTC and one out of three; convoluted nuclei, necrosis or increased mitosis (>3 per 10hpf) [168]. The MSKCC criteria identifies presence of follicular cell differentiation with presence of >5 mitosis/10HPF and/or necrosis [169]. PDTC and ATC often present with infiltrative growth and vascular invasion. The histopathological characteristics of ATC are spindle-shaped, epithelioid or giant cells with marked cytological pleomorphism and extensive necrosis. PDTC often still express some TTF-1 and Tg, reflecting their origin from thyroid epithelium, while ATCs often have completely lost expression of theses markers [67]. ATCs are in most cases infiltrated by immune cells; neutrophils, T-cells and tumour-associated macrophages, promoting an inflammatory and oncogenic microenvironment [67,170].
Kunstman et al. published the first whole exome sequencing of an ATC cohort showing distinct patterns of high frequency mutations in BRAF, P53 and RAS [171]. In addition, Landa et al. demonstrated a high TERT promoter mutation both in PDTC and ATC, as well as mutated targets in the PI3K/AKT signalling pathway; PIK3CA, mTOR, PTEN; transcriptions factors EIF1AX, NF1; cell cycle regulators CDKN1B; histone methyltransferases (HDAC, KMT) and increased CNA and ALK involved fusion-oncogenes [25]. Specifically, for ATC is the increased mutational burden in combination of epigenetic alterations enhancing the effect of genetic mutations. Co-existence of both constitutive MAPK- and PI3K/AKT signalling also augments genetic instability. Pozdeyev et al. demonstrated a pattern of three gene mutational clusters of ATC, suggesting the ATC tumours were derived from either PTC (BRAF-like), HCC or FTC (RAS-like) [172]. ATC are complex in their genetic and epigenetic signature, however, some ATC retain their dependence of its driver mutation [173,174].
5.2. Impairment of Iodine-Handling Machinery in Advanced Thyroid Cancer
The three cornerstones of treatment in DTC is surgery, adjuvant RAI-ablation, and TSH suppression. However, in PDTC and ATC these cornerstones are challenged with tumour biology promotes vascular and extrathyroidal invasion, downregulation of the expression of the TSH receptor gene and genes involved in the iodine-handling machinery, reducing RAI efficacy. Additional treatments with external beam radiation (EBR) and chemotherapy with taxanes in combination with carboplatin or doxorubicin can be useful in reducing tumour- and metastasis size and can sometimes enable debulking surgery to avoid airway obstruction and achieve local tumour control; however, response rates to chemotherapy are low, thereby limiting their use [175]. The prognosis for patients with advanced thyroid tumours with distant metastasis upon presentation is poor, around 60%, for 10-year survival and only 10% for RAI-refractory TC in comparison to the excellent prognosis of 95% for DTC making a better understanding of biology a strong impetus to improve outcomes for these patients [176,177].
The main and unique function of follicular cells is to use iodine to synthesise thyroid hormone. Iodide is transported into the cell by NIS which is the sodium-iodine symporter located in the basal membrane. At the apical membrane, pendrin transports iodine out of the cell into the lumen of the thyroid follicle. In the lumen, iodine undergoes oxidation by TPO and is incorporated into tyrosine residues of Tg, which is later cleaved through proteolysis to produce thyroid hormones, thyroxine (T4) and triiodothyronine (T3). This machinery is upregulated by TSH activation of the TSH-receptor (TSHR) and exploited in RAI-ablation and requires to be intact for RAI treatment to be effective [178].
BRAFV600E leads to decreased or inhibited expression of NIS, TSHR, TPO, TG as well as SLC26A4, the gene, which encodes for pendrin [34], which can lead to inability for the TC to incorporate iodine. Consequently, this causes TC RAI-refractory disease (RAIRD), and DTC with BRAFV600E mutations are significantly more likely to be RAI refractory [179] and, to a high extent, PDTC and especially ATC, are RAIRD. Additional genes, like PAX8 has also been shown to be a potent repressor of NIS in thyroid cells [180]. RAIRD is associated with FDG-PET avidity, which can therefore be used for diagnosis. RAIRD is clinically established when patients, appropriately TSH-stimulated, fail or loose ability to concentrate iodine in metastases or if there is tumour progression despite significant RAI uptake [181]. In these patients, there is no further role for RAI if their iodine refractory status cannot be reversed.
5.3. Tyrosine Kinase Inhibitors
The increased knowledge of the molecular biology of PDTC and ATC has enabled targeted treatments of the affected signalling pathways. Inhibitors of intracellular tyrosine kinase signalling pathways, tyrosine kinase inhibitors (TKIs), have shed new hope for the treatment of advanced TC. Although they do not cure, they can slow down growth, reverse adverse effects of the mutations, when traditional treatments are ineffective. TKIs have also shown results in reversing the TC oncogenesis, enabling for the traditional treatments to again become effective. This effect is demonstrated in the case report (Box 1, Figure 3) which shows how use of TKIs in a neoadjuvant setting can achieve local control and effective palliation for an aggressive advanced TC.
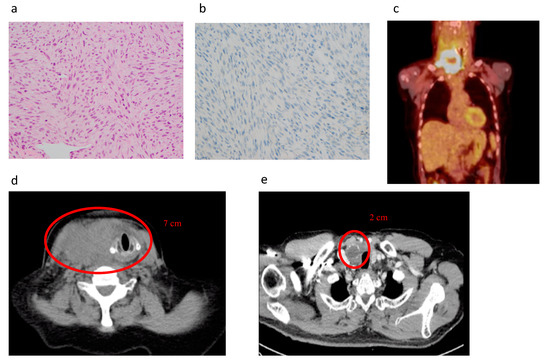
Figure 3.
Histopathology and images of case report described in Box 1. (a) Haematoxylin and eosin stain of tumour, (b) BRAFV600E stain of tumour, (c) PET scan at initial presentation, (d) CT at initial presentation, (e) CT after one month of daily Lenvatinib 20mg treatment and 50Gy external beam radiation.
The first clinically approved TKIs for RAIRD advanced TC were Lenvatinib and Sorafenib, which are both orally available multi-tyrosine kinase inhibitors. Lenvatinib inhibits vascular endothelial growth factor receptor 1-3 (VEGFR1-3), Fibroblast growth factor 1-4, RET, c-KIT and platelet derived growth factor receptor beta (PDGFRβ) [182]. The main tumour reducing effect is likely the anti-angiogenetic effect, but secondary effects of inhibiting additional kinases including RET and FGFR may have additional effects [183]. Sorafenib inhibits RAF, VEGFR 1-3, PDGF receptor and RET [184]. The SELECT trial, a major phase III study compared Lenvatinib to placebo in patients with RAIRD and showed a disease free survival of 18.3 months in the treatment arm compared to 3.6 months in the placebo group [185] which established the use of Lenvatinib as first line treatment of progressive systemic RAIRD. The DECISION study, a multicentre, randomised, double-blind phase III trial, comparing Sorafenib to placebo, showed progression-free survival of 10.5 months compared to 5.8 months with placebo [186]. TKIs usually require long-term use but diarrhoea, fatigue, hypertension and hand-foot skin reactions are frequent side effects [181] and in the SELECT trial, 14% of the patients treated with Lenvatinib have to cease treatment for this reason.
Since the entry of Lenvatinib and Sorafenib, there are several clinical trials evaluating effects of targeted TKI, tailored for a specific driver mutation. The BRAFV600E inhibitor Vemurafenib [187] and Dabrafenib are clinically approved for BRAFV600E positive RAIRD TC. Liu et al. in 2007 showed that by inhibiting MEK in TC cell cultures, the expression of genes involved in the iodine handling machinery could be restored [188] and the clinical available MEK inhibitor Selumetinib was shown to restore the NIS expression in TC [189]. In a study by Iravani et al. [190] with RAIRD, patients with either NRAS or BRAFV600E were treated with MEK inhibitor (Trametinib) and BRAF- and MEK inhibitors (Dabrafenib and Trametinib or Vemurafenib and Cobimetinib) for four weeks, showing that all three BRAFV600E-mutated patients and one NRAS patient demonstrated restoration of RAI uptake. Recently, the BRAF inhibitor Dabrafenib and the MEK inhibitor Trametinib was also approved for treatment in ATC, in patients not responding to locoregional treatment options [191]. Today there are several other available targeted TKIs; NTRK inhibitor Larotrectinib [192], mTOR inhibitor Everolimus [193], ALK inhibitor Crizotinib [194] and the RET inhibitor Selpercatinib was recently clinically approved for RAIRD metastatic advanced TC with RET-fusion genes [195,196]
Box 1. Case report.
