TIF1 Proteins in Genome Stability and Cancer


Abstract
1. Introduction
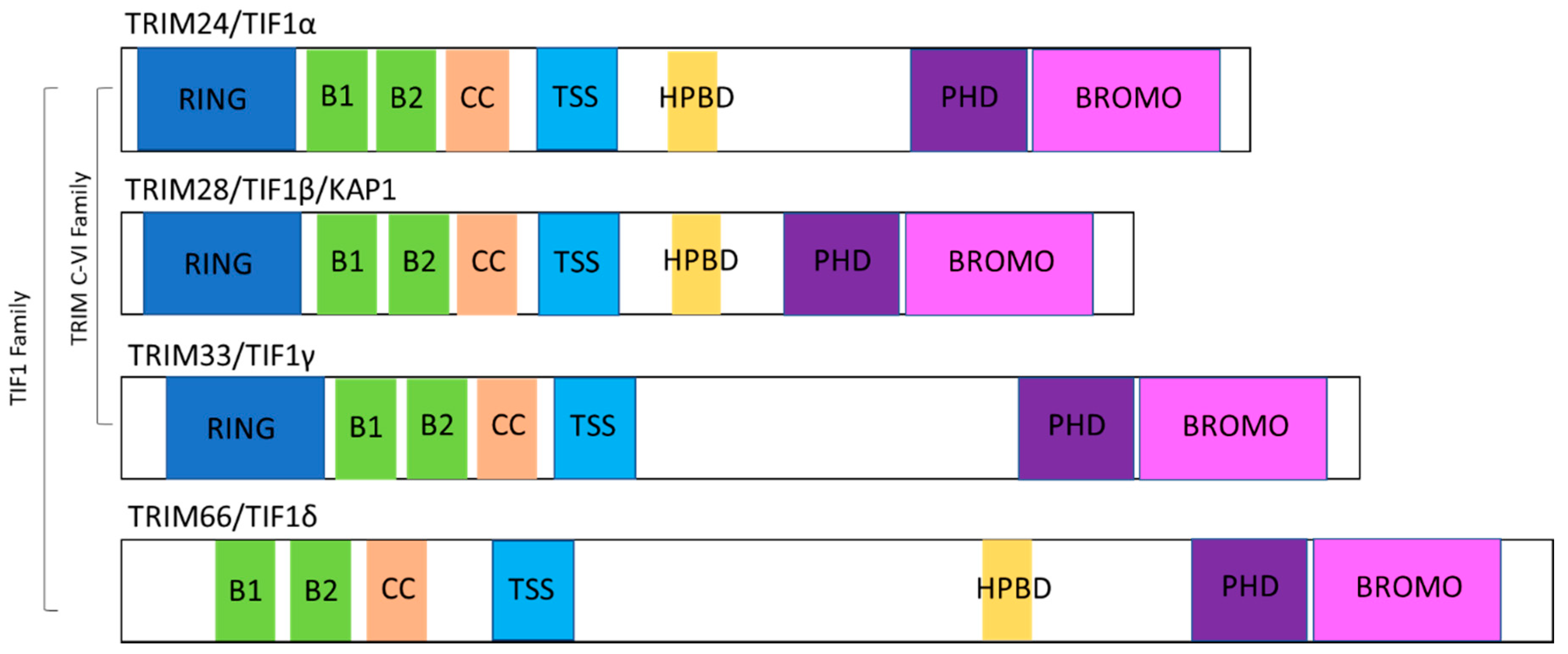
2. TIF1 Protein Family Overview
3. TIF1 Family Alterations in Cancer
3.1. TRIM24
3.2. TRIM28
3.3. TRIM33
3.4. TRIM66
4. TIF1 Proteins and DNA Damage Repair
4.1. Localisation of TIF1 Proteins to Sites of DNA Damage
4.2. Phosphorylation of TRIM24 and TRIM28
5. TIF1 Proteins, Histone Modifications and Chromatin Remodelling
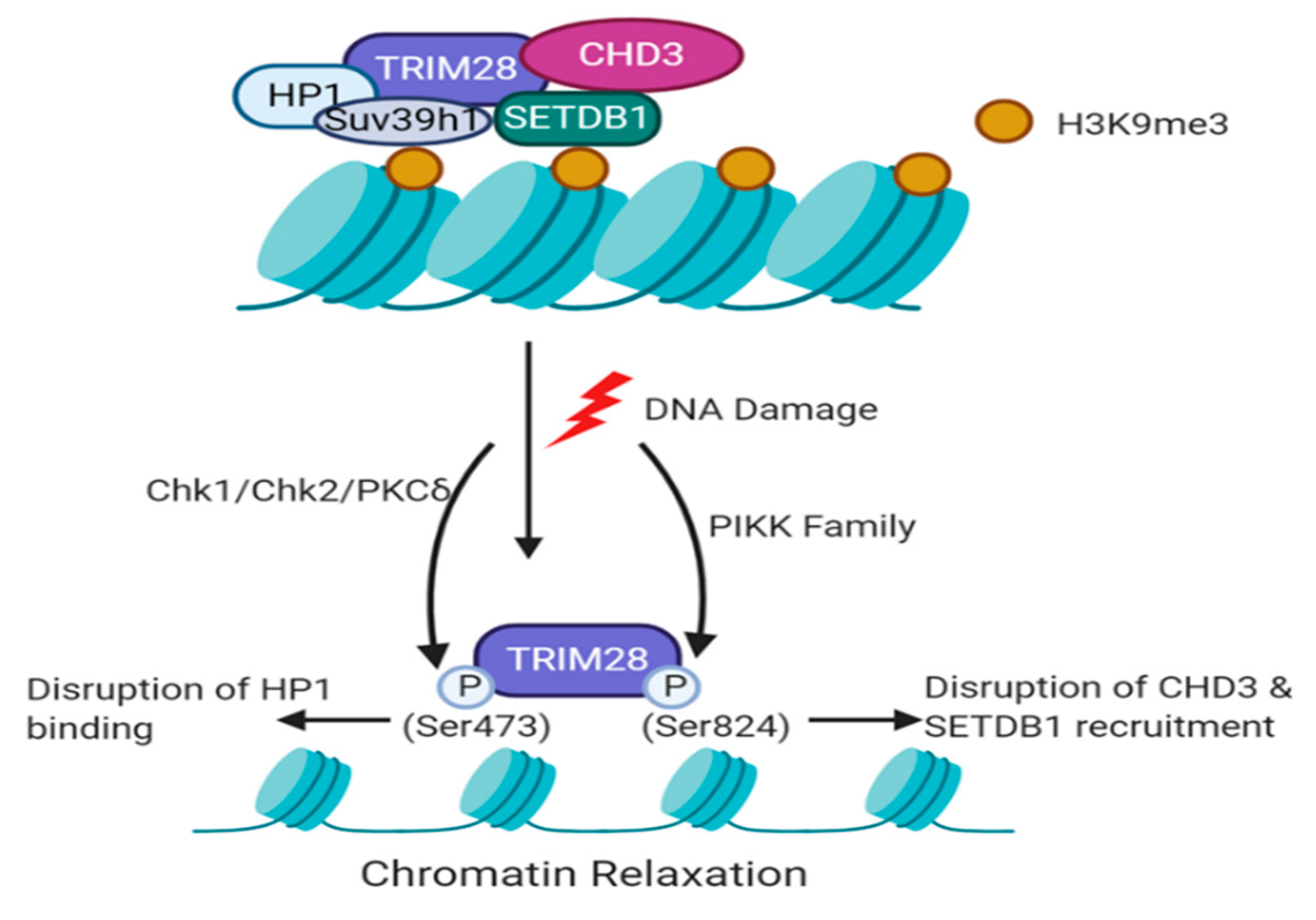
5.1. TRIM28-Dependent Recruitment of Histone Modifiers during the DDR
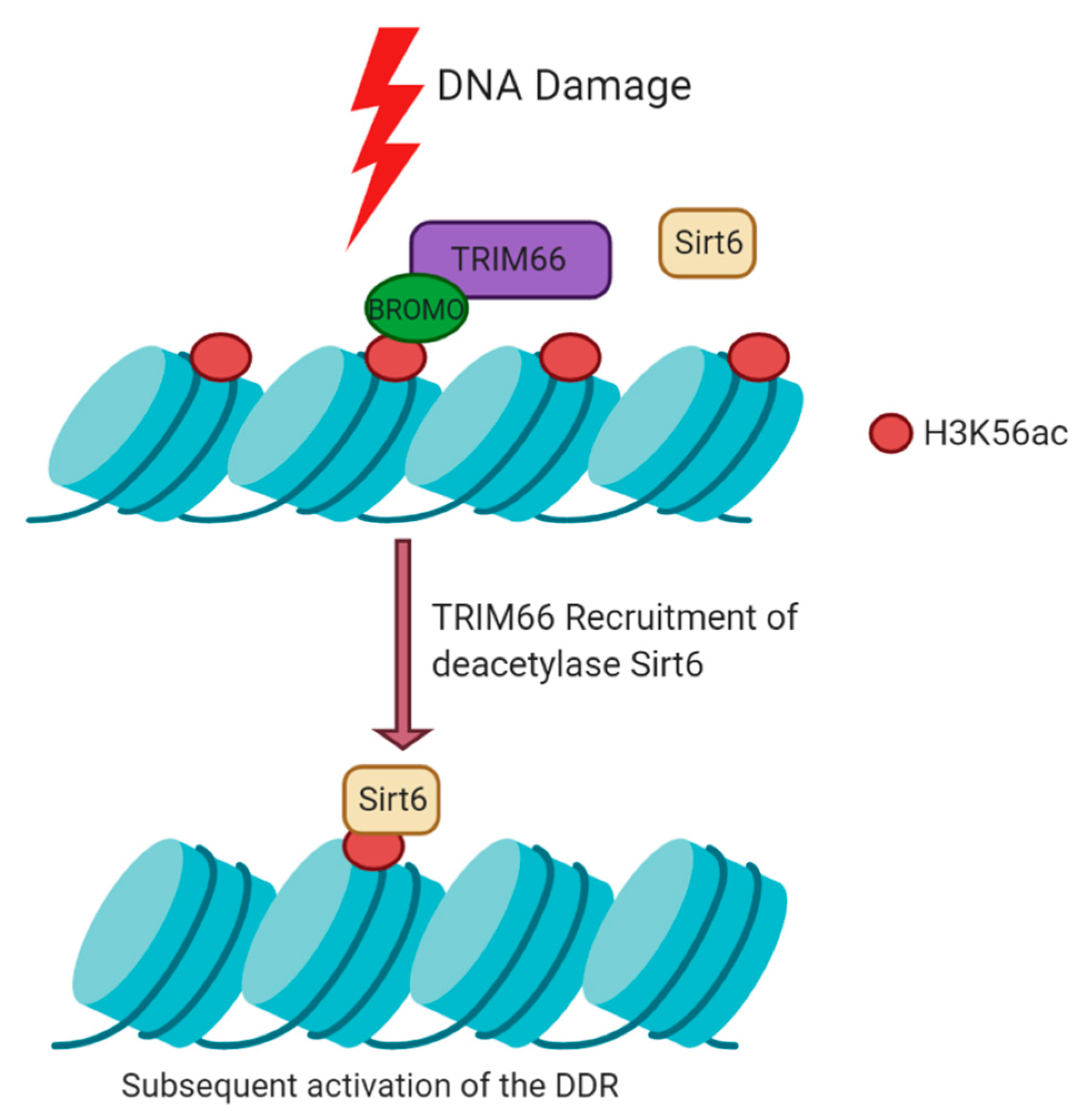
5.2. Histone Modifications Mediated by TRIM66 at DSBs
5.3. TRIM24 and TRIM33 as Histone Readers
6. TIF1 Proteins and Regulation of the Cell Cycle
6.1. G1 to S-Phase Transition
6.2. Regulation of p53
6.3. TRIM33 Regulation of Mitotic Checkpoints
7. Potential Therapeutic Exploitation of TIF1 Proteins in Cancer
8. Conclusions and Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2010, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Gallouet, A.-S.; Ferri, F.; Petit, V.; Parcelier, A.; Lewandowski, D.; Gault, N.; Barroca, V.; Le Gras, S.; Soler, E.; Grosveld, F.; et al. Macrophage production and activation are dependent on TRIM33. Oncotarget 2017, 8, 5111–5122. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Appikonda, S.; Thakkar, K.N.; Barton, M.C. Regulation of gene expression in human cancers by TRIM24. Drug Discov. Today Technol. 2016, 19, 57–63. [Google Scholar] [CrossRef]
- Yu, C.; Ding, Z.; Liang, H.; Zhang, B.; Chen, X. The Roles of TIF1γ in Cancer. Front. Oncol. 2019, 9, 979. [Google Scholar] [CrossRef]
- Cheng, C.-T. KAPtain in charge of multiple missions: Emerging roles of KAP1. World J. Biol. Chem. 2014, 5, 308–320. [Google Scholar] [CrossRef]
- Czerwińska, P.; Mazurek, S.; Wiznerowicz, M. The complexity of TRIM28 contribution to cancer. J. Biomed. Sci. 2017, 24, 63. [Google Scholar] [CrossRef]
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The tripartite motif family identifies cell compartments. EMBO J. 2001, 20, 2140–2151. [Google Scholar] [CrossRef]
- Stevens, R.V.; Esposito, D.; Rittinger, K. Characterisation of class VI TRIM RING domains: Linking RING activity to C-terminal domain identity. Life Sci. Alliance 2019, 2, e201900295. [Google Scholar] [CrossRef]
- Short, K.M.; Cox, T.C. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J. Biol. Chem. 2006, 281. [Google Scholar] [CrossRef]
- Sanchez, R.; Zhou, M.M. The PHD finger: A versatile epigenome reader. Trends Biochem. Sci. 2011, 36, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.; Zhou, M.-M. The role of human bromodomains in chromatin biology and gene transcription. Curr. Opin. Drug Discov. Dev. 2009, 12, 659–665. [Google Scholar] [CrossRef]
- Lin, L.; Zhao, W.; Sun, B.; Wang, X.; Liu, Q. Overexpression of TRIM24 is correlated with the progression of human cervical cancer. Am. J. Transl. Res. 2017, 9, 620–628. [Google Scholar] [PubMed]
- Zhang, L.H.; Yin, A.A.; Cheng, J.X.; Huang, H.Y.; Li, X.M.; Zhang, Y.Q.; Han, N.; Zhang, X. TRIM24 promotes glioma progression and enhances chemoresistance through activation of the PI3K/Akt signaling pathway. Oncogene 2015, 34, 600–610. [Google Scholar] [CrossRef]
