Germline Genetic Findings Which May Impact Therapeutic Decisions in Families with a Presumed Predisposition for Hereditary Breast and Ovarian Cancer

 , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Results
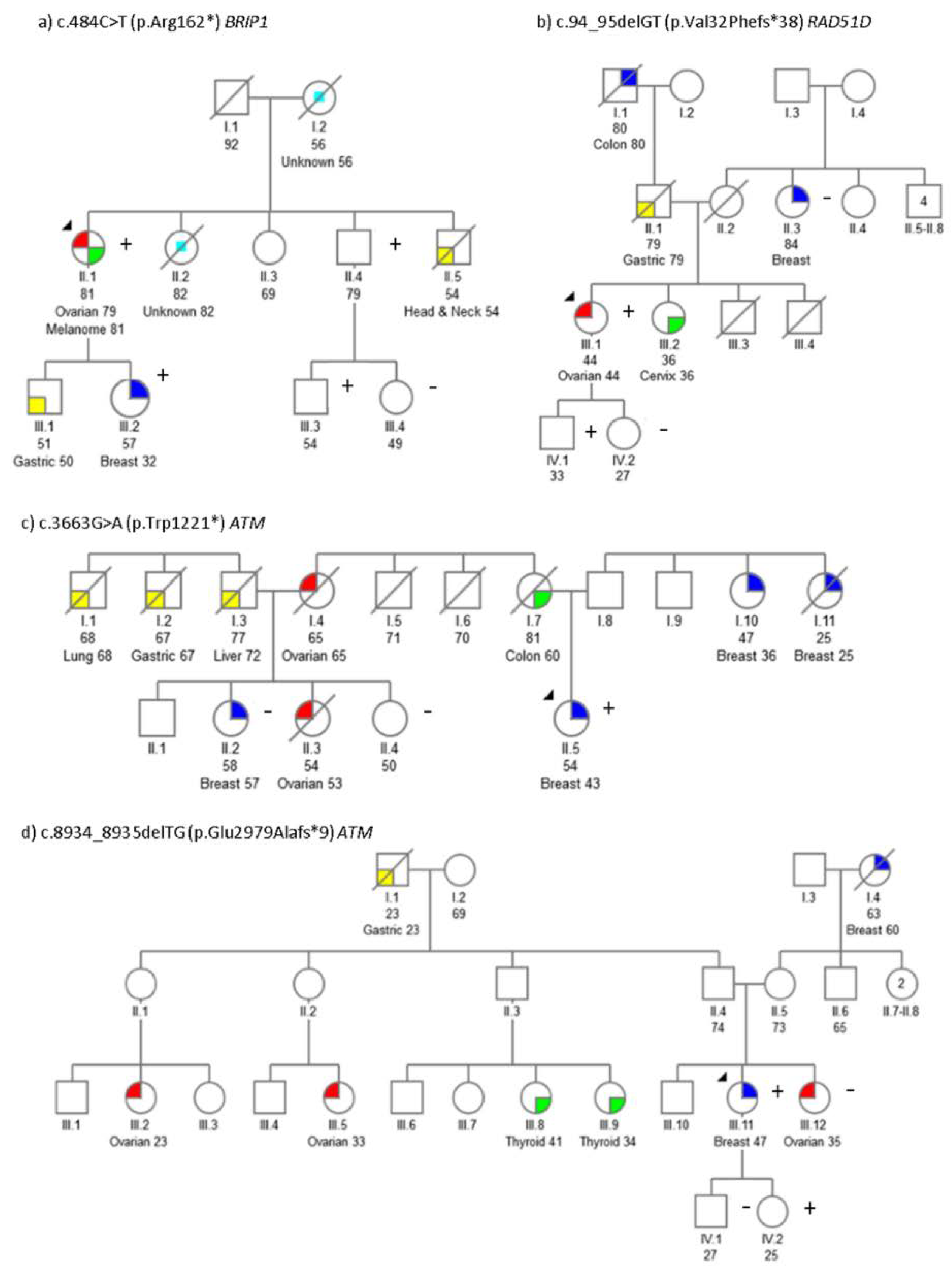
2.1. Mutation Screening
2.2. Segregation Analysis
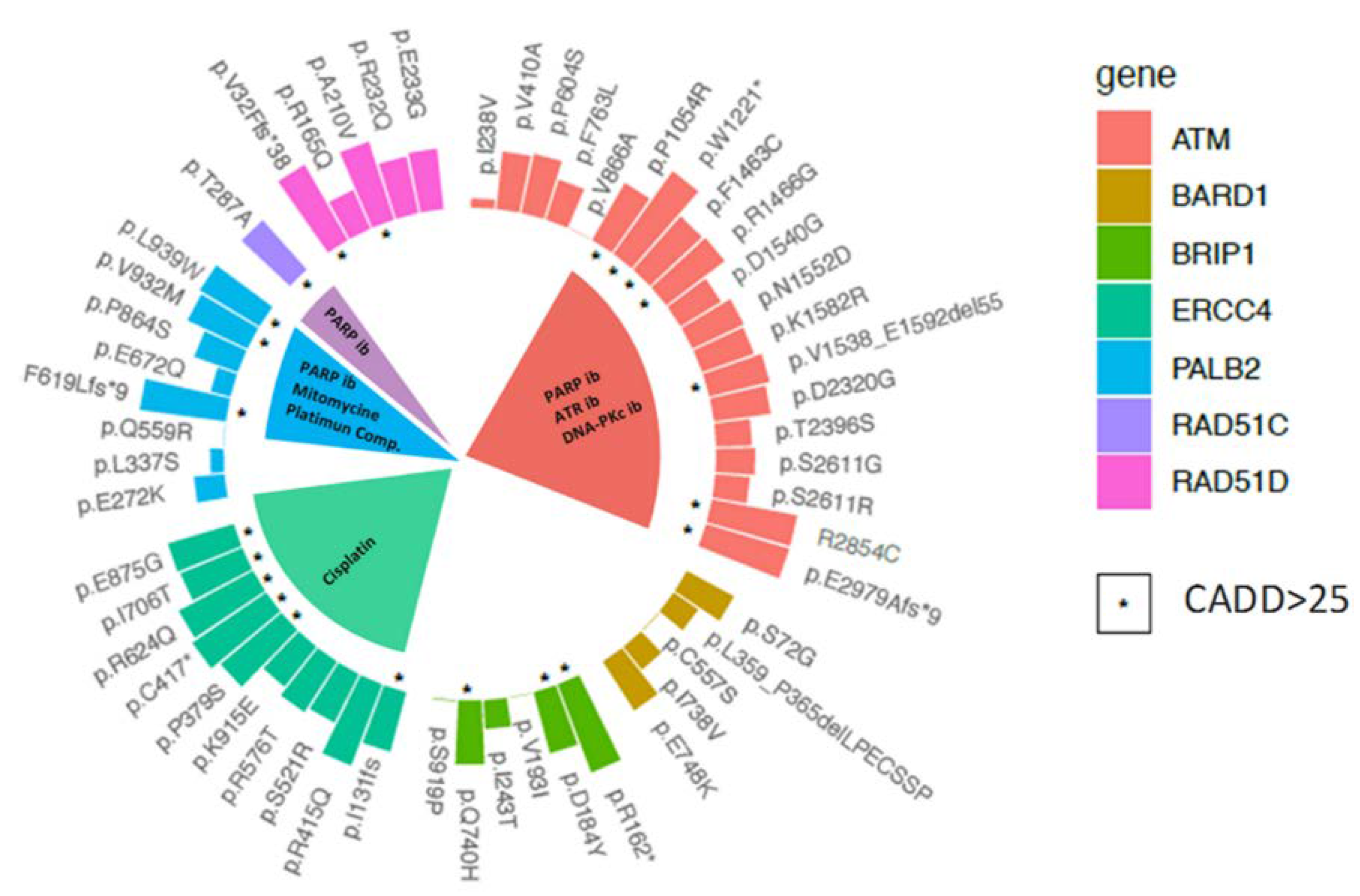
2.3. Variant Prioritisation
2.4. Therapeutic Implications
2.5. Genotype-Phenotypic Correlations
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. DNA and RNA Extraction
4.3. Mutation Screening
4.4. Sanger Sequencing
4.5. RT-PCR
4.6. In-Silico Analyses
4.7. Variant Classification
4.8. Genotype-Phenotypic Correlations and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Simon, B.; Zeichner, D.O. Prevention and Screening in Hereditary Breast and Ovarian Cancer. Available online: https://www.cancernetwork.com/review-article/prevention-and-screening-hereditary-breast-and-ovarian-cancer (accessed on 8 May 2020).