A man in his 60s presented to our clinic with one-month history of rapidly enlarging right neck mass and voice change. He had a family history of thyroid cancer. A core biopsy was performed that showed spindle cell type ATC which was BRAFV600E negative (Figure 3a,b). Laryngoscopy showed a right recurrent laryngeal nerve (RLN) palsy. FDG-PET scan was performed showing high uptake in the lesion on the neck as well as pulmonary and bone metastasis (Figure 3c). On imaging and clinical exam, the cancer was thought to unable to be resected without significant morbidity due to local invasion (Figure 3d). He was started on Lenvatinib and received external beam radiation to the neck and showed a response with regression of tumour from 7 to 3 cm within one month (Figure 3e). He developed side effects of Lenvatinib with ulcerations of feet and diarrhoea and due to the reduction in size of the cancer underwent a thyroidectomy where the Right RLN was invaded in the tumour and sacrificed. Histopathology showed a BRAFV600E negative, ALK negative, P53 positive in >50% of tumour. Postoperatively he was then restarted on lower dose Lenvatinib and three months later started a combination with Pembrolizumab due to the development of liver metastasis. He achieved good local control in his neck but unfortunately developed pulmonary infection and died 18 months following his initial surgery.
While many trials of TKIs were mutation agnostic it is possible identification of the driver mutation could improve response of advanced TC with TKIs. The advances of hybridisation capture-based NGS panels, Memorial Sloan Kettering integrated mutation profiling of actionable cancer targets (MSK-IMPACT), and Foundation One, which can detect somatic and germline mutations, are becoming more accessible for clinicians to customise treatment for patients with advanced TC [197,198].
5.4. Application of Molecular Markers to Decision Making for Advanced TC
The decision of when to start TKIs for advanced TC can be difficult with Tg doubling time, radiological progression or unresectable tumours indications to consider systemic treatment. As local, structural recurrence can be treated with surgery, EBR, radiofrequency-, cryo-, or alcohol ablation, it is suggested that TKIs should be reserved for radio-refractory patients with rapid tumour progression and severe symptoms that threaten life [15]. In addition, the use of TKIs have value in neo-adjuvant treatment in patients with advanced TC to reduce tumour size to enable complete macroscopic resection (R1), for symptomatic relief and also for enabling further RAI ablation [190,199].
How we apply this knowledge of biology to the clinic and choose which TKI to use initially is the subject of further assessment and research. While initial trials of Lenvatinib [185] and Sorafenib [186] did not suggest a difference in response according to driver mutation for the treatment of metastatic disease RAIRD, subsequent reports have suggested that redifferentiation may be higher in patients with RAS mutated compared to BRAFV600E mutated cancers [200].
In the setting of ATC, there is evidence of response specific to mutations such as the MEK inhibitor for BRAFV600E mutated cancers [191] and response to Lenvatinib for RAS mutated cancers [201] is an area of active research. In our experience, patients with BRAFV600E WT ATC have responded to Lenvatinib as shown in our case report (Box 1) and achieved remarkable responses allowing some ATCs to be down staged and proceed to surgery to achieve local control and effective palliation (Box 1, Figure 3). In addition, the availability of targeted therapies for rarer drivers such as NTRK inhibition with Larotrectinib [192], mTOR inhibition with Everolimus [193], ALK inhibition with Crizotinib [194] and the RET inhibition with Selpercatinib mean it is important to assess patients tumours with advanced disease for the genetic driver mutation.
Sequencing of all advanced TC would be of great value for increased understanding of the underlying driving mutations, modifying mutations, and to individually tailored targeted treatments; however, currently, it is too costly and the time taken for sequencing results (with a usual turnaround of 6–8 weeks) makes this approach not feasible for many endocrine centres and for patients presenting with advanced disease. One possible approach is to triage advanced tumours for further mutation testing using immunohistochemistry (IHC) which is the practice of our institution (Figure 4). In this approach, BRAFV600E mutation specific IHC, RASQ61R mutation specific IHC and ALK IHC is performed. For patients with high ALK expression, this is used as a surrogate for ALK gene rearrangement, which is then confirmed by ALK Fluorescence in situ hybridization (FISH). For patients who are BRAF wild type, RASQ61R negative and with normal ALK expression further genetic testing is performed using targeted next generation sequencing targeted DNA and RNA sequencing by NGS (Figure 4). An RNA panel is used to detects transcripts from 76 fusion variants from eight genes and include RET, ALK and NTRK3, BRAF, PPARG, THADA, and MAML2. Meanwhile, the DNA panel targets 2023 mutations in 14 key cancer genes and include AKT1, BRAF, CTNNB1, GNAS, HRAS, KRAS, NRAS, PIK3CA, PTEN, RET, TERT, PT53, and TSHR. Depending on the results, this information is used to consider the choice of targeted therapy (Figure 4).
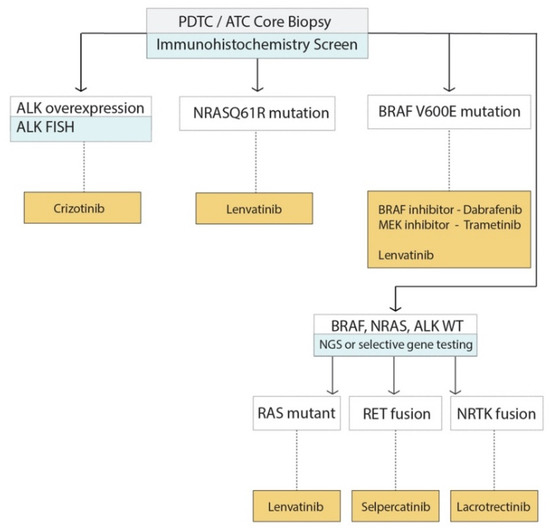
Figure 4.
Suggested flow chart of use of molecular testing in advanced thyroid cancer.
Molecular markers can be utilized to type advanced TC and identify the driving or modifying mutation, which can then be useful for choice of targeted treatment. This flowchart summarizes the management of molecular testing at our clinic and the selection of currently available therapies.
6. Conclusions
Molecular markers for diagnosis, prognosis, surveillance and treatment are becoming more important as knowledge of the molecular mechanisms of thyroid cancer increases. The potential of molecular testing in DTC is immense in terms of diagnosing cancer, assess prognosis and ultimately avoid surgical overtreatment. However, molecular testing still faces several challenges, which needs be addressed before a broad implementation into clinical practice can be achieved. There are currently several ongoing clinical trials, which evaluate new, targeted treatments for advanced thyroid cancers, where the prognosis was previously poor. This will allow clinicians to work with a larger arsenal of diagnostic and therapeutic tools to combat differentiated and advanced thyroid cancer. The key to success is molecular testing and personalised medicine, to obtain flexibility to tailor treatment for each unique cancer type and patient.
Author Contributions
Conceptualization, C.N., R.M., S.B.S., and A.R.G.; Writing—Original Draft preparation, C.N. and R.M.; Writing—Review & Editing: A.R.G., S.B.S., M.S.S., V.T., A.C., R.J.C.-B., B.G.R., and A.J.G.; Visualization: I.M.-R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Cancer Institute NSW Early Career Fellowship grant number 2019/ECF1081—Anthony Glover and by Bergholm/Eriksson foundation and Sigbrith Björkelund foundation—Carolina Nylén.
Conflicts of Interest
B.G.R. reports personal fees from Loxo Oncology, outside the submitted work. B.G.R., R.J.C.-B., and V.T. report personal fees from Eisai, outside the submitted work. The remaining authors have disclosed that they have no significant relationships with, or financial interest in, any commercial companies pertaining to this article.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Seib, C.D.; Sosa, J.A. Evolving understanding of the epidemiology of thyroid cancer. Endocrinol. Metab. Clin. N. Am. 2019, 48, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.; Welch, H.G. Current thyroid cancer trends in the united states. JAMA Otolaryngol. Head. Neck. Surg. 2014, 140, 317–322. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Howlader, N.; Noone, A.; Krapcho, M.; Miller, D.; Bishop, K.; Altekruse, S.F.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; et al. (Eds.) Seer Cancer Statistics Review, 1975–2013; National Cancer Institute: Bethesda, MD, USA, 2015. Available online: https://seer.cancer.gov/archive/csr/1975_2013/ (accessed on 14 May 2020).
