EMT-Inducing Transcription Factors, Drivers of Melanoma Phenotype Switching, and Resistance to Treatment

Abstract
1. Introduction
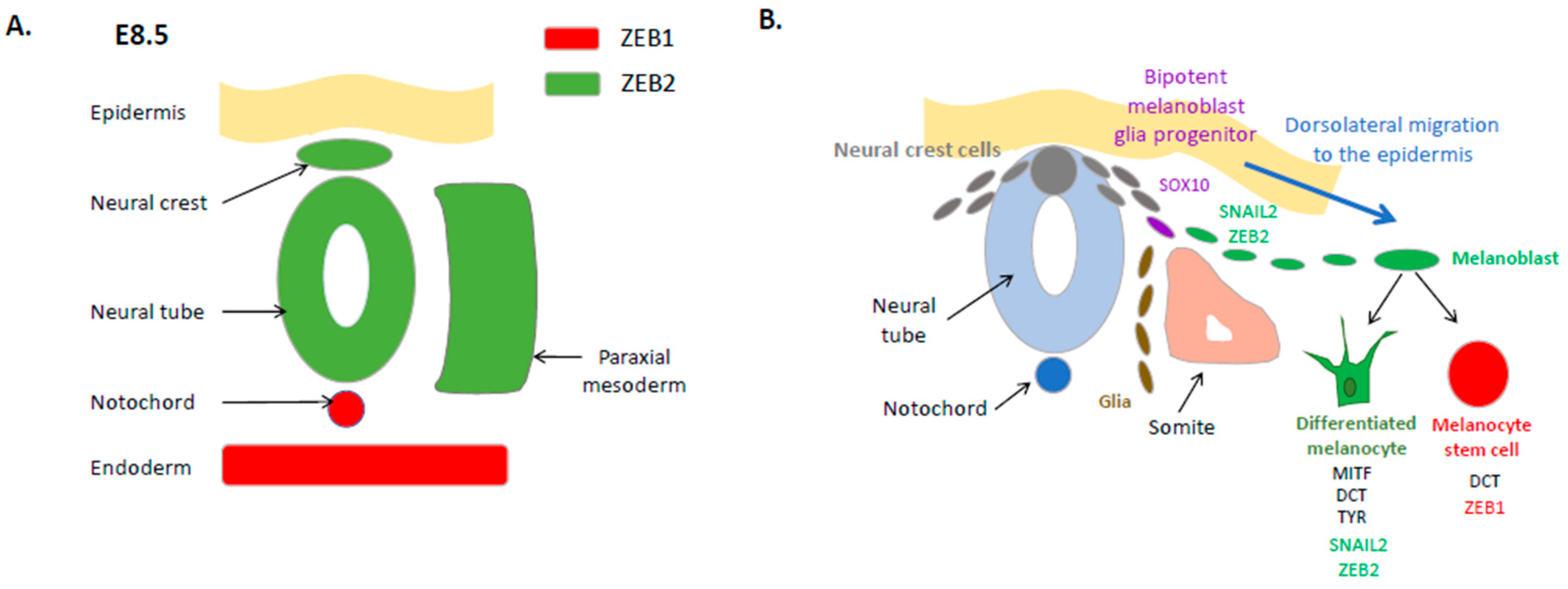
2. Expression and Function of EMT-TFs in the Embryonic Neural Crest and the Melanocyte Lineage
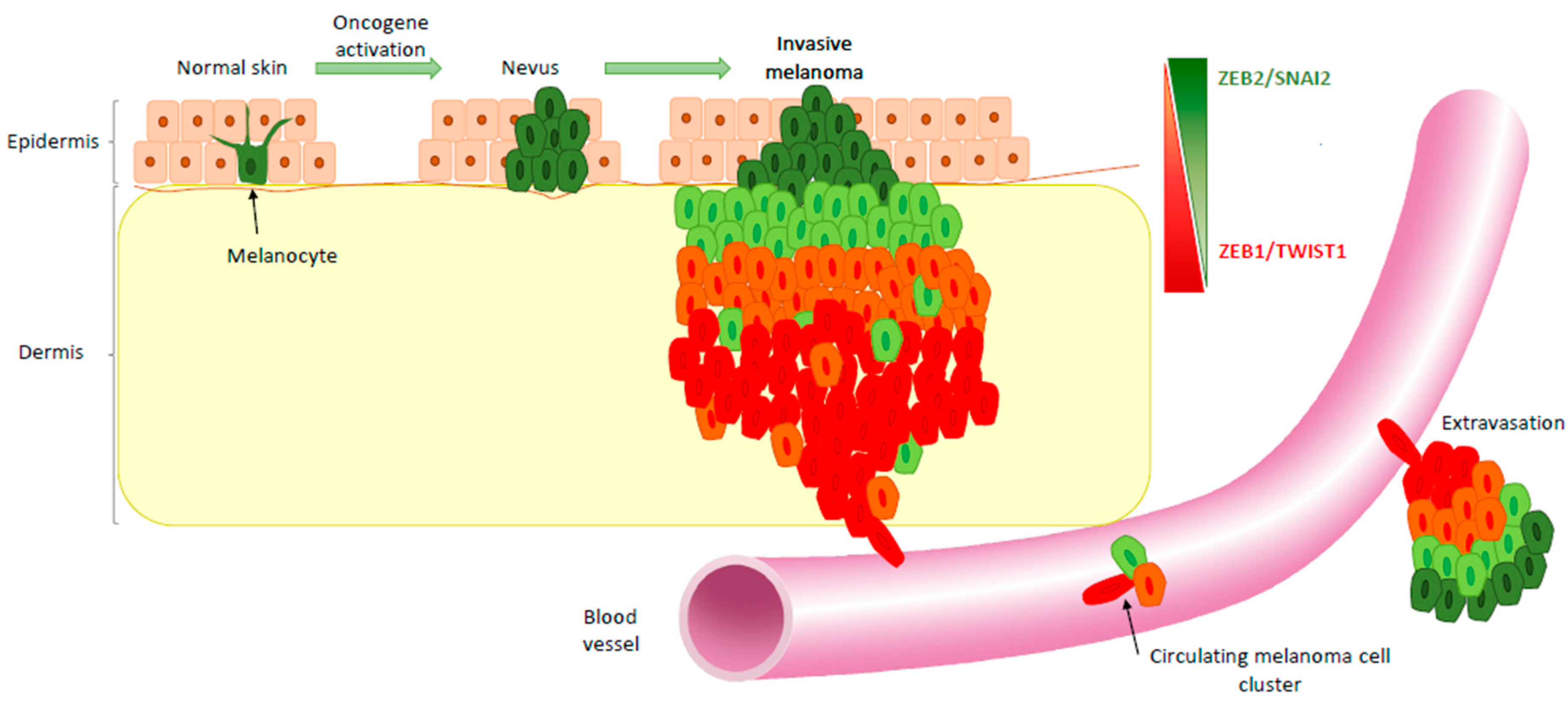
3. EMT-TF Expression Switch during Melanoma Development
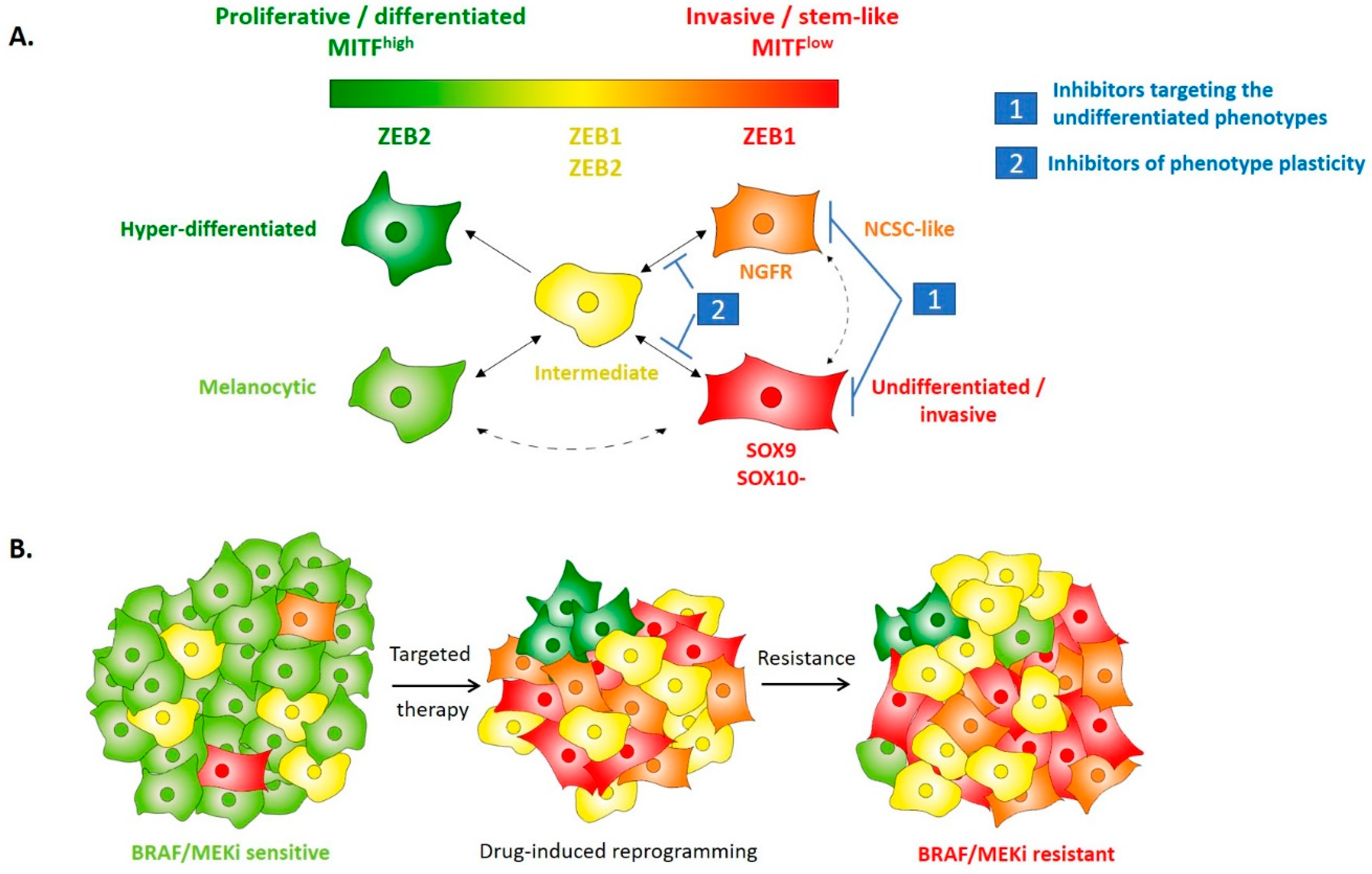
4. EMT-TFs Drive Phenotype Switching of Melanoma Cells and Intra-Tumor Heterogeneity
5. EMT-Like Cell Plasticity as a Driver of Resistance to Treatment in Melanoma
6. Perspectives: Targeting of Cell Plasticity to Prevent Resistance to Treatment
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 11, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Fu, R.; Lu, T.; Wu, Z.; Fu, R.; Li, Y.; Jiang, N.; Ren, B.; Zang, C.; Liu, L.; Lv, W.; et al. Inactivation of endothelial ZEB1 impedes tumor progression and sensitizes tumors to conventional therapies. J. Clin. Investig. 2020, 130, 1252–1270. [Google Scholar] [CrossRef]
- Mckinsey, G.L.; Lindtner, S.; Trzcinski, B.; Visel, A.; Pennacchio, L.A.; Huylebroeck, D.; Higashi, Y.; Rubenstein, J.L.R. Dlx1&2-Dependent Expression of Zfhx1b (Sip1, Zeb2) Regulates the Fate Switch between Cortical and Striatal Interneurons. Neuron 2013, 77, 83–98. [Google Scholar]
- Scott, C.L.; Omilusik, K.D. ZEBs: Novel Players in Immune Cell Development and Function. Trends Immunol. 2019, 40, 431–446. [Google Scholar] [CrossRef]
- Guan, T.; Dominguez, C.X.; Amezquita, R.A.; Laidlaw, B.J.; Cheng, J.; Henao-Mejia, J.; Williams, A.; Flavell, R.A.; Lu, J.; Kaech, S.M. ZEB1, ZEB2, and the miR-200 family form a counterregulatory network to regulate CD8+ T cell fates. J. Exp. Med. 2018, 215, 1153–1168. [Google Scholar] [CrossRef]
- Zhang, J.; Wencker, M.; Marliac, Q.; Berton, A.; Hasan, U.; Schneider, R.; Laubreton, D.; Cherrier, D.E.; Mathieu, A.; Rey, A.; et al. Zeb1 represses TCR signaling, promotes the proliferation of T cell progenitors and is essential for NK1.1 + T cell development. Cell. Mol. Immunol. 2020, 1–13. [Google Scholar] [CrossRef]
- Van Helden, M.J.; Goossens, S.; Daussy, C.; Mathieu, A.L.; Faure, F.; Marçais, A.; Vandamme, N.; Farla, N.; Mayol, K.; Viel, S.; et al. Terminal NK cell maturation is controlled by concerted actions of T-bet and Zeb2 and is essential for melanoma rejection. J. Exp. Med. 2015, 212, 2015–2025. [Google Scholar] [CrossRef]