- NCCN. Clinical Practice Guidelines in OncologyTM (2019). Available online: https://www.nccn.org/professionals/physician_gls/recently_updated.aspx (accessed on 15 March 2019).
- González-Santiago, S.; Ramón y Cajal, T.; Aguirre, E.; Alés-Martínez, J.E.; Andrés, R.; Balmaña, J.; Graña, B.; Herrero, A.; Llort, G.; González-Del-Alba, A. SEOM clinical guidelines in hereditary breast and ovarian cancer (2019). Clin. Transl. Oncol. 2020, 22, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Garber, J.E.; Offit, K. Hereditary Cancer Predisposition Syndromes. J. Clin. Oncol. 2016, 23. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Ohno, S.; Sasaki, Y.; Matsuura, M. Hereditary breast and ovarian cancer susceptibility genes (review). Oncol. Rep. 2013, 30, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Toss, A.; Tomasello, C.; Razzaboni, E.; Contu, G.; Grandi, G.; Cagnacci, A.; Schilder, R.J.; Cortesi, L. Hereditary ovarian cancer: Not only BRCA 1 and 2 genes. Biomed. Res. Int. 2015, 2015, 341723. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef]
- Seifert, B.A.; O’Daniel, J.M.; Amin, K.; Marchuk, D.S.; Patel, N.M.; Parker, J.S.; Hoyle, A.P.; Mose, L.E.; Marron, A.; Hayward, M.C.; et al. Germline Analysis from Tumor-Germline Sequencing Dyads to Identify Clinically Actionable Secondary Findings. Clin. Cancer Res. 2016, 22, 4087–4094. [Google Scholar] [CrossRef]
- Velázquez, C.; Esteban-Cardeñosa, E.M.; Lastra, E.; Abella, L.E.; de la Cruz, V.; Lobatón, C.D.; Durán, M.; Infante, M. A PALB2 truncating mutation: Implication in cancer prevention and therapy of Hereditary Breast and Ovarian Cancer. Breast 2019, 43, 91–96. [Google Scholar] [CrossRef]
- Damiola, F.; Schultz, I.; Barjhoux, L.; Sornin, V.; Dondon, M.-G.; Eon-Marchais, S.; Marcou, M.; Investigators, T.G.S.; Caron, O.; Gauthier-Villars, M.; et al. Mutation analysis of PALB2 gene in French breast cancer families. Breast Cancer Res. Treat. 2015, 154, 463–471. [Google Scholar] [CrossRef]
- Osorio, A.; Bogliolo, M.; Fernández, V.; Barroso, A.; de la Hoya, M.; Caldés, T.; Lasa, A.; Ramón y Cajal, T.; Santamariña, M.; Vega, A.; et al. Evaluation of rare variants in the new fanconi anemia gene ERCC4 (FANCQ) as familial breast/ovarian cancer susceptibility alleles. Hum. Mutat. 2013, 34, 1615–1618. [Google Scholar] [CrossRef]
- Gilad, S.; Chessa, L.; Khosravi, R.; Russell, P.; Galanty, Y.; Piane, M.; Gatti, R.A.; Jorgensen, T.J.; Shiloh, Y.; Bar-Shira, A. Genotype-Phenotype relationships in ataxia-telangiectasia and variants. Am. J. Hum. Genet. 1998, 62, 551–561. [Google Scholar] [CrossRef]
- Velasco, E.; Infante, M.; Durán, M.; Pérez-Cabornero, L.; Sanz, D.J.; Esteban-Cardeñosa, E.; Miner, C. Heteroduplex analysis by capillary array electrophoresis for rapid mutation detection in large multiexon genes. Nat. Protocols 2007, 2, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Aloraifi, F.; McDevitt, T.; Martiniano, R.; McGreevy, J.; McLaughlin, R.; Egan, C.M.; Cody, N.; Meany, M.; Kenny, E.; Green, A.J.; et al. Detection of novel germline mutations for breast cancer in non-BRCA1/2 families. FEBS J. 2015, 282, 3424–3437. [Google Scholar] [CrossRef] [PubMed]