- Noone, A.M.; Cronin, K.A.; Altekruse, S.F.; Howlader, N.; Lewis, D.R.; Petkov, V.I.; Penberthy, L. Cancer incidence and survival trends by subtype using data from the surveillance epidemiology and end results program, 1992–2013. Cancer Epidemiol. Biomark. Prev. 2017, 26, 632–641. [Google Scholar] [CrossRef]
- La Vecchia, C.; Malvezzi, M.; Bosetti, C.; Garavello, W.; Bertuccio, P.; Levi, F.; Negri, E. Thyroid cancer mortality and incidence: A global overview. Int. J. Cancer 2015, 136, 2187–2195. [Google Scholar] [CrossRef]
- Kim, J.; Gosnell, J.E.; Roman, S.A. Geographic influences in the global rise of thyroid cancer. Nat. Rev. Endocrinol. 2020, 16, 17–29. [Google Scholar] [CrossRef]
- Lim, H.; Devesa, S.S.; Sosa, J.A.; Check, D.; Kitahara, C.M. Trends in thyroid cancer incidence and mortality in the united states, 1974–2013. JAMA 2017, 317, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.S.; Kim, H.J.; Welch, H.G. Korea’s thyroid-cancer “epidemic”—Screening and overdiagnosis. N. Engl. J. Med. 2014, 371, 1765–1767. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.F.; Jonker, P.K.C.; Cunich, M.; Sidhu, S.B.; Delbridge, L.W.; Glover, A.R.; Learoyd, D.L.; Aniss, A.; Kruijff, S.; Sywak, M.S. Surgery alone for papillary thyroid microcarcinoma is less costly and more effective than long term active surveillance. Surgery 2020, 167, 110–116. [Google Scholar] [CrossRef]
- Aschebrook-Kilfoy, B.; Schechter, R.B.; Shih, Y.C.; Kaplan, E.L.; Chiu, B.C.; Angelos, P.; Grogan, R.H. The clinical and economic burden of a sustained increase in thyroid cancer incidence. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The i-predict study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 american thyroid association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: The american thyroid association guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Baloch, Z.; Bernet, V.; Chen, A.; Fahey, T.J., 3rd; Ganly, I.; Hodak, S.P.; Kebebew, E.; Patel, K.N.; Shaha, A.; et al. American thyroid association statement on surgical application of molecular profiling for thyroid nodules: Current impact on perioperative decision making. Thyroid 2015, 25, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.N.; Yip, L.; Lubitz, C.C.; Grubbs, E.G.; Miller, B.S.; Shen, W.; Angelos, P.; Chen, H.; Doherty, G.M.; Fahey, T.J., 3rd; et al. The american association of endocrine surgeons guidelines for the definitive surgical management of thyroid disease in adults. Ann. Surg. 2020, 271, e21–e93. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas, M.E.; McFadden, D.G.; Durante, C. Thyroid cancer. Lancet 2016, 388, 2783–2795. [Google Scholar] [CrossRef]
- Cohen, Y.; Xing, M.; Mambo, E.; Guo, Z.; Wu, G.; Trink, B.; Beller, U.; Westra, W.H.; Ladenson, P.W.; Sidransky, D. Braf mutation in papillary thyroid carcinoma. J. Natl. Cancer Inst. 2003, 95, 625–627. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef]
- Prete, A.; Borges de Souza, P.; Censi, S.; Muzza, M.; Nucci, N.; Sponziello, M. Update on fundamental mechanisms of thyroid cancer. Front Endocrinol. (Lausanne) 2020, 11, 102. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Bishop, J.; Zhu, G.; Zhang, T.; Ladenson, P.W.; Xing, M. Mortality risk stratification by combining braf v600e and tert promoter mutations in papillary thyroid cancer: Genetic duet of braf and tert promoter mutations in thyroid cancer mortality. JAMA Oncol. 2017, 3, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Acquaviva, G.; Visani, M.; Repaci, A.; Rhoden, K.J.; de Biase, D.; Pession, A.; Giovanni, T. Molecular pathology of thyroid tumours of follicular cells: A review of genetic alterations and their clinicopathological relevance. Histopathology 2018, 72, 6–31. [Google Scholar] [CrossRef] [PubMed]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef]
- Calabrese, G.; Dolcimascolo, A.; Caruso, G.; Forte, S. Mir-19a is involved in progression and malignancy of anaplastic thyroid cancer cells. Onco. Targets Ther. 2019, 12, 9571–9583. [Google Scholar] [CrossRef]
- Celano, M.; Rosignolo, F.; Maggisano, V.; Pecce, V.; Iannone, M.; Russo, D.; Bulotta, S. Micrornas as biomarkers in thyroid carcinoma. Int. J. Genom. 2017, 2017, 6496570. [Google Scholar] [CrossRef]
- Santiago, K.; Chen Wongworawat, Y.; Khan, S. Differential microrna-signatures in thyroid cancer subtypes. J. Oncol. 2020, 2020, 2052396. [Google Scholar] [CrossRef]
- Marini, F.; Luzi, E.; Brandi, M.L. Microrna role in thyroid cancer development. J. Thyroid Res. 2011, 2011, 407123. [Google Scholar] [CrossRef]
- Calabrese, G.; Dolcimascolo, A.; Torrisi, F.; Zappala, A.; Gulino, R.; Parenti, R. Mir-19a overexpression in ftc-133 cell line induces a more de-differentiated and aggressive phenotype. Int. J. Mol. Sci. 2018, 19, 3944. [Google Scholar] [CrossRef]
- Lee, J.C.; Gundara, J.S.; Glover, A.; Serpell, J.; Sidhu, S.B. Microrna expression profiles in the management of papillary thyroid cancer. Oncologist 2014, 19, 1141–1147. [Google Scholar] [CrossRef]
- Lee, J.C.; Zhao, J.T.; Clifton-Bligh, R.J.; Gill, A.; Gundara, J.S.; Ip, J.C.; Glover, A.; Sywak, M.S.; Delbridge, L.W.; Robinson, B.G.; et al. Microrna-222 and microrna-146b are tissue and circulating biomarkers of recurrent papillary thyroid cancer. Cancer 2013, 119, 4358–4365. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Li, C.; Liu, C.; Zhao, S.; Wang, Y.; Fu, Z. Expressions of mirnas in papillary thyroid carcinoma and their associations with the clinical characteristics of ptc. Cancer Biomark. 2017, 18, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Giordano, T.J. Follicular cell thyroid neoplasia: Insights from genomics and the cancer genome atlas research network. Curr. Opin. Oncol. 2016, 28, 1–4. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A comprehensive review on mapk: A promising therapeutic target in cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, A.R. Signal integration by jnk and p38 mapk pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Adeniran, A.J.; Zhu, Z.; Gandhi, M.; Steward, D.L.; Fidler, J.P.; Giordano, T.J.; Biddinger, P.W.; Nikiforov, Y.E. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am. J. Surg. Pathol. 2006, 30, 216–222. [Google Scholar] [CrossRef]
- Chakravarty, D.; Santos, E.; Ryder, M.; Knauf, J.A.; Liao, X.H.; West, B.L.; Bollag, G.; Kolesnick, R.; Thin, T.H.; Rosen, N.; et al. Small-molecule mapk inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional braf activation. J. Clin. Investig. 2011, 121, 4700–4711. [Google Scholar] [CrossRef]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the raf-erk signaling pathway by oncogenic mutations of b-raf. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Rusinek, D.; Swierniak, M.; Chmielik, E.; Kowal, M.; Kowalska, M.; Cyplinska, R.; Czarniecka, A.; Piglowski, W.; Korfanty, J.; Chekan, M.; et al. Brafv600e-associated gene expression profile: Early changes in the transcriptome, based on a transgenic mouse model of papillary thyroid carcinoma. PLoS ONE 2015, 10, e0143688. [Google Scholar] [CrossRef]
- Fagin, J.A. Challenging dogma in thyroid cancer molecular genetics--role of ret/ptc and braf in tumor initiation. J. Clin. Endocrinol. Metab. 2004, 89, 4264–4266. [Google Scholar] [CrossRef]
- Ikenoue, T.; Hikiba, Y.; Kanai, F.; Tanaka, Y.; Imamura, J.; Imamura, T.; Ohta, M.; Ijichi, H.; Tateishi, K.; Kawakami, T.; et al. Functional analysis of mutations within the kinase activation segment of b-raf in human colorectal tumors. Cancer Res. 2003, 63, 8132–8137. [Google Scholar] [PubMed]
- Lee, S.J.; Lee, M.H.; Kim, D.W.; Lee, S.; Huang, S.; Ryu, M.J.; Kim, Y.K.; Kim, S.J.; Kim, S.J.; Hwang, J.H.; et al. Cross-regulation between oncogenic braf(v600e) kinase and the mst1 pathway in papillary thyroid carcinoma. PLoS ONE 2011, 6, e16180. [Google Scholar]