- Cui, Z.; Cao, W.; Li, J.; Song, X.; Mao, L.; Chen, W. TRIM24 Overexpression Is Common in Locally Advanced Head and Neck Squamous Cell Carcinoma and Correlates with Aggressive Malignant Phenotypes. PLoS ONE 2013, 8, e63887. [Google Scholar] [CrossRef]
- Khetchoumian, K.; Teletin, M.; Tisserand, J.; Mark, M.; Herquel, B.; Ignat, M.; Zucman-Rossi, J.; Cammas, F.; Lerouge, T.; Thibault, C.; et al. Loss of Trim24 (Tif1α) gene function confers oncogenic activity to retinoic acid receptor alpha. Nat. Genet. 2007, 39, 1500–1506. [Google Scholar] [CrossRef]
- Herquel, B.; Ouararhni, K.; Khetchoumian, K.; Ignat, M.; Teletin, M.; Mark, M.; Béchade, G.; Van Dorsselaer, A.; Sanglier-Cianférani, S.; Hamiche, A.; et al. Transcription cofactors TRIM24, TRIM28, and TRIM33 associate to form regulatory complexes that suppress murine hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 8212–8217. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sun, L.; Tang, Z.; Fu, L.; Xu, Y.; Li, Z.; Luo, W.; Qiu, X.; Wang, E. Overexpression of TRIM24 correlates with tumor progression in non-small cell lung cancer. PLoS ONE 2012, 7, e37657. [Google Scholar] [CrossRef]
- Belloni, E.; Trubia, M.; Gasparini, P.; Micucci, C.; Tapinassi, C.; Confalonieri, S.; Nuciforo, P.; Martino, B.; Lo-Coco, F.; Pelicci, P.G.; et al. 8p11 myeloproliferative syndrome with a novel t(7;8) translocation leading to fusion of the FGFR1 and TIF1 genes. Genes Chromosom. Cancer 2005, 42, 320–325. [Google Scholar] [CrossRef]
- Wang, P.; Shen, N.; Liu, D.; Ning, X.; Wu, D.; Huang, X. TRIM24 siRNA induced cell apoptosis and reduced cell viability in human nasopharyngeal carcinoma cells. Mol. Med. Rep. 2018, 18, 369–376. [Google Scholar] [CrossRef]
- Offermann, A.; Roth, D.; Hupe, M.C.; Hohensteiner, S.; Becker, F.; Joerg, V.; Carlsson, J.; Kuempers, C.; Ribbat-Idel, J.; Tharun, L.; et al. TRIM24 as an independent prognostic biomarker for prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2019, 37, 576.e1–576.e10. [Google Scholar] [CrossRef]
- Addison, J.B.; Koontz, C.; Fugett, J.H.; Creighton, C.J.; Chen, D.; Farrugia, M.K.; Padon, R.R.; Voronkova, M.A.; McLaughlin, S.L.; Livengood, R.H.; et al. KAP1 promotes proliferation and metastatic progression of breast cancer cells. Cancer Res. 2015, 75, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Czerwinska, P.; Shah, P.K.; Tomczak, K.; Klimczak, M.; Mazurek, S.; Sozanska, B.; Biecek, P.; Korski, K.; Filas, V.; Mackiewicz, A.; et al. TRIM28 multi-domain protein regulates cancer stem cell population in breast tumor development. Oncotarget 2017, 8, 863–882. [Google Scholar] [CrossRef] [PubMed]
- Yokoe, T.; Toiyama, Y.; Okugawa, Y.; Tanaka, K.; Ohi, M.; Inoue, Y.; Mohri, Y.; Miki, C.; Kusunoki, M. KAP1 is associated with peritoneal carcinomatosis in gastric cancer. Ann. Surg. Oncol. 2010. [Google Scholar] [CrossRef]
- Qi, Z.X.; Cai, J.J.; Chen, L.C.; Yue, Q.; Gong, Y.; Yao, Y.; Mao, Y. TRIM28 as an independent prognostic marker plays critical roles in glioma progression. J. Neurooncol. 2016, 126, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, J.; Li, Q.; Ma, H.; Xu, Z.; Gao, Y. KAP1 is overexpressed in hepatocellular carcinoma and its clinical significance. Int. J. Clin. Oncol. 2016, 927–933. [Google Scholar] [CrossRef]