- Scott, C.L.; T’Jonck, W.; Martens, L.; Todorov, H.; Sichien, D.; Soen, B.; Bonnardel, J.; De Prijck, S.; Vandamme, N.; Cannoodt, R.; et al. The Transcription Factor ZEB2 Is Required to Maintain the Tissue-Specific Identities of Macrophages. Immunity 2018, 49, 312–325.e5. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T. To differentiate or not-routes towards metastasis. Nat. Rev. Cancer 2012, 12, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Brabletz, T.; Kang, Y.; Longmore, G.D.; Nieto, M.A.; Stanger, B.Z.; Yang, J.; Weinberg, R.A. Upholding a role for EMT in breast cancer metastasis. Nature 2017, 547, E1–E3. [Google Scholar] [CrossRef] [PubMed]
- Krebs, A.M.; Mitschke, J.; Losada, M.L.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef]
- Beck, B.; Lapouge, G.; Rorive, S.; Drogat, B.; Desaedelaere, K.; Delafaille, S.; Dubois, C.; Salmon, I.; Willekens, K.; Marine, J.C.; et al. Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell 2015, 16, 67–79. [Google Scholar] [CrossRef]
- Morel, A.-P.; Hinkal, G.W.; Thomas, C.; Fauvet, F.; Courtois-Cox, S.; Wierinckx, A.; Devouassoux-Shisheboran, M.; Treilleux, I.; Tissier, A.; Gras, B.; et al. EMT inducers catalyze malignant transformation of mammary epithelial cells and drive tumorigenesis towards claudin-low tumors in transgenic mice. PLoS Genet. 2012, 8, e1002723. [Google Scholar] [CrossRef]
- Ansieau, S.; Bastid, J.; Doreau, A.; Morel, A.P.; Bouchet, B.P.; Thomas, C.; Fauvet, F.; Puisieux, I.; Doglioni, C.; Piccinin, S.; et al. Induction of EMT by Twist Proteins as a Collateral Effect of Tumor-Promoting Inactivation of Premature Senescence. Cancer Cell 2008, 14, 79–89. [Google Scholar] [CrossRef]
- Morel, A.-P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Siebzehnrubl, F.A.; Silver, D.J.; Tugertimur, B.; Deleyrolle, L.P.; Siebzehnrubl, D.; Sarkisian, M.R.; Devers, K.G.; Yachnis, A.T.; Kupper, M.D.; Neal, D.; et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol. Med. 2013, 5, 1196–1212. [Google Scholar] [CrossRef] [PubMed]
- Boeva, V.; Louis-Brennetot, C.; Peltier, A.; Durand, S.; Pierre-Eugène, C.; Raynal, V.; Etchevers, H.C.; Thomas, S.; Lermine, A.; Daudigeos-Dubus, E.; et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 2017, 49, 1408–1413. [Google Scholar] [CrossRef]
- Van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef]
- Goding, C.R.; Arnheiter, H. MITF—The first 25 years. Genes Dev. 2019, 33, 983–1007. [Google Scholar] [CrossRef]
- Hoek, K.S.; Goding, C.R. Cancer stem cells versus phenotype-switching in melanoma. Pigment. Cell Melanoma Res. 2010, 23, 746–759. [Google Scholar] [CrossRef]
- Hoek, K.S.; Eichhoff, O.M.; Schlegel, N.C.; Döbbeling, U.; Kobert, N.; Schaerer, L.; Hemmi, S.; Dummer, R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008, 68, 650–656. [Google Scholar] [CrossRef]