- Liu, X.; Bishop, J.; Shan, Y.; Pai, S.; Liu, D.; Murugan, A.K.; Sun, H.; El-Naggar, A.K.; Xing, M. Highly prevalent tert promoter mutations in aggressive thyroid cancers. Endocr. Relat. Cancer 2013, 20, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent tert promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Qu, S.; Liu, R.; Sheng, C.; Shi, X.; Zhu, G.; Murugan, A.K.; Guan, H.; Yu, H.; Wang, Y.; et al. Tert promoter mutations and their association with braf v600e mutation and aggressive clinicopathological characteristics of thyroid cancer. J. Clin. Endocrinol. Metab. 2014, 99, E1130–E1136. [Google Scholar] [CrossRef]
- Shinohara, M.; Chung, Y.J.; Saji, M.; Ringel, M.D. Akt in thyroid tumorigenesis and progression. Endocrinology 2007, 148, 942–947. [Google Scholar] [CrossRef]
- Liu, Z.; Hou, P.; Ji, M.; Guan, H.; Studeman, K.; Jensen, K.; Vasko, V.; El-Naggar, A.K.; Xing, M. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J. Clin. Endocrinol. Metab. 2008, 93, 3106–3116. [Google Scholar] [CrossRef]
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef]
- Miller, K.A.; Yeager, N.; Baker, K.; Liao, X.H.; Refetoff, S.; Di Cristofano, A. Oncogenic kras requires simultaneous pi3k signaling to induce erk activation and transform thyroid epithelial cells in vivo. Cancer Res. 2009, 69, 3689–3694. [Google Scholar] [CrossRef]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacocke, M.; et al. Germline mutations of the pten gene in cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 1997, 16, 64–67. [Google Scholar] [CrossRef]
- Fusco, A.; Grieco, M.; Santoro, M.; Berlingieri, M.T.; Pilotti, S.; Pierotti, M.A.; Della Porta, G.; Vecchio, G. A new oncogene in human thyroid papillary carcinomas and their lymph-nodal metastases. Nature 1987, 328, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, M.I.; Junit, S.M.; Ng, K.L.; Jayapalan, J.J.; Karikalan, B.; Hashim, O.H. Papillary thyroid cancer: Genetic alterations and molecular biomarker investigations. Int. J. Med. Sci. 2019, 16, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Fagin, J.A.; Mitsiades, N. Molecular pathology of thyroid cancer: Diagnostic and clinical implications. Best Pract. Res. Clin. Endocrinol. Metab. 2008, 22, 955–969. [Google Scholar] [CrossRef] [PubMed]
- Nikiforova, M.N.; Lynch, R.A.; Biddinger, P.W.; Alexander, E.K.; Dorn, G.W., 2nd; Tallini, G.; Kroll, T.G.; Nikiforov, Y.E. Ras point mutations and pax8-ppar gamma rearrangement in thyroid tumors: Evidence for distinct molecular pathways in thyroid follicular carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 2318–2326. [Google Scholar] [CrossRef] [PubMed]
- Zafon, C.; Gil, J.; Pérez-González, B.; Jordà, M. DNA methylation in thyroid cancer. Endocr. Relat. Cancer 2019, 26, R415–R439. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Liu, D.; Xing, M. Genome-wide alterations in gene methylation by the braf v600e mutation in papillary thyroid cancer cells. Endocr. Relat. Cancer 2011, 18, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Bisarro Dos Reis, M.; Barros-Filho, M.C.; Marchi, F.A.; Beltrami, C.M.; Kuasne, H.; Pinto, C.A.L.; Ambatipudi, S.; Herceg, Z.; Kowalski, L.P.; Rogatto, S.R. Prognostic classifier based on genome-wide DNA methylation profiling in well-differentiated thyroid tumors. J. Clin. Endocrinol. Metab. 2017, 102, 4089–4099. [Google Scholar] [CrossRef]
- Audia, J.E.; Campbell, R.M. Histone modifications and cancer. Cold Spring Harb. Perspect Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Marks, P.; Rifkind, R.A.; Richon, V.M.; Breslow, R.; Miller, T.; Kelly, W.K. Histone deacetylases and cancer: Causes and therapies. Nat. Rev. Cancer 2001, 1, 194–202. [Google Scholar] [CrossRef]
- He, H.; Jazdzewski, K.; Li, W.; Liyanarachchi, S.; Nagy, R.; Volinia, S.; Calin, G.A.; Liu, C.G.; Franssila, K.; Suster, S.; et al. The role of microrna genes in papillary thyroid carcinoma. Proc. Natl. Acad. Sci. USA 2005, 102, 19075–19080. [Google Scholar] [CrossRef]
- Sui, F.; Ji, M.; Hou, P. Long non-coding rnas in thyroid cancer: Biological functions and clinical significance. Mol. Cell Endocrinol. 2018, 469, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Sedaghati, M.; Kebebew, E. Long noncoding rnas in thyroid cancer. Curr. Opin. Endocrinol. Diabetes Obes. 2019, 26, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Hauptman, N.; Glavac, D. Long non-coding rna in cancer. Int. J. Mol. Sci. 2013, 14, 4655–4669. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.Y. Thyroid cancer stem cells. Nat. Rev. Endocrinol. 2011, 7, 609–616. [Google Scholar] [CrossRef]
- Hunt, J.L.; Tometsko, M.; LiVolsi, V.A.; Swalsky, P.; Finkelstein, S.D.; Barnes, E.L. Molecular evidence of anaplastic transformation in coexisting well-differentiated and anaplastic carcinomas of the thyroid. Am. J. Surg. Pathol. 2003, 27, 1559–1564. [Google Scholar] [CrossRef]
- Molinaro, E.; Romei, C.; Biagini, A.; Sabini, E.; Agate, L.; Mazzeo, S.; Materazzi, G.; Sellari-Franceschini, S.; Ribechini, A.; Torregrossa, L.; et al. Anaplastic thyroid carcinoma: From clinicopathology to genetics and advanced therapies. Nat. Rev. Endocrinol. 2017, 13, 644–660. [Google Scholar] [CrossRef]
- Kondo, T.; Ezzat, S.; Asa, S.L. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat. Rev. Cancer 2006, 6, 292–306. [Google Scholar] [CrossRef]
- Cibas, E.S.; Ali, S.Z. The bethesda system for reporting thyroid cytopathology. Thyroid 2009, 19, 1159–1165. [Google Scholar] [CrossRef]
- Bongiovanni, M.; Spitale, A.; Faquin, W.C.; Mazzucchelli, L.; Baloch, Z.W. The bethesda system for reporting thyroid cytopathology: A meta-analysis. Acta Cytol. 2012, 56, 333–339. [Google Scholar] [CrossRef]
- Bongiovanni, M.; De Saussure, B.; Kumar, N.; Pache, J.C.; Cibas, E.S. A quality control study on cytotechnologist-cytopathologist concordance and its relationship to the number of dots on the slide. Acta Cytol. 2009, 53, 653–658. [Google Scholar] [CrossRef]
- Cibas, E.S.; Baloch, Z.W.; Fellegara, G.; LiVolsi, V.A.; Raab, S.S.; Rosai, J.; Diggans, J.; Friedman, L.; Kennedy, G.C.; Kloos, R.T.; et al. A prospective assessment defining the limitations of thyroid nodule pathologic evaluation. Ann. Intern. Med. 2013, 159, 325–332. [Google Scholar] [CrossRef]
- Iskandar, M.E.; Bonomo, G.; Avadhani, V.; Persky, M.; Lucido, D.; Wang, B.; Marti, J.L. Evidence for overestimation of the prevalence of malignancy in indeterminate thyroid nodules classified as bethesda category iii. Surgery 2015, 157, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Seethala, R.R.; Tallini, G.; Baloch, Z.W.; Basolo, F.; Thompson, L.D.; Barletta, J.A.; Wenig, B.M.; Al Ghuzlan, A.; Kakudo, K.; et al. Nomenclature revision for encapsulated follicular variant of papillary thyroid carcinoma: A paradigm shift to reduce overtreatment of indolent tumors. JAMA Oncol. 2016, 2, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.V.; Osamura, R.; Klöppel, G.; Rosai, J. WHO Classification of Tumours of Endocrine Organs from the World Health Organization; IARC WHO Classification of Tumours; WHO: Geneva, Switzerland, 2017; Volume 10. [Google Scholar]
- Cibas, E.S.; Ali, S.Z. The 2017 bethesda system for reporting thyroid cytopathology. Thyroid 2017, 27, 1341–1346. [Google Scholar] [CrossRef] [PubMed]