- Chen, L.; Chen, D.T.; Chen, T.; Kurtyka, C.; Rawal, B.; Fulp, W.J.; Haura, E.B.; Cress, W.D. Tripartite motif containing 28 (Trim28) can regulate cell proliferation by bridging HDAC1/E2F interactions. J. Biol. Chem. 2012, 287, 40106–40118. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Yang, S.; Fu, X.; Feng, J.; Xu, S.; Ying, G. High levels of KAP1 expression are associated with aggressive clinical features in ovarian cancer. Int. J. Mol. Sci. 2015, 16, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Zhan, L.; Jiang, J.; Pan, Y.; Zhang, H.; Li, X.; Pen, F.; Wang, M.; Qin, R.; Sun, C. KAP-1 is overexpressed and correlates with increased metastatic ability and tumorigenicity in pancreatic cancer. Med. Oncol. 2014, 31, 25. [Google Scholar] [CrossRef]
- Diets, I.J.; Hoyer, J.; Ekici, A.B.; Popp, B.; Hoogerbrugge, N.; van Reijmersdal, S.V.; Bhaskaran, R.; Hadjihannas, M.; Vasileiou, G.; Thiel, C.T.; et al. TRIM28 haploinsufficiency predisposes to Wilms tumor. Int. J. Cancer 2019, 145, 941–951. [Google Scholar] [CrossRef]
- Kassem, L.; Deygas, M.; Fattet, L.; Lopez, J.; Goulvent, T.; Lavergne, E.; Chabaud, S.; Carrabin, N.; Chopin, N.; Bachelot, T.; et al. TIF1γ interferes with TGFβ1/SMAD4 signaling to promote poor outcome in operable breast cancer patients. BMC Cancer 2015, 15, 453. [Google Scholar] [CrossRef] [PubMed]
- Cai, F.; Cai, L.U.; Zhou, Z.; Pan, X.I.N.; Wang, M.; Chen, S.U.; Antonio, M.; Luis, F.; Cen, C.; Biskup, E. Prognostic role of Tif1 γ expression and circulating tumor cells in patients with breast cancer. Mol. Med. Rep. 2019, 19, 3685–3695. [Google Scholar] [CrossRef]
- Pommier, R.M.; Gout, J.; Vincent, D.F.; Alcaraz, L.B.; Chuvin, N.; Arfi, V.; Martel, S.; Kaniewski, B.; Devailly, G.; Fourel, G.; et al. TIF1γ Suppresses Tumor Progression by Regulating Mitotic Checkpoints and Chromosomal Stability. Cancer Res. 2015, 75, 4335–4350. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Kawaoka, S.; Roe, J.S.; Shi, J.; Hohmann, A.F.; Xu, Y.; Bhagwat, A.S.; Suzuki, Y.; Kinney, J.B. The transcriptional cofactor TRIM33 prevents apoptosis in B lymphoblastic leukemia by deactivating a single enhancer. eLife 2015, 4, e06377. [Google Scholar] [CrossRef]
- Aucagne, R.; Droin, N.; Paggetti, J.; Lagrange, B.; Largeot, A.; Hammann, A.; Bataille, A.; Martin, L.; Yan, K.P.; Fenaux, P.; et al. Transcription intermediary factor 1γ is a tumor suppressor in mouse and human chronic myelomonocytic leukemia. J. Clin. Investig. 2011, 121, 2361–2370. [Google Scholar] [CrossRef]
- Jain, S.; Singhal, S.; Francis, F.; Hajdu, C.; Wang, J.H.; Suriawinata, A.; Wang, Y.Q.; Zhang, M.; Weinshel, E.H.; Francois, F.; et al. Association of overexpression of TIF1γ with colorectal carcinogenesis and advanced colorectal adenocarcinoma. World. J. Gastroenterol. 2011, 17, 3994–4000. [Google Scholar] [CrossRef]
- Ding, Z.Y.; Jin, G.N.; Wang, W.; Chen, W.X.; Wu, Y.H.; Ai, X.; Chen, L.; Zhang, W.G.; Liang, H.F.; Laurence, A.; et al. Reduced expression of transcriptional intermediary factor 1 gamma promotes metastasis and indicates poor prognosis of hepatocellular carcinoma. Hepatology 2014, 60, 1620–1636. [Google Scholar] [CrossRef]
- Crawford, L.J.; Johnston, C.K.; Irvine, A.E. TRIM proteins in blood cancers. J. Cell Commun. Signal. 2017, 12, 21–29. [Google Scholar] [CrossRef]