- Cheli, Y.; Guiliano, S.; Botton, T.; Rocchi, S.; Hofman, V.; Hofman, P.; Bahadoran, P.; Bertolotto, C.; Ballotti, R. Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene 2011, 30, 2307–2318. [Google Scholar] [CrossRef]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Van De Putte, T.; Maruhashi, M.; Francis, A.; Nelles, L.; Kondoh, H.; Huylebroeck, D.; Higashi, Y. Mice lacking Zfhx1b, the gene that codes for Smad-interacting protein-1, reveal a role for multiple neural crest cell defects in the etiology of hirschsprung disease-mental retardation syndrome. Am. J. Hum. Genet. 2003, 72, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Van de Putte, T.; Francis, A.; Nelles, L.; van Grunsven, L.A.; Huylebroeck, D. Neural crest-specific removal of Zfhx1b in mouse leads to a wide range of neurocristopathies reminiscent of Mowat-Wilson syndrome. Hum. Mol. Genet. 2007, 16, 1423–1436. [Google Scholar] [CrossRef]
- Miyoshi, T.; Maruhashi, M.; Van De Putte, T.; Kondoh, H.; Huylebroeck, D.; Higashi, Y. Complementary expression pattern of Zfhx1 genes Sip1 and δEF1 in the mouse embryo and their genetic interaction revealed by compound mutants. Dev. Dyn. 2006, 235, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Mort, R.L.; Jackson, I.J.; Elizabeth Patton, E. The melanocyte lineage in development and disease. Development 2015, 142, 620–632. [Google Scholar] [CrossRef]
- Pérez-Losada, J.; Sánchez-Martin, M.; Rodríguez-García, A.; Sánchez, M.L.; Orfao, A.; Flores, T.; Sánchez-García, I. Zinc-finger transcription factor slug contributes to the function of the stem cell factor c-kit signaling pathway. Blood 2002, 100, 1274–1286. [Google Scholar] [CrossRef]
- Sanchez-Martin, M.; Rodriguez-Garcia, A.; Perez-Losada, J.; Sagrera, A.; Read, A.P.; Sanchez-Garcia, I. SLUG (SNAI2) deletions in patients with Waardenburg disease. Hum. Mol. Genet. 2002, 11, 3231–3236. [Google Scholar] [CrossRef]
- Sánchez-Martín, M.; Pérez-Losada, J.; Rodríguez-García, A.; González-Sánchez, B.; Korf, B.R.; Kuster, W.; Moss, C.; Spritz, R.A.; Sánchez-García, I. Deletion of the SLUG (SNAI2) gene results in human piebaldism. Am. J. Med. Genet. 2003, 122A, 125–132. [Google Scholar] [CrossRef]
- Denecker, G.; Vandamme, N.; Akay, Ö.; Koludrovic, D.; Taminau, J.; Lemeire, K.; Gheldof, A.; De Craene, B.; Van Gele, M.; Brochez, L.; et al. Identification of a ZEB2-MITF-ZEB1 transcriptional network that controls melanogenesis and melanoma progression. Cell Death Differ. 2014, 21, 1250–1261. [Google Scholar] [CrossRef]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Sborne, J.; Hutchinson, P.; Tse, G.; et al. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef]