- Mallick, R.; Stevens, T.M.; Winokur, T.S.; Asban, A.; Wang, T.N.; Lindeman, B.M.; Porterfield, J.R.; Chen, H. Is frozen-section analysis during thyroid operation useful in the era of molecular testing? J. Am. Coll Surg. 2019, 228, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Guevara, N.; Lassalle, S.; Benaim, G.; Sadoul, J.L.; Santini, J.; Hofman, P. Role of frozen section analysis in nodular thyroid pathology. Eur. Ann. Otorhinolaryngol. Head. Neck. Dis. 2015, 132, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.S.; Sarti, E.E.; Jain, K.S.; Wang, H.; Nixon, I.J.; Shaha, A.R.; Shah, J.P.; Kraus, D.H.; Ghossein, R.; Fish, S.A.; et al. Malignancy rate in thyroid nodules classified as bethesda category iii (aus/flus). Thyroid 2014, 24, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Nikiforova, M. Update on molecular testing for cytologically indeterminate thyroid nodules. Arch Pathol. Lab Med. 2018, 142, 446–457. [Google Scholar] [CrossRef]
- Ali, S.; Cibas, E.S. The Bethesda System for Reporting Thyroid Cytopathology: Definitions, Criteria, and Explanatory Notes, 2nd ed.; Springer: New York, NY, USA, 2018. [Google Scholar]
- Muzza, M.; Colombo, C.; Pogliaghi, G.; Karapanou, O.; Fugazzola, L. Molecular markers for the classification of cytologically indeterminate thyroid nodules. J. Endocrinol. Investig. 2020, 43, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.; Yip, L. Decision making in indeterminate thyroid nodules and the role of molecular testing. Surg. Clin. N. Am. 2019, 99, 587–598. [Google Scholar] [CrossRef]
- Patel, K.N.; Angell, T.E.; Babiarz, J.; Barth, N.M.; Blevins, T.; Duh, Q.Y.; Ghossein, R.A.; Harrell, R.M.; Huang, J.; Kennedy, G.C.; et al. Performance of a genomic sequencing classifier for the preoperative diagnosis of cytologically indeterminate thyroid nodules. JAMA Surg. 2018, 153, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Labourier, E.; Shifrin, A.; Busseniers, A.E.; Lupo, M.A.; Manganelli, M.L.; Andruss, B.; Wylie, D.; Beaudenon-Huibregtse, S. Molecular testing for mirna, mrna, and DNA on fine-needle aspiration improves the preoperative diagnosis of thyroid nodules with indeterminate cytology. J. Clin. Endocrinol. Metab. 2015, 100, 2743–2750. [Google Scholar] [CrossRef] [PubMed]
- Steward, D.L.; Carty, S.E.; Sippel, R.S.; Yang, S.P.; Sosa, J.A.; Sipos, J.A.; Figge, J.J.; Mandel, S.; Haugen, B.R.; Burman, K.D.; et al. Performance of a multigene genomic classifier in thyroid nodules with indeterminate cytology: A prospective blinded multicenter study. JAMA Oncol. 2019, 5, 204–212. [Google Scholar] [CrossRef]
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Khatami, F.; Larijani, B.; Nikfar, S.; Hasanzad, M.; Fendereski, K.; Tavangar, S.M. Personalized treatment options for thyroid cancer: Current perspectives. Pharmgenom. Pers. Med. 2019, 12, 235–245. [Google Scholar] [CrossRef]
- Zhu, X.; Yao, J.; Tian, W. Microarray technology to investigate genes associated with papillary thyroid carcinoma. Mol. Med. Rep. 2015, 11, 3729–3733. [Google Scholar] [CrossRef] [PubMed]
- Grogan, R.H.; Kaplan, S.P.; Cao, H.; Weiss, R.E.; Degroot, L.J.; Simon, C.A.; Embia, O.M.; Angelos, P.; Kaplan, E.L.; Schechter, R.B. A study of recurrence and death from papillary thyroid cancer with 27 years of median follow-up. Surgery 2013, 154, 1436–1446. [Google Scholar] [CrossRef]
- Vuong, H.G.; Duong, U.N.; Altibi, A.M.; Ngo, H.T.; Pham, T.Q.; Tran, H.M.; Gandolfi, G.; Hassell, L. A meta-analysis of prognostic roles of molecular markers in papillary thyroid carcinoma. Endocr. Connect 2017, 6, R8–R17. [Google Scholar] [CrossRef]
- Xing, M.; Alzahrani, A.S.; Carson, K.A.; Viola, D.; Elisei, R.; Bendlova, B.; Yip, L.; Mian, C.; Vianello, F.; Tuttle, R.M.; et al. Association between braf v600e mutation and mortality in patients with papillary thyroid cancer. JAMA 2013, 309, 1493–1501. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, E.S.; Kim, Y.S. Clinicopathologic significance of braf v600e mutation in papillary carcinomas of the thyroid: A meta-analysis. Cancer 2007, 110, 38–46. [Google Scholar] [CrossRef]
- Yip, L.; Nikiforova, M.N.; Yoo, J.Y.; McCoy, K.L.; Stang, M.T.; Armstrong, M.J.; Nicholson, K.J.; Ohori, N.P.; Coyne, C.; Hodak, S.P.; et al. Tumor genotype determines phenotype and disease-related outcomes in thyroid cancer: A study of 1510 patients. Ann. Surg. 2015, 262, 519–525. [Google Scholar] [CrossRef]
- Zatelli, M.C.; Trasforini, G.; Leoni, S.; Frigato, G.; Buratto, M.; Tagliati, F.; Rossi, R.; Cavazzini, L.; Roti, E.; degli Uberti, E.C. Braf v600e mutation analysis increases diagnostic accuracy for papillary thyroid carcinoma in fine-needle aspiration biopsies. Eur. J. Endocrinol. 2009, 161, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Park, Y.J.; Lim, J.A.; Ahn, H.Y.; Lee, E.K.; Lee, Y.J.; Kim, K.W.; Hahn, S.K.; Youn, Y.K.; Kim, K.H.; et al. The association of the braf(v600e) mutation with prognostic factors and poor clinical outcome in papillary thyroid cancer: A meta-analysis. Cancer 2012, 118, 1764–1773. [Google Scholar] [CrossRef]
- Fnais, N.; Soobiah, C.; Al-Qahtani, K.; Hamid, J.S.; Perrier, L.; Straus, S.E.; Tricco, A.C. Diagnostic value of fine needle aspiration braf(v600e) mutation analysis in papillary thyroid cancer: A systematic review and meta-analysis. Hum. Pathol. 2015, 46, 1443–1454. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, E.K.; Kwak, J.Y.; Moon, H.J.; Yoon, J.H. What to do with thyroid nodules showing benign cytology and braf(v600e) mutation? A study based on clinical and radiologic features using a highly sensitive analytic method. Surgery 2015, 157, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Antonello, Z.A.; Hsu, N.; Bhasin, M.; Roti, G.; Joshi, M.; Van Hummelen, P.; Ye, E.; Lo, A.S.; Karumanchi, S.A.; Bryke, C.R.; et al. Vemurafenib-resistance via de novo rbm genes mutations and chromosome 5 aberrations is overcome by combined therapy with palbociclib in thyroid carcinoma with braf(v600e). Oncotarget 2017, 8, 84743–84760. [Google Scholar] [CrossRef]
- Finkel, A.; Liba, L.; Simon, E.; Bick, T.; Prinz, E.; Sabo, E.; Ben-Izhak, O.; Hershkovitz, D. Subclonality for braf mutation in papillary thyroid carcinoma is associated with earlier disease stage. J. Clin. Endocrinol. Metab. 2016, 101, 1407–1413. [Google Scholar] [CrossRef]
- Vuong, H.G.; Altibi, A.M.A.; Duong, U.N.P.; Hassell, L. Prognostic implication of braf and tert promoter mutation combination in papillary thyroid carcinoma-a meta-analysis. Clin. Endocrinol. (Oxf.) 2017, 87, 411–417. [Google Scholar] [CrossRef]
- Howell, G.M.; Nikiforova, M.N.; Carty, S.E.; Armstrong, M.J.; Hodak, S.P.; Stang, M.T.; McCoy, K.L.; Nikiforov, Y.E.; Yip, L. Braf v600e mutation independently predicts central compartment lymph node metastasis in patients with papillary thyroid cancer. Ann. Surg. Oncol. 2013, 20, 47–52. [Google Scholar] [CrossRef]
- Afkhami, M.; Karunamurthy, A.; Chiosea, S.; Nikiforova, M.N.; Seethala, R.; Nikiforov, Y.E.; Coyne, C. Histopathologic and clinical characterization of thyroid tumors carrying the braf(k601e) mutation. Thyroid 2016, 26, 242–247. [Google Scholar] [CrossRef]
- Ohori, N.P.; Singhal, R.; Nikiforova, M.N.; Yip, L.; Schoedel, K.E.; Coyne, C.; McCoy, K.L.; LeBeau, S.O.; Hodak, S.P.; Carty, S.E.; et al. Braf mutation detection in indeterminate thyroid cytology specimens: Underlying cytologic, molecular, and pathologic characteristics of papillary thyroid carcinoma. Cancer Cytopathol. 2013, 121, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Grogan, R.H.; Mitmaker, E.J.; Clark, O.H. The evolution of biomarkers in thyroid cancer-from mass screening to a personalized biosignature. Cancers 2010, 2, 885–912. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Qadri, Q.; Makhdoomi, M.J.; Wani, M.A.; Malik, A.A.; Niyaz, M.; Masoodi, S.R.; Andrabi, K.I.; Ahmad, R.; Mudassar, S. Ret/ptc gene rearrangements in thyroid carcinogenesis: Assessment and clinico-pathological correlations. Pathol. Oncol. Res. 2020, 26, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Rowland, J.M.; Bove, K.E.; Monforte-Munoz, H.; Fagin, J.A. Distinct pattern of ret oncogene rearrangements in morphological variants of radiation-induced and sporadic thyroid papillary carcinomas in children. Cancer Res. 1997, 57, 1690–1694. [Google Scholar]