- Shaughnessy, J.D.; Zhan, F.; Burington, B.E.; Huang, Y.; Colla, S.; Hanamura, I.; Stewart, J.P.; Kordsmeier, B.; Randolph, C.; Williams, D.R.; et al. Avalidated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood 2007, 109, 2276–2284. [Google Scholar] [CrossRef]
- Vincent, D.F.; Yan, K.P.; Treilleux, I.; Gay, F.; Arfi, V.; Kaniewsky, B.; Marie, J.C.; Lepinasse, F.; Martel, S.; Goddard-Leon, S.; et al. Inactivation of TIF1γ cooperates with KrasG12D to induce cystic tumors of the pancreas. PLoS Genet. 2009, 5, e1000575. [Google Scholar] [CrossRef]
- Jingushi, K.; Ueda, Y.; Kitae, K.; Hase, H.; Egawa, H.; Ohshio, I.; Kawakami, R.; Kashiwagi, Y.; Tsukada, Y.; Kobayashi, T.; et al. MiR-629 targets TRIM33 to promote TGFβ/smad signaling and metastatic phenotypes in ccRCC. Mol. Cancer Res. 2015, 13, 565–574. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Cui, J.; Wu, Y.; Sun, X.; Chen, N. Knockdown of TRIM66 inhibits cell proliferation, migration and invasion in colorectal cancer through JAK2/STAT3 pathway. Life Sci. 2019, 116799. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Y.; Ye, Q.; Zhang, L.J.; Song, Y.N.; Zhang, M.; Wang, W.H.; Zhang, H. TRIM66 knockdown inhibits cell growth, but induces cell cycle arrest and apoptosis of hepatocellular carcinoma cells. Int. J. Clin. Exp. Pathol. 2017, 10, 1030–1040. [Google Scholar]
- Ma, Y.; Dai, H.Y.; Zhang, F.; Zhao, D. TRIM66 expression in non-small cell lung cancer: A new predictor of prognosis. Cancer Biomark. 2017, 20, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guo, Y.; Yang, H.; Shi, G.; Xu, G.; Shi, J.; Yin, N.; Chen, D. TRIM66 overexpresssion contributes to osteosarcoma carcinogenesis and indicates poor survival outcome. Oncotarget 2015, 6, 23708–23719. [Google Scholar] [CrossRef]
- Cao, H.; Gao, R.; Chen, L.; Feng, Y. TRIM66 promotes malignant progression of prostate carcinoma through the JAK/STAT pathway. FEBS Open Bio 2020, 10, 515–524. [Google Scholar] [CrossRef]
- Wang, H.; Xue, W.; Jiang, X. Overexpression of TRIM24 Stimulates Proliferation and Glucose Metabolism of Head and Neck Squamous Cell Carcinoma. Biomed Res. Int. 2018. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Li, L.; Zhao, Z.S.; Wang, H.J. Clinical utility of measuring expression levels of KAP1, TIMP1 and STC2 in peripheral blood of patients with gastric cancer. Worldj. Surg. Oncol. 2013, 11. [Google Scholar] [CrossRef]
- Shi, X.; Mihaylova, V.T.; Kuruvilla, L.; Chen, F.; Viviano, S.; Baldassarre, M.; Sperandio, D.; Martinez, R.; Yue, P.; Bates, J.G.; et al. Loss of TRIM33 causes resistance to BET bromodomain inhibitors through MYC- and TGF-beta-dependent mechanisms. Proc. Natl. Acad. Sci. USA 2016, 113, E4558–E4566. [Google Scholar] [CrossRef]
- Dai, H.Y.; Ma, Y.; Da, Z.; Hou, X.M. Knockdown of TRIM66 inhibits malignant behavior and epithelial-mesenchymal transition in non-small cell lung cancer. Pathol. Res. Pract. 2018, 214, 1130–1135. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.G.; Boulton, S.J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA repair in cancer: Beyond PARP inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Allton, K.; Duncan, A.D.; Barton, M.C. TRIM24 Is a p53-Induced E3-Ubiquitin Ligase That Undergoes ATM-Mediated Phosphorylation and Autodegradation during DNA Damage. Mol. Cell. Biol. 2014, 34, 2695–2709. [Google Scholar] [CrossRef]