- Liu, Y.; Ye, F.; Li, Q.; Tamiya, S.; Darling, D.S.; Kaplan, H.J.; Dean, D.C. Zeb1 represses Mitf and regulates pigment synthesis, cell proliferation, and epithelial morphology. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5080–5088. [Google Scholar] [CrossRef] [PubMed]
- Karreth, F.A.; Tay, Y.; Perna, D.; Ala, U.; Tan, S.M.; Rust, A.G.; Denicola, G.; Webster, K.A.; Weiss, D.; Perez-Mancera, P.A.; et al. In vivo identification of tumor- suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell 2011, 147, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Shirley, S.H.; Greene, V.R.; Duncan, L.M.; Cabala, C.A.T.; Grimm, E.A.; Kusewitt, D.F. Slug Expression during Melanoma Progression. Am. J. Pathol. 2012, 180, 2479–2489. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, N.; Denecker, G.; Bruneel, K.; Blancke, G.; Akay, Ö.; Taminau, J.; De Coninck, J.; De Smedt, E.; Skrypek, N.; Van Loocke, W.; et al. The EMT transcription factor ZEB2 promotes proliferation of primary and metastatic melanoma while suppressing an invasive, mesenchymal-like phenotype. Cancer Res. 2020, 80, 2983–2995. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop—A motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef]
- Richard, G.; Dalle, S.; Monet, M.-A.; Ligier, M.; Boespflug, A.; Pommier, R.M.; de la Fouchardière, A.; Perier-Muzet, M.; Depaepe, L.; Barnault, R.; et al. ZEB1-mediated melanoma cell plasticity enhances resistance to MAPK inhibitors. EMBO Mol. Med. 2016, 8, 1143–1161. [Google Scholar] [CrossRef]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal Regulation of Epithelial-Mesenchymal Transition Is Essential for Squamous Cell Carcinoma Metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef]
- Ocaña, O.H.; Córcoles, R.; Fabra, Á.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic Colonization Requires the Repression of the Epithelial-Mesenchymal Transition Inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef]
- Gupta, P.B.; Kuperwasser, C.; Brunet, J.P.; Ramaswamy, S.; Kuo, W.L.; Gray, J.W.; Naber, S.P.; Weinberg, R.A. The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat. Genet. 2005, 37, 1047–1054. [Google Scholar] [CrossRef]
- Khoja, L.; Shenjere, P.; Hodgson, C.; Hodgetts, J.; Clack, G.; Hughes, A.; Lorigan, P.; Dive, C. Prevalence and Heterogeneity of Circulating Tumour Cells in Metastatic Cutaneous Melanoma. Melanoma Res. 2014, 1, 40–46. [Google Scholar] [CrossRef]