- Su, X.; He, C.; Ma, J.; Tang, T.; Zhang, X.; Ye, Z.; Long, Y.; Shao, Q.; Shao, J.; Yang, A. Ret/ptc rearrangements are associated with elevated postoperative tsh levels and multifocal lesions in papillary thyroid cancer without concomitant thyroid benign disease. PLoS ONE 2016, 11, e0165596. [Google Scholar] [CrossRef]
- Paulson, V.A.; Rudzinski, E.R.; Hawkins, D.S. Thyroid cancer in the pediatric population. Genes 2019, 10, 723. [Google Scholar] [CrossRef]
- Howell, G.M.; Hodak, S.P.; Yip, L. Ras mutations in thyroid cancer. Oncologist 2013, 18, 926–932. [Google Scholar] [CrossRef]
- Gupta, N.; Dasyam, A.K.; Carty, S.E.; Nikiforova, M.N.; Ohori, N.P.; Armstrong, M.; Yip, L.; LeBeau, S.O.; McCoy, K.L.; Coyne, C.; et al. Ras mutations in thyroid fna specimens are highly predictive of predominantly low-risk follicular-pattern cancers. J. Clin. Endocrinol. Metab. 2013, 98, E914–E922. [Google Scholar] [CrossRef]
- Sabra, M.M.; Dominguez, J.M.; Grewal, R.K.; Larson, S.M.; Ghossein, R.A.; Tuttle, R.M.; Fagin, J.A. Clinical outcomes and molecular profile of differentiated thyroid cancers with radioiodine-avid distant metastases. J. Clin. Endocrinol. Metab. 2013, 98, E829–E836. [Google Scholar] [CrossRef]
- Nikiforov, Y.E. Molecular diagnostics of thyroid tumors. Arch. Pathol. Lab. Med. 2011, 135, 569–577. [Google Scholar]
- Brandler, T.C.; Liu, C.Z.; Cho, M.; Zhou, F.; Cangiarella, J.; Yee-Chang, M.; Shi, Y.; Simsir, A.; Sun, W. Does noninvasive follicular thyroid neoplasm with papillary-like nuclear features (niftp) have a unique molecular profile? Am. J. Clin. Pathol. 2018, 150, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Xing, M. Clinical utility of ras mutations in thyroid cancer: A blurred picture now emerging clearer. BMC Med. 2016, 14, 12. [Google Scholar] [CrossRef] [PubMed]
- Giannini, R.; Ugolini, C.; Poma, A.M.; Urpi, M.; Niccoli, C.; Elisei, R.; Chiarugi, M.; Vitti, P.; Miccoli, P.; Basolo, F. Identification of two distinct molecular subtypes of non-invasive follicular neoplasm with papillary-like nuclear features by digital rna counting. Thyroid 2017, 27, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- D’Cruz, A.K.; Vaish, R.; Vaidya, A.; Nixon, I.J.; Williams, M.D.; Vander Poorten, V.; Lopez, F.; Angelos, P.; Shaha, A.R.; Khafif, A.; et al. Molecular markers in well-differentiated thyroid cancer. Eur. Arch. Otorhinolaryngol. 2018, 275, 1375–1384. [Google Scholar] [CrossRef]
- Hodak, S.; Tuttle, R.M.; Maytal, G.; Nikiforov, Y.E.; Randolph, G. Changing the cancer diagnosis: The case of follicular variant of papillary thyroid cancer-primum non nocere and niftp. Thyroid 2016, 26, 869–871. [Google Scholar] [CrossRef]
- Patel, S.G.; Carty, S.E.; McCoy, K.L.; Ohori, N.P.; LeBeau, S.O.; Seethala, R.R.; Nikiforova, M.N.; Nikiforov, Y.E.; Yip, L. Preoperative detection of ras mutation may guide extent of thyroidectomy. Surgery 2017, 161, 168–175. [Google Scholar] [CrossRef]
- Yang, J.; Gong, Y.; Yan, S.; Chen, H.; Qin, S.; Gong, R. Association between tert promoter mutations and clinical behaviors in differentiated thyroid carcinoma: A systematic review and meta-analysis. Endocrine 2020, 67, 44–57. [Google Scholar] [CrossRef]
- Grani, G.; Lamartina, L.; Durante, C.; Filetti, S.; Cooper, D.S. Follicular thyroid cancer and hurthle cell carcinoma: Challenges in diagnosis, treatment, and clinical management. Lancet Diabetes Endocrinol. 2018, 6, 500–514. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Yang, H.; Yip, L.; Ohori, N.P.; McCoy, K.L.; Stang, M.T.; Hodak, S.P.; Nikiforova, M.N.; Carty, S.E.; Nikiforov, Y.E. Pax8/ppargamma rearrangement in thyroid nodules predicts follicular-pattern carcinomas, in particular the encapsulated follicular variant of papillary carcinoma. Thyroid 2014, 24, 1369–1374. [Google Scholar] [CrossRef]
- Boos, L.A.; Dettmer, M.; Schmitt, A.; Rudolph, T.; Steinert, H.; Moch, H.; Sobrinho-Simoes, M.; Komminoth, P.; Perren, A. Diagnostic and prognostic implications of the pax8-ppargamma translocation in thyroid carcinomas-a tma-based study of 226 cases. Histopathology 2013, 63, 234–241. [Google Scholar] [CrossRef]
- Nicolson, N.G.; Murtha, T.D.; Dong, W.; Paulsson, J.O.; Choi, J.; Barbieri, A.L.; Brown, T.C.; Kunstman, J.W.; Larsson, C.; Prasad, M.L.; et al. Comprehensive genetic analysis of follicular thyroid carcinoma predicts prognosis independent of histology. J. Clin. Endocrinol. Metab. 2018, 103, 2640–2650. [Google Scholar] [CrossRef] [PubMed]
- Ganly, I.; Makarov, V.; Deraje, S.; Dong, Y.; Reznik, E.; Seshan, V.; Nanjangud, G.; Eng, S.; Bose, P.; Kuo, F.; et al. Integrated genomic analysis of hurthle cell cancer reveals oncogenic drivers, recurrent mitochondrial mutations, and unique chromosomal landscapes. Cancer Cell 2018, 34, 256–270.e255. [Google Scholar] [CrossRef] [PubMed]
- Stokowy, T.; Gawel, D.; Wojtas, B. Differences in mirna and mrna profile of papillary thyroid cancer variants. Int. J. Endocrinol. 2016, 2016, 1427042. [Google Scholar] [CrossRef] [PubMed]
- Stokowy, T.; Wojtas, B.; Jarzab, B.; Krohn, K.; Fredman, D.; Dralle, H.; Musholt, T.; Hauptmann, S.; Lange, D.; Hegedus, L.; et al. Two-mirna classifiers differentiate mutation-negative follicular thyroid carcinomas and follicular thyroid adenomas in fine needle aspirations with high specificity. Endocrine 2016, 54, 440–447. [Google Scholar] [CrossRef]
- Kwok, G.T.; Zhao, J.T.; Weiss, J.; Mugridge, N.; Brahmbhatt, H.; MacDiarmid, J.A.; Robinson, B.G.; Sidhu, S.B. Translational applications of micrornas in cancer, and therapeutic implications. Noncoding RNA Res. 2017, 2, 143–150. [Google Scholar] [CrossRef]
- Larrea, E.; Sole, C.; Manterola, L.; Goicoechea, I.; Armesto, M.; Arestin, M.; Caffarel, M.M.; Araujo, A.M.; Araiz, M.; Fernandez-Mercado, M.; et al. New concepts in cancer biomarkers: Circulating mirnas in liquid biopsies. Int. J. Mol. Sci. 2016, 17, 627. [Google Scholar] [CrossRef]
- Rosignolo, F.; Memeo, L.; Monzani, F.; Colarossi, C.; Pecce, V.; Verrienti, A.; Durante, C.; Grani, G.; Lamartina, L.; Forte, S.; et al. Microrna-based molecular classification of papillary thyroid carcinoma. Int. J. Oncol. 2017, 50, 1767–1777. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Shirvani-Farsani, Z.; Taheri, M. The role of micrornas in the pathogenesis of thyroid cancer. Noncoding RNA Res. 2020, 5, 88–98. [Google Scholar] [CrossRef]
- Ramirez-Moya, J.; Wert-Lamas, L.; Santisteban, P. Microrna-146b promotes pi3k/akt pathway hyperactivation and thyroid cancer progression by targeting pten. Oncogene 2018, 37, 3369–3383. [Google Scholar] [CrossRef]
- Riesco-Eizaguirre, G.; Wert-Lamas, L.; Perales-Paton, J.; Sastre-Perona, A.; Fernandez, L.P.; Santisteban, P. The mir-146b-3p/pax8/nis regulatory circuit modulates the differentiation phenotype and function of thyroid cells during carcinogenesis. Cancer Res. 2015, 75, 4119–4130. [Google Scholar] [CrossRef]
- Deng, X.; Wu, B.; Xiao, K.; Kang, J.; Xie, J.; Zhang, X.; Fan, Y. Mir-146b-5p promotes metastasis and induces epithelial-mesenchymal transition in thyroid cancer by targeting znrf3. Cell Physiol. Biochem. 2015, 35, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Jin, M.; Li, Y.; Ren, P.; Liu, J. Mir-137 acts as a tumor suppressor in papillary thyroid carcinoma by targeting cxcl12. Oncol. Rep. 2016, 35, 2151–2158. [Google Scholar] [CrossRef] [PubMed]