- White, D.E.; Negorev, D.; Peng, H.; Ivanov, A.V.; Maul, G.G.; Rauscher, F.J. KAP1, a novel substrate for PIKK family members, colocalizes with numerous damage response factors at DNA lesions. Cancer Res. 2006, 66, 11594–11599. [Google Scholar] [CrossRef]
- Gong, F.; Chiu, L.Y.; Cox, B.; Aymard, F.; Clouaire, T.; Leung, J.W.; Cammarata, M.; Perez, M.; Agarwal, P.; Brodbelt, J.S.; et al. Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination. Genes Dev. 2015, 29, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Lee, S.Y.; Gong, F.; Battenhouse, A.M.; Boutz, D.R.; Bashyal, A.; Refvik, S.T.; Chiang, C.M.; Xhemalce, B.; Paull, T.T.; et al. Systematic bromodomain protein screens identify homologous recombination and R-loop suppression pathways involved in genome integrity. Genes Dev. 2019, 33, 1751–1774. [Google Scholar] [CrossRef]
- Lin, Y.H.; Yuan, J.; Pei, H.; Liu, T.; Ann, D.K.; Lou, Z. KAP1 deacetylation by SIRT1 promotes non-homologous end-joining repair. PLoS ONE 2015, 10, e123935. [Google Scholar] [CrossRef]
- Kulkarni, A.; Oza, J.; Yao, M.; Sohail, H.; Ginjala, V.; Tomas-Loba, A.; Horejsi, Z.; Tan, A.R.; Boulton, S.J.; Ganesan, S. Tripartite motif-containing 33 (TRIM33) protein functions in the poly(ADP-ribose) polymerase (PARP)-dependent DNA damage response through interaction with amplified in liver cancer 1 (ALC1) protein. J. Biol. Chem. 2013, 288, 32357–32369. [Google Scholar] [CrossRef]
- McAvera, R.; Morgan, J.; Johnston, C.; Mills, K.; Crawford, L. Loss of TRIM33 in Multiple Myeloma correlates with increased genomic instability. Clin. Lymphoma Myeloma Leuk. 2019. [Google Scholar] [CrossRef]
- Oza, J.; Ganguly, B.; Kulkarni, A.; Ginjala, V.; Yao, M.; Ganesan, S. A novel role of chromodomain protein CBX8 in DNA damage response. J. Biol. Chem. 2016, 291, 22881–22893. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Z.; Guo, X.; Li, F.; Wei, Q.; Chen, X.; Gong, D.; Xu, Y.; Chen, W.; Liu, Y.; et al. TRIM66 reads unmodified H3R2K4 and H3K56ac to respond to DNA damage in embryonic stem cells. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- White, D.E.; Ivanov, A.V.; Corsinotti, A.; Peng, H.; Lee, S.C. The ATM substrate KAP1 controls DNA repair in heterochromatin: Regulation by HP1 proteins and Serine 473/824 phosphorylation. Mol. Cancer Res. 2012, 10, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Ziv, Y.; Bielopolski, D.; Galanty, Y.; Lukas, C.; Taya, Y.; Schultz, D.C.; Lukas, J.; Bekker-Jensen, S.; Bartek, J.; Shiloh, Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM-and KAP-1 dependent pathway. Nat. Cell Biol. 2006, 8, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Löbrich, M.; Jeggo, P.A. ATM Signaling Facilitates Repair of DNA Double-Strand Breaks Associated with Heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Noon, A.T.; Shibata, A.; Rief, N.; Löbrich, M.; Stewart, G.S.; Jeggo, P.A.; Goodarzi, A.A. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat. Cell Biol. 2010, 12, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Goodarzi, A.A.; Adelmant, G.O.; Pan, Y.; Jeggo, P.A.; Marto, J.A.; Chowdhury, D. Phosphoproteomic analysis reveals that PP4 dephosphorylates KAP-1 impacting the DNA damage response. EMBO J. 2012, 31, 2403–2415. [Google Scholar] [CrossRef]