- Hong, X.; Sullivan, R.J.; Kalinich, M.; Kwan, T.T.; Giobbie-Hurder, A.; Pan, S.; LiCausi, J.A.; Milner, J.D.; Nieman, L.T.; Wittner, B.S.; et al. Molecular signatures of circulating melanoma cells for monitoring early response to immune checkpoint therapy. Proc. Natl. Acad. Sci. USA 2018, 115, 2467–2472. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Hoek, K.S.; Schlegel, N.C.; Sucker, A.; Ugurel, S.; Weber, B.L.; Katherine, L.; Phillips, D.J.; Schadendorf, D. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment. Cell Melanoma Res. 2006, 19, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Jerby-arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997. [Google Scholar] [CrossRef]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.M.; Foppen, M.H.G.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef]
- Postigo, A.A. Opposing functions of ZEB proteins in the regulation of the TGF beta/BMP signaling pathway. Embo J. 2003, 22, 2443–2452. [Google Scholar] [CrossRef]
- Restivo, G.; Diener, J.; Cheng, P.F.; Kiowski, G.; Bonalli, M.; Biedermann, T.; Reichmann, E.; Levesque, M.P.; Dummer, R.; Sommer, L. Low Neurotrophin receptor CD271 regulates phenotype switching in Melanoma. Nat. Commun. 2017, 8, 1988. [Google Scholar] [CrossRef]
- Köhler, C.; Nittner, D.; Rambow, F.; Radaelli, E.; Stanchi, F.; Vandamme, N.; Baggiolini, A.; Sommer, L.; Berx, G.; van den Oord, J.J.; et al. Mouse Cutaneous Melanoma Induced by Mutant BRaf Arises from Expansion and Dedifferentiation of Mature Pigmented Melanocytes. Cell Stem Cell 2017, 21, 679–693.e6. [Google Scholar] [CrossRef]
- Perrot, C.Y.; Gilbert, C.; Marsaud, V.; Postigo, A.; Javelaud, D.; Mauviel, A. GLI2 Cooperates With ZEB1 for Transcriptional Repression of CDH1 Expression in Human Melanoma Cells. Pigment. Cell Melanoma Res. 2013, 26, 861–873. [Google Scholar] [CrossRef]
- Ennen, M.; Keime, C.; Gambi, G.; Kieny, A.; Coassolo, S.; Thibault-carpentier, C.; Margerin-schaller, F.; Davidson, G.; Lipsker, D.; Davidson, I. MITF-high and MITF-low cells and a novel subpopulation expressing genes of both cell states contribute to intra and inter-tumoral heterogeneity of primary melanoma. Clin. cancer Res. 2017, 23, 7097–7107. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Tuncer, E.; Calçada, R.R.; Zingg, D.; Varum, S.; Cheng, P.; Freiberger, S.N.; Deng, C.X.; Kleiter, I.; Levesque, M.P.; Dummer, R.; et al. SMAD signaling promotes melanoma metastasis independently of phenotype switching. J. Clin. Investig. 2019, 129, 2702–2716. [Google Scholar] [CrossRef] [PubMed]