- Tavera-Tapia, A.; Pérez-Cabornero, L.; Macías, J.A.; Ceballos, M.I.; Roncador, G.; de la Hoya, M.; Barroso, A.; Felipe-Ponce, V.; Serrano-Blanch, R.; Hinojo, C.; et al. Almost 2% of Spanish breast cancer families are associated to germline pathogenic mutations in the ATM gene. Breast Cancer Res. Treat. 2017, 161, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Minion, L.E.; Dolinsky, J.S.; Chase, D.M.; Dunlop, C.L.; Chao, E.C.; Monk, B.J. Hereditary predisposition to ovarian cancer, looking beyond BRCA1/BRCA2. Gynecol. Oncol. 2015, 137, 86–92. [Google Scholar] [CrossRef]
- Carranza, D.; Vega, A.K.; Torres-Rusillo, S.; Montero, E.; Martinez, L.J.; Santamaría, M.; Santos, J.L.; Molina, I.J. Molecular and Functional Characterisation of a Cohort of Spanish Patients with Ataxia-Telangiectasia. Neuromol. Med. 2017, 19, 161–174. [Google Scholar] [CrossRef]
- Gu, Y.; Yu, Y.; Ai, L.; Shi, J.; Liu, X.; Sun, H.; Liu, Y. Association of the ATM gene polymorphisms with papillary thyroid cancer. Endocrine 2014, 45, 454–461. [Google Scholar] [CrossRef]
- Graña, B.; Fachal, L.; Darder, E.; Balmaña, J.; Ramón Y Cajal, T.; Blanco, I.; Torres, A.; Lázaro, C.; Diez, O.; Alonso, C.; et al. Germline ATM mutational analysis in BRCA1/BRCA2 negative hereditary breast cancer families by MALDI-TOF mass spectrometry. Breast Cancer Res. Treat. 2011, 128, 573–579. [Google Scholar] [CrossRef]
- Helgason, H.; Rafnar, T.; Olafsdottir, H.S.; Jonasson, J.G.; Sigurdsson, A.; Stacey, S.N.; Jonasdottir, A.; Tryggvadottir, L.; Alexiusdottir, K.; Haraldsson, A.; et al. Loss-Of-Function variants in ATM confer risk of gastric cancer. Nat. Genet. 2015, 47, 906–910. [Google Scholar] [CrossRef]
- Roberts, N.J.; Jiao, Y.; Yu, J.; Kopelovich, L.; Petersen, G.M.; Bondy, M.L.; Gallinger, S.; Schwartz, A.G.; Syngal, S.; Cote, M.L.; et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Dis. 2012, 2, 41–46. [Google Scholar] [CrossRef]
- AlDubayan, S.H.; Giannakis, M.; Moore, N.D.; Han, G.C.; Reardon, B.; Hamada, T.; Mu, X.J.; Nishihara, R.; Qian, Z.; Liu, L.; et al. Inherited DNA-Repair Defects in Colorectal Cancer. Am. J. Hum. Genet. 2018, 102, 401–414. [Google Scholar] [CrossRef]
- Suszynska, M.; Klonowska, K.; Jasinska, A.J.; Kozlowski, P. Large-Scale meta-analysis of mutations identified in panels of breast/ovarian cancer-related genes—Providing evidence of cancer predisposition genes. Gynecol. Oncol. 2019, 153, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.M.; Weiss, N.S.; Ulrich, C.M.; Doherty, J.A.; Voigt, L.F.; Chen, C. Interindividual variation in nucleotide excision repair genes and risk of endometrial cancer. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2524–2530. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, M.; Yang, J.-X.; Cao, D.-Y.; Zhang, Y.; Zhou, H.-M.; Yuan, Z.; Shen, K. Genomic Comparison of Endometrioid Endometrial Carcinoma and its Precancerous Lesions in Chinese Patients by High-Depth Next Generation Sequencing. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Ow, S.G.W.; Tan, K.T.; Yang, H.; Yap, H.-L.; Sapari, N.S.B.; Ong, P.Y.; Soong, R.; Lee, S.-C. Next Generation Sequencing Reveals Novel Mutations in Mismatch Repair Genes and Other Cancer Predisposition Genes in Asian Patients with Suspected Lynch Syndrome. Clin. Colorectal. Cancer 2019, 18, e324–e334. [Google Scholar] [CrossRef]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-Perry, M.; Snape, K.; et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef]