- Bhatia, N.; Xiao, T.Z.; Rosenthal, K.A.; Diddiqui, I.A.; Thiyagarajan, S.; Smart, B.; Meng, Q.; Zuleger, C.L.; Mukhtar, H.; Kenney, S.C.; et al. MAGE-C2 Promotes Growth and Tumorigenicity of Melanoma Cells, Phosphorylation of KAP1, and DNA Damage Repair. J. Investig. Dermatol. 2013, 133, 759–767. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, S.; Gao, X.; Gao, X.; Xu, X.; Lv, Y.; Zhang, Y.; Zhu, Z.; Zhang, C.; Li, Q.; et al. Roles of Kruppel-associated box (KRAB)-associated co-repressor KAP1 Ser-473 phosphorylation in DNA damage response. J. Biol. Chem. 2012, 287, 18937–18952. [Google Scholar] [CrossRef]
- Blasius, M.; Forment, J.V.; Thakkar, N.; Wagner, S.A.; Choudhary, C.; Jackson, S.P. A phospho-proteomic screen identifies substrates of the checkpoint kinase Chk1. Genome Biol. 2011, 12, R78. [Google Scholar] [CrossRef]
- Chang, C.W.; Chou, H.Y.; Lin, Y.S.; Huang, K.H.; Chang, C.J.; Hsu, T.C.; Lee, S.C. Phosphorylation at Ser473 regulates heterochromatin protein 1 binding and corepressor function of TIF1beta/KAP1. BMC Mol. Biol. 2008, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ayrapetov, M.K.; Gursoy-Yuzugullu, O.; Xu, C.; Xu, Y.; Price, B.D. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc. Natl. Acad. Sci. USA 2014, 111, 9169–9174. [Google Scholar] [CrossRef] [PubMed]
- Downs, J.A.; Nussenzweig, M.C.; Nussenzweig, A. Chromatin dynamics and the preservation of genetic information. Nature 2007, 447, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Kurka, T.; Jeggo, P.A. KAP-1 phosphorylation regulates CHD3 nucleosome remodeling during the DNA double-strand break response. Nat. Struct. Mol. Biol. 2011, 18, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef]
- Kalousi, A.; Hoffbeck, A.S.; Selemenakis, P.N.; Pinder, J.; Savage, K.I.; Khanna, K.K.; Brino, L.; Dellaire, G.; Gorgoulis, V.G.; Soutoglou, E. The nuclear oncogene SET controls DNA repair by KAP1 and HP1 retention to chromatin. Cell Rep. 2015, 11, 149–163. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Peng, H.; Yurchenko, V.; Yap, K.L.; Negorev, D.G.; Schultz, D.C.; Psulkowski, E.; Fredericks, W.J.; White, D.E.; Maul, G.G.; et al. PHD Domain-Mediated E3 Ligase Activity Directs Intramolecular Sumoylation of an Adjacent Bromodomain Required for Gene Silencing. Mol. Cell 2007, 28, 823–837. [Google Scholar] [CrossRef]
- Allouch, A.; Di Primio, C.; Alpi, E.; Lusic, M.; Arosio, D.; Giacca, M.; Cereseto, A. The TRIM family protein KAP1 inhibits HIV-1 integration. Cell Host Microbe 2011, 9, 484–495. [Google Scholar] [CrossRef]
- Kepkay, R.; Attwood, K.M.; Ziv, Y.; Shiloh, Y.; Dellaire, G. KAP1 depletion increases PML nuclear body number in concert with ultrastructural changes in chromatin. Cell Cycle 2011, 10, 308–322. [Google Scholar] [CrossRef]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 2010, 17, 1144–1151. [Google Scholar] [CrossRef]