- Rambow, F.; Rogiers, A.; Marin-bejar, O.; Aibar, S.; Femel, J.; Dewaele, M. Toward Minimal Residual Disease-Directed Therapy in Melanoma Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19. [Google Scholar] [CrossRef]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904.e5. [Google Scholar] [CrossRef]
- Rambow, F.; Marine, J.C.; Goding, C.R. Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 2019, 33, 1295–1318. [Google Scholar] [CrossRef]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Opinion—Migrating cancer stem cells—An integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef]
- Lito, P.; Rosen, N.; Solit, D.B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013, 19, 1401–1409. [Google Scholar] [CrossRef]
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015, 162, 1271–1285. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Boussemart, L.; Malka-Mahieu, H.; Girault, I.; Allard, D.; Hemmingsson, O.; Tomasic, G.; Thomas, M.; Basmadjian, C.; Ribeiro, N.; Thuaud, F.; et al. eIF4F is a nexus of resistance to anti-BRAF and anti-MEK cancer therapies. Nature 2014, 513, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Ohanna, M.; Cerezo, M.; Nottet, N.; Bille, K.; Didier, R.; Nottet, N.; Beranger, G.; Mograbi, B.; Rocchi, S.; Yvan-charvet, L.; et al. Pivotal role of NAMPT in the switch of melanoma cells toward an invasive and drug-resistant phenotype. Genes Dev. 2018, 32, 448–461. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A Gene Expression Signature Associated with “K-Ras Addiction” Reveals Regulators of EMT and Tumor Cell Survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef]
- Queirolo, P.; Boutros, A.; Tanda, E.; Spagnolo, F.; Quaglino, P. Immune-checkpoint inhibitors for the treatment of metastatic melanoma: A model of cancer immunotherapy. Semin. Cancer Biol. 2019, 59, 290–297. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wölfel, T.; Hölzel, M.; et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Abril-Rodriguez, G.; Torrejon, D.Y.; Liu, W.; Zaretsky, J.M.; Nowicki, T.S.; Tsoi, J.; Puig-Saus, C.; Baselga-Carretero, I.; Medina, E.; Quist, M.J.; et al. PAK4 inhibition improves PD-1 blockade immunotherapy. Nat. Cancer 2020, 1, 46–58. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to T cell–mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Kim, Y.J.; Robert, L.; Tsoi, J.; Comin-Anduix, B.; Berent-Maoz, B.; Cochran, A.J.; Economou, J.S.; Tumeh, P.C.; Puig-Saus, C.; et al. Immunotherapy resistance by inflammation-induced dedifferentiation. Cancer Discov. 