- Paskas, S.; Jankovic, J.; Zivaljevic, V.; Tatic, S.; Bozic, V.; Nikolic, A.; Radojkovic, D.; Savin, S.; Cvejic, D. Malignant risk stratification of thyroid fna specimens with indeterminate cytology based on molecular testing. Cancer Cytopathol. 2015, 123, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Zheng, X.; Hu, M.; Cui, Y.; Zhong, Q.; Wang, S.; Huang, F. Mirna-221/222 in thyroid cancer: A meta-analysis. Clin. Chim. Acta 2018, 484, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.J.; Shen, C.T.; Song, H.J.; Qiu, Z.L.; Luo, Q.Y. Micrornas as a potential tool in the differential diagnosis of thyroid cancer: A systematic review and meta-analysis. Clin. Endocrinol. (Oxf.) 2016, 84, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Wang, P.; Hurst, Z.A.; Chang, Y.S.; Chen, G. Active surveillance for papillary thyroid microcarcinoma: Challenges and prospects. Front. Endocrinol. (Lausanne) 2018, 9, 736. [Google Scholar] [CrossRef]
- Hardin, H.; Helein, H.; Meyer, K.; Robertson, S.; Zhang, R.; Zhong, W.; Lloyd, R.V. Thyroid cancer stem-like cell exosomes: Regulation of emt via transfer of lncrnas. Lab Invest. 2018, 98, 1133–1142. [Google Scholar] [CrossRef]
- Zhang, R.; Hardin, H.; Huang, W.; Chen, J.; Asioli, S.; Righi, A.; Maletta, F.; Sapino, A.; Lloyd, R.V. Malat1 long non-coding rna expression in thyroid tissues: Analysis by in situ hybridization and real-time pcr. Endocr. Pathol. 2017, 28, 7–12. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, X.; Fu, X.; Yan, W.; Lin, F.; Kuang, P.; Luo, Y.; Lin, E.; Hong, X.; Wu, G. Long non-coding rna bancr regulates cancer stem cell markers in papillary thyroid cancer via the raf/mek/erk signaling pathway. Oncol. Rep. 2018, 40, 859–866. [Google Scholar] [CrossRef]
- Lan, X.; Sun, W.; Dong, W.; Wang, Z.; Zhang, T.; He, L.; Zhang, H. Downregulation of long noncoding rna h19 contributes to the proliferation and migration of papillary thyroid carcinoma. Gene 2018, 646, 98–105. [Google Scholar] [CrossRef]
- Zhang, R.; Hardin, H.; Chen, J.; Guo, Z.; Lloyd, R.V. Non-coding rnas in thyroid cancer. Endocr. Pathol. 2016, 27, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Arita, T.; Ichikawa, D.; Konishi, H.; Komatsu, S.; Shiozaki, A.; Shoda, K.; Kawaguchi, T.; Hirajima, S.; Nagata, H.; Kubota, T.; et al. Circulating long non-coding rnas in plasma of patients with gastric cancer. Anticancer Res. 2013, 33, 3185–3193. [Google Scholar] [PubMed]
- Martinez-Aguilar, J.; Clifton-Bligh, R.; Molloy, M.P. Proteomics of thyroid tumours provides new insights into their molecular composition and changes associated with malignancy. Sci. Rep. 2016, 6, 23660. [Google Scholar] [CrossRef] [PubMed]
- Navas-Carrillo, D.; Rodriguez, J.M.; Montoro-Garcia, S.; Orenes-Pinero, E. High-resolution proteomics and metabolomics in thyroid cancer: Deciphering novel biomarkers. Crit. Rev. Clin. Lab. Sci. 2017, 54, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E. Role of molecular markers in thyroid nodule management: Then and now. Endocr. Pract. 2017, 23, 979–988. [Google Scholar] [CrossRef]
- Yip, L.; Sosa, J.A. Molecular-directed treatment of differentiated thyroid cancer: Advances in diagnosis and treatment. JAMA Surg. 2016, 151, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Steward, D.L.; Robinson-Smith, T.M.; Haugen, B.R.; Klopper, J.P.; Zhu, Z.; Fagin, J.A.; Falciglia, M.; Weber, K.; Nikiforova, M.N. Molecular testing for mutations in improving the fine-needle aspiration diagnosis of thyroid nodules. J. Clin. Endocrinol. Metab. 2009, 94, 2092–2098. [Google Scholar] [CrossRef]
- Sistrunk, J.W.; Shifrin, A.; Frager, M.; Bardales, R.H.; Thomas, J.; Fishman, N.; Goldberg, P.; Guttler, R.; Grant, E. Clinical performance of multiplatform mutation panel and microrna risk classifier in indeterminate thyroid nodules. J. Am. Soc. Cytopathol. 2020. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Mercurio, S.; Wald, A.I.; Barbi de Moura, M.; Callenberg, K.; Santana-Santos, L.; Gooding, W.E.; Yip, L.; Ferris, R.L.; Nikiforov, Y.E. Analytical performance of the thyroseq v3 genomic classifier for cancer diagnosis in thyroid nodules. Cancer 2018, 124, 1682–1690. [Google Scholar] [CrossRef]
- Ohori, N.P.; Landau, M.S.; Carty, S.E.; Yip, L.; LeBeau, S.O.; Manroa, P.; Seethala, R.R.; Schoedel, K.E.; Nikiforova, M.N.; Nikiforov, Y.E. Benign call rate and molecular test result distribution of thyroseq v3. Cancer Cytopathol. 2019, 127, 161–168. [Google Scholar] [CrossRef]
- Nikiforov, Y.E.; Baloch, Z.W. Clinical validation of the thyroseq v3 genomic classifier in thyroid nodules with indeterminate fna cytology. Cancer Cytopathol. 2019, 127, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Chudova, D.; Wilde, J.I.; Wang, E.T.; Wang, H.; Rabbee, N.; Egidio, C.M.; Reynolds, J.; Tom, E.; Pagan, M.; Rigl, C.T.; et al. Molecular classification of thyroid nodules using high-dimensionality genomic data. J. Clin. Endocrinol. Metab. 2010, 95, 5296–5304. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Song, Y.S.; Kim, Y.A.; Lim, J.A.; Cho, S.W.; Moon, J.H.; Hahn, S.; Park, D.J.; Park, Y.J. Effects of coexistent braf(v600e) and tert promoter mutations on poor clinical outcomes in papillary thyroid cancer: A meta-analysis. Thyroid 2017, 27, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Aragon Han, P.; Olson, M.T.; Fazeli, R.; Prescott, J.D.; Pai, S.I.; Schneider, E.B.; Tufano, R.P.; Zeiger, M.A. The impact of molecular testing on the surgical management of patients with thyroid nodules. Ann. Surg. Oncol. 2014, 21, 1862–1869. [Google Scholar] [CrossRef] [PubMed]
- Noureldine, S.I.; Najafian, A.; Aragon Han, P.; Olson, M.T.; Genther, D.J.; Schneider, E.B.; Prescott, J.D.; Agrawal, N.; Mathur, A.; Zeiger, M.A.; et al. Evaluation of the effect of diagnostic molecular testing on the surgical decision-making process for patients with thyroid nodules. JAMA Otolaryngol. Head. Neck. Surg. 2016, 142, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Noureldine, S.I.; Zeiger, M.A.; Tufano, R.P. Gene expression classifier testing and the surgical decision-making process for patients with thyroid nodules-reply. JAMA Otolaryngol. Head. Neck. Surg. 2016, 142, 807. [Google Scholar] [CrossRef]
- Sahli, Z.T.; Smith, P.W.; Umbricht, C.B.; Zeiger, M.A. Preoperative molecular markers in thyroid nodules. Front. Endocrinol. (Lausanne) 2018, 9, 179. [Google Scholar] [CrossRef]
- Nicholson, K.J.; Roberts, M.S.; McCoy, K.L.; Carty, S.E.; Yip, L. Molecular testing versus diagnostic lobectomy in bethesda iii/iv thyroid nodules: A cost-effectiveness analysis. Thyroid 2019, 29, 1237–1243. [Google Scholar] [CrossRef]
- Wang, L.Y.; Ganly, I. Post-treatment surveillance of thyroid cancer. Eur. J. Surg. Oncol. 2018, 44, 357–366. [Google Scholar] [CrossRef]
- Spencer, C.A.; Takeuchi, M.; Kazarosyan, M.; Wang, C.C.; Guttler, R.B.; Singer, P.A.; Fatemi, S.; LoPresti, J.S.; Nicoloff, J.T. Serum thyroglobulin autoantibodies: Prevalence, influence on serum thyroglobulin measurement, and prognostic significance in patients with differentiated thyroid carcinoma. J. Clin. Endocrinol. Metab. 1998, 83, 1121–1127. [Google Scholar] [CrossRef]
- Lee, Z.J.O.; Eslick, G.D.; Edirimanne, S. Investigating anti-thyroglobulin antibody as a prognostic marker for differentiated thyroid cancer: A meta-analysis and systematic review. Thyroid 2020. [Google Scholar] [CrossRef]
- Durante, C.; Tognini, S.; Montesano, T.; Orlandi, F.; Torlontano, M.; Puxeddu, E.; Attard, M.; Costante, G.; Tumino, S.; Meringolo, D.; et al. Clinical aggressiveness and long-term outcome in patients with papillary thyroid cancer and circulating anti-thyroglobulin autoantibodies. Thyroid 2014, 24, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Rosignolo, F.; Sponziello, M.; Giacomelli, L.; Russo, D.; Pecce, V.; Biffoni, M.; Bellantone, R.; Lombardi, C.P.; Lamartina, L.; Grani, G.; et al. Identification of thyroid-associated serum microrna profiles and their potential use in thyroid cancer follow-up. J. Endocr. Soc. 2017, 1, 3–13. [Google Scholar] [PubMed]