- Khetchoumian, K.; Teletin, M.; Mark, M.; Lerouge, T.; Cerviño, M.; Oulad-Abdelghani, M.; Chambon, P.; Losson, R. TIF1δ, a novel HP1-interacting member of the transcriptional intermediary factor 1 (TIF1) family expressed by elongating spermatids. J. Biol. Chem. 2004, 279, 48329–48341. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.W.; Wang, Z.; Yiu, T.T.; Akdemir, K.C.; Xia, W.; Winter, S.; Tsai, C.Y.; Shi, X.; Schwarzer, D.; Plunkett, W.; et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature 2010, 468, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Xi, Q.; Wang, Z.; Zaromytidou, A.; Zhang, X.H.; Liu, J.X.; Kim, H.; Barlas, A.; Manova-todorova, K.; Studer, L.; Mark, W.; et al. A poised chromatin platform for TGF-β access to master regulators. Cell 2013, 147, 1511–1524. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, B.N.; Thackray, J.K.; Simonet, N.G.; Kane-Goldsmith, N.; Martinez-Redondo, P.; Nguyen, T.; Bunting, S.; Vaquero, A.; Tischfield, J.A.; Serrano, L. SIRT 7 promotes genome integrity and modulates non-homologous end joining DNA repair. EMBO J. 2016, 35, 1488–1503. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 93–115. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.; Morgan, D.O. Finishing mitosis, one step at a time. Nat. Rev. Mol. Cell Biol. 2007, 8, 894–903. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of p53 in cell death and human cancers. Cancers (Basel) 2011, 3, 994–1013. [Google Scholar] [CrossRef]
- Gatz, S.A.; Wiesmüller, L. p53 in recombination and repair. Cell Death Differ. 2006, 13, 1003–1016. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiong, Y. Control of p53 ubiquitination and nuclear export by MDM2 and ARF. Cell Growth Differ. 2001, 12, 175–186. [Google Scholar]
- Ito, A.; Lai, C.H.; Zhao, X.; Saito, S.; Hamilton, M.H.; Appella, E.; Yao, T.P. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001, 20, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ivanov, A.; Chen, L.; Fredericks, W.J.; Seto, E.; Rauscher, F.J.; Chen, J. MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO J. 2005, 24, 3279–3290. [Google Scholar] [CrossRef]
- Okamoto, K.; Kitabayashi, I.; Taya, Y. KAP1 dictates p53 response induced by chemotherapeutic agents via Mdm2 interaction. Biochem. Biophys. Res. Commun. 2006, 351, 216–222. [Google Scholar] [CrossRef]
- Allton, K.; Jain, A.K.; Herz, H.M.; Tsai, W.W.; Sung, Y.J.; Qin, J.; Bergmann, A.; Johnson, R.L.; Barton, M.C. Trim24 targets endogenous p53 for degradation. Proc. Natl. Acad. Sci. USA 2009, 106, 11612–11616. [Google Scholar] [CrossRef] [PubMed]
- Lara-Gonzalez, P.; Westhorpe, F.G.; Taylor, S.S. The spindle assembly checkpoint. Curr. Biol. 2012, 22, R966–R980. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, G.G.; Townsend, K.; Martin, A.; Shimwell, N.J.; Roger, J.A.; Stewart, G.S.; Nilsson, J.; Turnell, A.S. Transcriptional Intermediary Factor 1 γ binds to the Anaphase- Promoting Complex/Cyclosome and promotes mitosis. Oncogene 2013, 32, 8970–8980. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lawrence, K.S.; Chau, T.; Engebrecht, J.A. DNA Damage Response and Spindle Assembly Checkpoint Function throughout the Cell Cycle to Ensure Genomic Integrity. PLoS Genet. 2015, 11, e1005150. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Bertoni, F. BET proteins as targets for anticancer treatment. Cancer Discov. 2018, 8, 24–26. [Google Scholar] [CrossRef]