2018, 8, 935–943. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.-H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer Metastasis Is Accelerated through Immunosuppression during Snail-Induced EMT of Cancer Cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Meidhof, S.; Brabletz, S.; Lehmann, W.; Preca, B.-T.; Mock, K.; Ruh, M.; Schüler, J.; Berthold, M.; Weber, A.; Burk, U.; et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. 2015, 7, 831–847. [Google Scholar] [CrossRef]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I study of GC1008 (Fresolimumab): A human anti-transforming growth factor-beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef] [PubMed]
- Boshuizen, J.; Koopman, L.A.; Krijgsman, O.; Shahrabi, A.; Van Den Heuvel, E.G.; Ligtenberg, M.A.; Vredevoogd, D.W.; Kemper, K.; Kuilman, T.; Song, J.; et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF / MEK inhibitors. Nat. Med. 2018, 24, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Woods, D.M.; Sodré, A.L.; Villagra, A.; Sarnaik, A.; Sotomayor, E.M.; Weber, J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol. Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Maheswaran, S.; Mcdermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; Wong, K.; et al. A chromatin-mediated reversible drug tolerant state in cancer cell subpopulations. Cell 2011, 141, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; De Oliveira, R.L.; Huijberts, S.; Beijnen, J.H.; Schellens, J.H.M.; Bernards, R. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell 2018, 173, 1413–1425. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, V.; Brabletz, T.; Ceppi, P. Targeting EMT in Cancer with Repurposed Metabolic Inhibitors. Trends Cancer 2020. [Google Scholar] [CrossRef]
- Davila-Gonz alez, D.; Choi, D.S.; Rosato, R.R.; Granados-Principal, S.M.; Kuhn, J.G.; Li, W.F.; Qian, W.; Chen, W.; Kozielski, A.J.; Wong, H.; et al. Pharmacological inhibition of NOS activates ASK1/JNK pathway augmenting docetaxel-mediated apoptosis in triple-negative breast cancer. Clin. Cancer Res. 2018, 24, 1152–1162. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Saez-Ayala, M.; Montenegro, M.F.; Sanchez-del-Campo, L.; Fernandez-Perez, M.P.; Chazarra, S.; Freter, R.; Middleton, M.; Pinero-Madrona, A.; Cabezas-Herrera, J.; Goding, C.R.; et al. Directed Phenotype Switching as an Effective Antimelanoma Strategy. Cancer Cell 2013, 24, 105–119. [Google Scholar] [CrossRef]
- Skrypek, N.; Goossens, S.; De Smedt, E.; Vandamme, N.; Berx, G. Epithelial-to-Mesenchymal Transition: Epigenetic Reprogramming Driving Cellular Plasticity. Trends Genet. 2017, 33, 943–959. [Google Scholar] [CrossRef]