- Nixon, A.M.; Provatopoulou, X.; Kalogera, E.; Zografos, G.N.; Gounaris, A. Circulating thyroid cancer biomarkers: Current limitations and future prospects. Clin. Endocrinol. (Oxf.) 2017, 87, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Volante, M.; Collini, P.; Nikiforov, Y.E.; Sakamoto, A.; Kakudo, K.; Katoh, R.; Lloyd, R.V.; LiVolsi, V.A.; Papotti, M.; Sobrinho-Simoes, M.; et al. Poorly differentiated thyroid carcinoma: The turin proposal for the use of uniform diagnostic criteria and an algorithmic diagnostic approach. Am. J. Surg. Pathol. 2007, 31, 1256–1264. [Google Scholar] [CrossRef]
- Ibrahimpasic, T.; Ghossein, R.; Shah, J.P.; Ganly, I. Poorly differentiated carcinoma of the thyroid gland: Current status and future prospects. Thyroid 2019, 29, 311–321. [Google Scholar] [CrossRef]
- Chintakuntlawar, A.V.; Foote, R.L.; Kasperbauer, J.L.; Bible, K.C. Diagnosis and management of anaplastic thyroid cancer. Endocrinol. Metab. Clin. N. Am. 2019, 48, 269–284. [Google Scholar] [CrossRef]
- Kunstman, J.W.; Juhlin, C.C.; Goh, G.; Brown, T.C.; Stenman, A.; Healy, J.M.; Rubinstein, J.C.; Choi, M.; Kiss, N.; Nelson-Williams, C.; et al. Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum. Mol. Genet 2015, 24, 2318–2329. [Google Scholar] [CrossRef]
- Pozdeyev, N.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Deaver, K.E.; Davis, S.; French, J.D.; Borre, P.V.; LaBarbera, D.V.; Tan, A.C.; et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin. Cancer Res. 2018, 24, 3059–3068. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in multiple nonmelanoma cancers with braf v600 mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Godbert, Y.; Henriques de Figueiredo, B.; Bonichon, F.; Chibon, F.; Hostein, I.; Pérot, G.; Dupin, C.; Daubech, A.; Belleannée, G.; Gros, A.; et al. Remarkable response to crizotinib in woman with anaplastic lymphoma kinase-rearranged anaplastic thyroid carcinoma. J. Clin. Oncol. 2015, 33, e84–e87. [Google Scholar] [CrossRef] [PubMed]
- Bible, K.C. Treating advanced radioresistant differentiated thyroid cancer. Lancet Oncol. 2012, 13, 854–855. [Google Scholar] [CrossRef]
- Fagin, J.A.; Wells, S.A., Jr. Biologic and clinical perspectives on thyroid cancer. N. Engl. J. Med. 2016, 375, 1054–1067. [Google Scholar] [CrossRef]
- Durante, C.; Haddy, N.; Baudin, E.; Leboulleux, S.; Hartl, D.; Travagli, J.P.; Caillou, B.; Ricard, M.; Lumbroso, J.D.; De Vathaire, F.; et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: Benefits and limits of radioiodine therapy. J. Clin. Endocrinol. Metab. 2006, 91, 2892–2899. [Google Scholar] [CrossRef] [PubMed]
- Clark, O.; Duh, Q.-Y.; Kebebew, E. Textbook of Endocrine Surgery, 3rd ed.; JP Medical Ltd.: New Delhi, India, 2016. [Google Scholar]
- Ricarte-Filho, J.C.; Ryder, M.; Chitale, D.A.; Rivera, M.; Heguy, A.; Ladanyi, M.; Janakiraman, M.; Solit, D.; Knauf, J.A.; Tuttle, R.M.; et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for braf, pik3ca, and akt1. Cancer Res. 2009, 69, 4885–4893. [Google Scholar] [CrossRef] [PubMed]
- Costamagna, E.; García, B.; Santisteban, P. The functional interaction between the paired domain transcription factor pax8 and smad3 is involved in transforming growth factor-beta repression of the sodium/iodide symporter gene. J. Biol. Chem. 2004, 279, 3439–3446. [Google Scholar] [CrossRef]
- Gild, M.L.; Topliss, D.J.; Learoyd, D.; Parnis, F.; Tie, J.; Hughes, B.; Walsh, J.P.; McLeod, D.S.A.; Clifton-Bligh, R.J.; Robinson, B.G. Clinical guidance for radioiodine refractory differentiated thyroid cancer. Clin. Endocrinol. (Oxf.) 2018, 88, 529–537. [Google Scholar] [CrossRef]
- Okamoto, K.; Kodama, K.; Takase, K.; Sugi, N.H.; Yamamoto, Y.; Iwata, M.; Tsuruoka, A. Antitumor activities of the targeted multi-tyrosine kinase inhibitor lenvatinib (e7080) against ret gene fusion-driven tumor models. Cancer Lett. 2013, 340, 97–103. [Google Scholar] [CrossRef]
- Tohyama, O.; Matsui, J.; Kodama, K.; Hata-Sugi, N.; Kimura, T.; Okamoto, K.; Minoshima, Y.; Iwata, M.; Funahashi, Y. Antitumor activity of lenvatinib (e7080): An angiogenesis inhibitor that targets multiple receptor tyrosine kinases in preclinical human thyroid cancer models. J. Thyroid Res. 2014, 2014, 638747. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both raf and vegf and pdgf receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef]
- Brose, M.S.; Cabanillas, M.E.; Cohen, E.E.; Wirth, L.J.; Riehl, T.; Yue, H.; Sherman, S.I.; Sherman, E.J. Vemurafenib in patients with braf(v600e)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: A non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1272–1282. [Google Scholar] [CrossRef]
- Liu, D.; Hu, S.; Hou, P.; Jiang, D.; Condouris, S.; Xing, M. Suppression of braf/mek/map kinase pathway restores expression of iodide-metabolizing genes in thyroid cells expressing the v600e braf mutant. Clin. Cancer Res. 2007, 13, 1341–1349. [Google Scholar] [CrossRef]
- Ho, A.L.; Grewal, R.K.; Leboeuf, R.; Sherman, E.J.; Pfister, D.G.; Deandreis, D.; Pentlow, K.S.; Zanzonico, P.B.; Haque, S.; Gavane, S.; et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N. Engl. J. Med. 2013, 368, 623–632. [Google Scholar] [CrossRef]
- Iravani, A.; Solomon, B.; Pattison, D.A.; Jackson, P.; Ravi Kumar, A.; Kong, G.; Hofman, M.S.; Akhurst, T.; Hicks, R.J. Mitogen-activated protein kinase pathway inhibition for redifferentiation of radioiodine refractory differentiated thyroid cancer: An evolving protocol. Thyroid 2019, 29, 1634–1645. [Google Scholar] [CrossRef]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; Cabanillas, M.E.; Urbanowitz, G.; et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic braf v600-mutant anaplastic thyroid cancer. J. Clin. Oncol. 2018, 36, 7–13. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. Ntrk fusion-positive cancers and trk inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Harris, E.J.; Hanna, G.J.; Chau, N.; Rabinowits, G.; Haddad, R.; Margalit, D.N.; Schoenfeld, J.; Tishler, R.B.; Barletta, J.A.; Nehs, M.; et al. Everolimus in anaplastic thyroid cancer: A case series. Front. Oncol. 2019, 9, 106. [Google Scholar] [CrossRef]
- Gambacorti-Passerini, C.; Orlov, S.; Zhang, L.; Braiteh, F.; Huang, H.; Esaki, T.; Horibe, K.; Ahn, J.S.; Beck, J.T.; Edenfield, W.J.; et al. Long-term effects of crizotinib in alk-positive tumors (excluding nsclc): A phase 1b open-label study. Am. J. Hematol. 2018, 93, 607–614. [Google Scholar] [CrossRef]
- Subbiah, V.; Velcheti, V.; Tuch, B.B.; Ebata, K.; Busaidy, N.L.; Cabanillas, M.E.; Wirth, L.J.; Stock, S.; Smith, S.; Lauriault, V.; et al. Selective ret kinase inhibition for patients with ret-altered cancers. Ann. Oncol. 2018, 29, 1869–1876. [Google Scholar] [CrossRef] [PubMed]
- Registrational Results of Loxo-292 in Patients with Ret-altered Thyroid Cancers. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-selpercatinib-lung-and-thyroid-cancers-ret-gene-mutations-or-fusions (accessed on 10 May 2020).
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Sheu, N.W.; Jiang, H.J.; Wu, C.W.; Chiang, F.Y.; Chiou, H.C.; Hsiao, P.J. Lenvatinib complementary with radioiodine therapy for patients with advanced differentiated thyroid carcinoma: Case reports and literature review. World J. Surg. Oncol. 2019, 17, 84. [Google Scholar] [CrossRef]
- Nikiforov, Y.E.; Erickson, L.A.; Nikiforova, M.N.; Caudill, C.M.; Lloyd, R.V. Solid variant of papillary thyroid carcinoma: Incidence, clinical-pathologic characteristics, molecular analysis, and biologic behavior. Am. J. Surg. Pathol. 2001, 25, 1478–1484. [Google Scholar] [CrossRef]
- Tiedje, V.; Stuschke, M.; Weber, F.; Dralle, H.; Moss, L.; Fuhrer, D. Anaplastic thyroid carcinoma: Review of treatment protocols. Endocr. Relat. Cancer 2018, 25, R153–R161. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).