- Thompson, E.R.; Rowley, S.M.; Sawyer, S.; ConFab, K.; Eccles, D.M.; Trainer, A.H.; Mitchell, G.; James, P.A.; Campbell, I.G. Analysis of RAD51D in Ovarian Cancer Patients and Families with a History of Ovarian or Breast Cancer. PLoS ONE 2013, 8, e54772. [Google Scholar] [CrossRef]
- Song, H.; Dicks, E.; Ramus, S.J.; Tyrer, J.P.; Intermaggio, M.P.; Hayward, J.; Edlund, C.K.; Conti, D.; Harrington, P.; Fraser, L.; et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J. Clin. Oncol. 2015, 33, 2901–2907. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Paluch-Shimon, S.; Cardoso, F.; Sessa, C.; Balmana, J.; Cardoso, M.J.; Gilbert, F.; Senkus, E. ESMO Guidelines Committee Prevention and screening in BRCA mutation carriers and other breast/ovarian hereditary cancer syndromes: ESMO Clinical Practice Guidelines for cancer prevention and screening. Ann. Oncol. 2016, 27, v103–v110. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucl. Acid. Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- van der Velde, K.J.; Kuiper, J.; Thompson, B.A.; Plazzer, J.-P.; van Valkenhoef, G.; de Haan, M.; Jongbloed, J.D.H.; Wijmenga, C.; de Koning, T.J.; Abbott, K.M.; et al. Evaluation of CADD Scores in Curated Mismatch Repair Gene Variants Yields a Model for Clinical Validation and Prioritisation. Hum. Mutat. 2015, 36, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, H.; Mochizuki, H.; Inoue, M.; Hirotsu, Y.; Amemiya, K.; Sakamoto, I.; Nakagomi, S.; Kubota, T.; Omata, M. Combined annotation-dependent depletion score for BRCA1/2 variants in patients with breast and/or ovarian cancer. Cancer Sci. 2018, 109, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Hintzsche, J.; Kim, J.; Yadav, V.; Amato, C.; Robinson, S.E.; Seelenfreund, E.; Shellman, Y.; Wisell, J.; Applegate, A.; McCarter, M.; et al. IMPACT: A whole-exome sequencing analysis pipeline for integrating molecular profiles with actionable therapeutics in clinical samples. J. Am. Med. Inf. Assoc. 2016, 23, 721–730. [Google Scholar] [CrossRef][Green Version]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Smith, M.A.; Hampton, O.A.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Gorlick, R.; Kolb, E.A.; Lock, R.; Carol, H.; Keir, S.T.; et al. Initial Testing (Stage 1) of the PARP Inhibitor BMN 673 by the Pediatric Preclinical Testing Program: PALB2 Mutation Predicts Exceptional in Vivo Response to BMN 673. Pediatra Blood Cancer 2015, 62, 91–98. [Google Scholar] [CrossRef]
- Villarroel, M.C.; Rajeshkumar, N.V.; Garrido-Laguna, I.; De Jesus-Acosta, A.; Jones, S.; Maitra, A.; Hruban, R.H.; Eshleman, J.R.; Klein, A.; Laheru, D.; et al. Personalising cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol. Cancer Ther. 2011, 10, 3–8. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef]