- Zingg, D.; Arenas-ramirez, N.; Sahin, D.; Haeusel, J.; Sommer, L.; Boyman, O.; Zingg, D.; Arenas-ramirez, N.; Sahin, D.; Rosalia, R.A.; et al. The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Article The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. Cell Rep. 2017, 20, 854–867. [Google Scholar] [CrossRef] [PubMed]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours—Past lessons and future promise. Nat. Rev. Clin. Oncol. 2020, 17, 91–107. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Strategy | References | Target | Drug/Compound | Clinical Trial | Trial ID | Cancer Type |
|---|---|---|---|---|---|---|
| TGFβ pathway inhibitors | [91] | TGFβ monoclonal antibody | GC1008 (fresolimumab) | Phase I | NCT00356460 | Renal Cell Carcinoma and Malignant Melanoma |
| AXL targeting | [92] | AXL small molecule inhibitor | BGB324 | Phase I/II in combination with either dabrafenib/trametinib or pembrolizumab | NCT02872259 | Metastatic Melanoma |
| Epigenetic drugs | [93,94] | HDAC inhibitors | Entinostat | Phase II in combination with erlotinib | NCT00602030 | Non–Small-Cell Lung Cancer |
| [95] | Vorinostat | Phase I | NCT02836548 | Resistant BRAFV600 melanoma | ||
| Metabolic inhibitors | [96,97] | Inducible nitric oxide synthase (iNOS) | L-NMMA | Phase Ib/2 in combination with Taxane | NCT02834403 | Triple Negative Breast Cancer |
| Ferroptosis inductors | [65,98] | GPX4 (through gluthatione depletion) | PRLX 93936 (Erastin analog) | Phase I | NCT00528047 | Advanced Solid Tumors |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, Y.; Durand, S.; Dalle, S.; Caramel, J. EMT-Inducing Transcription Factors, Drivers of Melanoma Phenotype Switching, and Resistance to Treatment. Cancers 2020, 12, 2154. https://doi.org/10.3390/cancers12082154
Tang Y, Durand S, Dalle S, Caramel J. EMT-Inducing Transcription Factors, Drivers of Melanoma Phenotype Switching, and Resistance to Treatment. Cancers. 2020; 12(8):2154. https://doi.org/10.3390/cancers12082154
Chicago/Turabian StyleTang, Yaqi, Simon Durand, Stéphane Dalle, and Julie Caramel. 2020. "EMT-Inducing Transcription Factors, Drivers of Melanoma Phenotype Switching, and Resistance to Treatment" Cancers 12, no. 8: 2154. https://doi.org/10.3390/cancers12082154
APA StyleTang, Y., Durand, S., Dalle, S., & Caramel, J. (2020). EMT-Inducing Transcription Factors, Drivers of Melanoma Phenotype Switching, and Resistance to Treatment. Cancers, 12(8), 2154. https://doi.org/10.3390/cancers12082154