- Liu, J.; Li, F.; Bao, H.; Jiang, Y.; Zhang, S.; Ma, R.; Gao, J.; Wu, J.; Ruan, K. The polar warhead of a TRIM24 bromodomain inhibitor rearranges a water-mediated interaction network. FEBS J. 2017, 284, 1082–1095. [Google Scholar] [CrossRef]
- Roxburgh, P.; Hock, A.K.; Dickens, M.P.; Mezna, M.; Fischer, P.M.; Vousden, K.H. Small molecules that bind the Mdm2 RING stabilize and activate p53. Carcinogenesis 2012, 33, 791–798. [Google Scholar] [CrossRef]
- Wu, W.; Xu, C.; Ling, X.; Fan, C.; Buckley, B.P.; Chernov, M.V.; Ellis, L.; Li, F.; Muñoz, I.G.; Wang, X. Targeting RING domains of Mdm2-MdmX E3 complex activates apoptotic arm of the p53 pathway in leukemia/lymphoma cells. Cell Death Dis. 2015, 6, e2035. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TIF1 Member | Cancer Type | Alteration | Observed Function | Reference |
|---|---|---|---|---|
| TRIM24 | Cervical | High expression | Oncogene | [13] |
| Glioma | High expression | Oncogene | [14] | |
| Head & Neck | High expression | Oncogene | [15] | |
| Liver | Gene deletion | Tumour suppressor | [16] | |
| Liver | N/A | Tumour suppressor | [17] | |
| Lung | High expression | Oncogene | [18] | |
| Myeloproliferative syndrome | Chromosome translocation: FGFR1 5 | Oncogene | [19] | |
| Nasopharyngeal | High expression | Oncogene | [20] | |
| Prostate | High expression | Oncogene | [21] | |
| TRIM28 | Breast | High expression | Oncogene | [22,23] |
| Gastric | High expression | Oncogene | [24] | |
| Glioma | High expression | Oncogene | [25] | |
| Liver | High expression | Oncogene | [26] | |
| Liver | N/A | Tumour suppressor | [17] | |
| Lung (early stage) | High expression | Tumour suppressor | [27] | |
| Ovarian | High expression | Oncogene | [28] | |
| Pancreatic | High expression | Oncogene | [29] | |
| Renal (WT 1) | Loss-of-function mutation | Tumour suppressor | [30] | |
| TRIM33 | Breast | High expression | Oncogene | [31] |
| Breast | Reduced expression | Tumour suppressor | [32,33] | |
| B-ALL 2 | N/A | Oncogene | [34] | |
| CMML 3 | Reduced expression | Tumour suppressor | [35] | |
| Colorectal | High expression | Oncogene | [36] | |
| Liver | Reduced expression | Tumour suppressor | [37] | |
| MM 4 | Reduced expression | Tumour suppressor | [38,39] | |
| Pancreatic | Reduced expression | Tumour suppressor | [33,40] | |
| Renal | Reduced expression | Tumour suppressor | [33,41] | |
| TRIM66 | Colorectal | High expression | Oncogene | [42] |
| Liver | High expression | Oncogene | [43] | |
| Lung | High expression | Oncogene | [44] | |
| Osteosarcoma | High expression | Oncogene | [45] | |
| Prostate | N/A | Oncogene | [46] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McAvera, R.M.; Crawford, L.J. TIF1 Proteins in Genome Stability and Cancer. Cancers 2020, 12, 2094. https://doi.org/10.3390/cancers12082094
McAvera RM, Crawford LJ. TIF1 Proteins in Genome Stability and Cancer. Cancers. 2020; 12(8):2094. https://doi.org/10.3390/cancers12082094
Chicago/Turabian StyleMcAvera, Roisin M., and Lisa J. Crawford. 2020. "TIF1 Proteins in Genome Stability and Cancer" Cancers 12, no. 8: 2094. https://doi.org/10.3390/cancers12082094
APA StyleMcAvera, R. M., & Crawford, L. J. (2020). TIF1 Proteins in Genome Stability and Cancer. Cancers, 12(8), 2094. https://doi.org/10.3390/cancers12082094