- Ceccaldi, R.; O’Connor, K.W.; Mouw, K.W.; Li, A.Y.; Matulonis, U.A.; D’Andrea, A.D.; Konstantinopoulos, P.A. A unique subset of epithelial ovarian cancers with platinum sensitivity and PARP inhibitor resistance. Cancer Res. 2015, 75, 628–634. [Google Scholar] [CrossRef]
- McNeish, I.A.; Oza, A.M.; Coleman, R.L.; Scott, C.L.; Konecny, G.E.; Tinker, A.; O’Malley, D.M.; Brenton, J.; Kristeleit, R.S.; Bell-McGuinn, K.; et al. Results of ARIEL2: A Phase 2 trial to prospectively identify ovarian cancer patients likely to respond to rucaparib using tumor genetic analysis. JCO 2015, 33, 5508. [Google Scholar] [CrossRef]
- Plimack, E.R.; Dunbrack, R.L.; Brennan, T.A.; Andrake, M.D.; Zhou, Y.; Serebriiskii, I.G.; Slifker, M.; Alpaugh, K.; Dulaimi, E.; Palma, N.; et al. Defects in DNA Repair Genes Predict Response to Neoadjuvant Cisplatin-based Chemotherapy in Muscle-invasive Bladder Cancer. Eur. Urol. 2015, 68, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Menezes, D.L.; Holt, J.; Tang, Y.; Feng, J.; Barsanti, P.; Pan, Y.; Ghoddusi, M.; Zhang, W.; Thomas, G.; Holash, J.; et al. A synthetic lethal screen reveals enhanced sensitivity to ATR inhibitor treatment in mantle cell lymphoma with ATM loss-of-function. Mol. Cancer Res. 2015, 13, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Knittel, G.; Welcker, D.; Yang, T.-P.; George, J.; Nowak, M.; Leeser, U.; Büttner, R.; Perner, S.; Peifer, M.; et al. ATM Deficiency is Associated with Sensitivity to PARP1 and ATR Inhibitors in Lung Adenocarcinoma. Cancer Res. 2017, 77, 3040–3056. [Google Scholar] [CrossRef] [PubMed]
- Riabinska, A.; Daheim, M.; Herter-Sprie, G.S.; Winkler, J.; Fritz, C.; Hallek, M.; Thomas, R.K.; Kreuzer, K.-A.; Frenzel, L.P.; Monfared, P.; et al. Therapeutic targeting of a robust non-oncogene addiction to PRKDC in ATM-defective tumors. Sci. Transl. Med. 2013, 5, 189ra78. [Google Scholar] [CrossRef]
- Berger, N.A. Young Adult Cancer: Influence of the Obesity Pandemic. Obesity 2018, 26, 641–650. [Google Scholar] [CrossRef]
- Assi, H.A.; Khoury, K.E.; Dbouk, H.; Khalil, L.E.; Mouhieddine, T.H.; El Saghir, N.S. Epidemiology and prognosis of breast cancer in young women. J. Thorac. Dis. 2013, 5. [Google Scholar] [CrossRef]
- Hampel, H.; Sweet, K.; Westman, J.A.; Offit, K.; Eng, C. Referral for cancer genetics consultation: A review and compilation of risk assessment criteria. J. Med. Genet. 2004, 41, 81. [Google Scholar] [CrossRef]
- Dwight, Z.; Palais, R.; Wittwer, C.T. uMELT: Prediction of high-resolution melting curves and dynamic melting profiles of PCR products in a rich web application. Bioinformatics 2011, 27, 1019–1020. [Google Scholar] [CrossRef]
- Leeneer, K.D.; Hellemans, J.; Steyaert, W.; Lefever, S.; Vereecke, I.; Debals, E.; Crombez, B.; Baetens, M.; Heetvelde, M.V.; Coppieters, F.; et al. Flexible, Scalable, and Efficient Targeted Resequencing on a Benchtop Sequencer for Variant Detection in Clinical Practice. Hum. Mutat. 2015, 36, 379–387. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucl. Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Nono, A.D.; Chen, K.; Liu, X. Comparison of different functional prediction scores using a gene-based permutation model for identifying cancer driver genes. BMC Med. Genom. 2019, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant graphics for data analysis. In Use R! Springer: New York, NY, USA, 2009. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | c.DNA Change | Protein Change | ACMG Rules | Proband Cancer Type (Age) | Family History Cancer Type (Age) | ||
|---|---|---|---|---|---|---|---|
| First-Degree-Relatives | Second-Degree-Relatives | Third-Degree-Relatives | |||||
| ATM | c.3663G>A | p.Trp1221 * | PVS1, PM2 | Br (43) | Col (50) | Br (36), Br (25), Ov (62) | Br (57), Ov (53) |
| ATM | c.4776+2T>C | - | PVS1, PM2 | Br (52) Ov (60) | Pan (29), Pan (58), Br (47), Col (67), Col + Br (66,74) | CUP (78) | Gas (58), Gas (65), Gas (70), Gas (72), Br + Col (66,74) |
| ATM | c.8934_8935delTG | p.Glu2979Alafs*9 | PVS1, PM2 | Br (47) | Ov (35) | Gas (23), Br (60) | Ov (23), Ov (33), Thy (34), Thy (41) |
| BRIP1 | c.484C>T | p.Arg162 * | PVS1, PM1, PM2 | Ov (79)Mel (81) | Gas (50), Br (32), H&N (54), CUP (82), CUP (56) | ||
| ERCC4 | c.1251T>A | p.Cys417 * | PVS1, PM1, PM2 | BilBr (59-59) | End (47), End (51), End + Col (57,59), Gas (80) | Br (70), Gas (75) | Br (72) |
| ERCC4 | c.584+1G >A | - | PVS1, PM2 | Br (53) | Br (55) | Col (45), Br (54), Br (33), Br (57), Pr (84) | Col (36), Ute (47), Br (53), Br (55) |
| PALB2 | c.1857delT | p.Phe619Leufs*9 | PVS1, PM1, PM2 | Ov (33) | Br (36), Liv (78) | End (55), Lun (62) | Leu (n.a) |
| RAD51D | c.94_95delGT | p.Val32Phefs*38 | PVS1, PM1, PM2 | Ov (44) | Cvx (36), Gas (79) | BilBr (84) | Col (80) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velázquez, C.; K., D.L.; Esteban-Cardeñosa, E.M.; Avila Cobos, F.; Lastra, E.; Abella, L.E.; de la Cruz, V.; Lobatón, C.D.; Claes, K.B.; Durán, M.; et al. Germline Genetic Findings Which May Impact Therapeutic Decisions in Families with a Presumed Predisposition for Hereditary Breast and Ovarian Cancer. Cancers 2020, 12, 2151. https://doi.org/10.3390/cancers12082151
Velázquez C, K. DL, Esteban-Cardeñosa EM, Avila Cobos F, Lastra E, Abella LE, de la Cruz V, Lobatón CD, Claes KB, Durán M, et al. Germline Genetic Findings Which May Impact Therapeutic Decisions in Families with a Presumed Predisposition for Hereditary Breast and Ovarian Cancer. Cancers. 2020; 12(8):2151. https://doi.org/10.3390/cancers12082151
Chicago/Turabian StyleVelázquez, Carolina, De Leeneer K., Eva M. Esteban-Cardeñosa, Francisco Avila Cobos, Enrique Lastra, Luis E. Abella, Virginia de la Cruz, Carmen D. Lobatón, Kathleen B. Claes, Mercedes Durán, and et al. 2020. "Germline Genetic Findings Which May Impact Therapeutic Decisions in Families with a Presumed Predisposition for Hereditary Breast and Ovarian Cancer" Cancers 12, no. 8: 2151. https://doi.org/10.3390/cancers12082151
APA StyleVelázquez, C., K., D. L., Esteban-Cardeñosa, E. M., Avila Cobos, F., Lastra, E., Abella, L. E., de la Cruz, V., Lobatón, C. D., Claes, K. B., Durán, M., & Infante, M. (2020). Germline Genetic Findings Which May Impact Therapeutic Decisions in Families with a Presumed Predisposition for Hereditary Breast and Ovarian Cancer. Cancers, 12(8), 2151. https://doi.org/10.3390/cancers12082151