Distinct Molecular Landscape of Epstein–Barr Virus Associated Pulmonary Lymphoepithelioma-Like Carcinoma Revealed by Genomic Sequencing

 , , ,
, , , 

Abstract
1. Introduction
2. Result
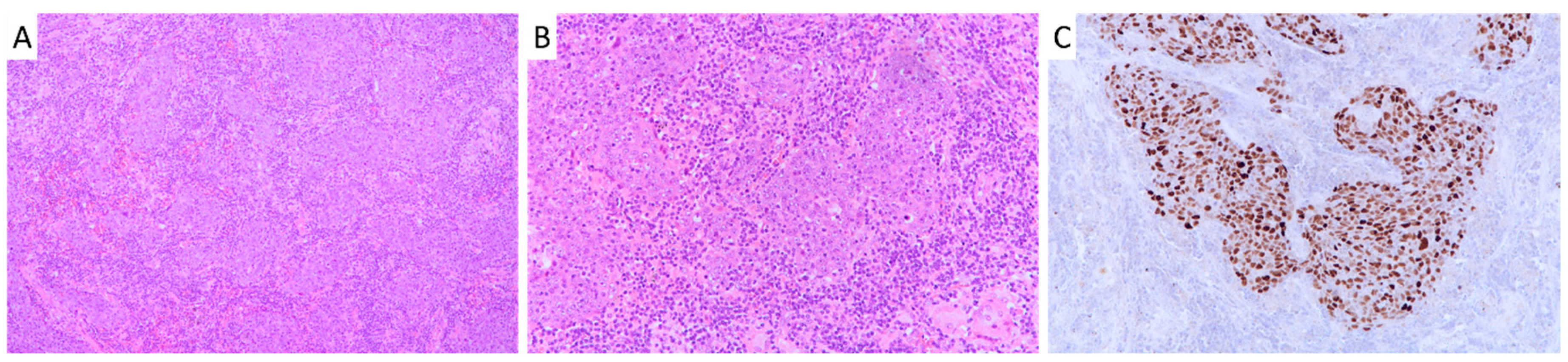
2.1. Patient Cohort Characteristics and Survival Analysis
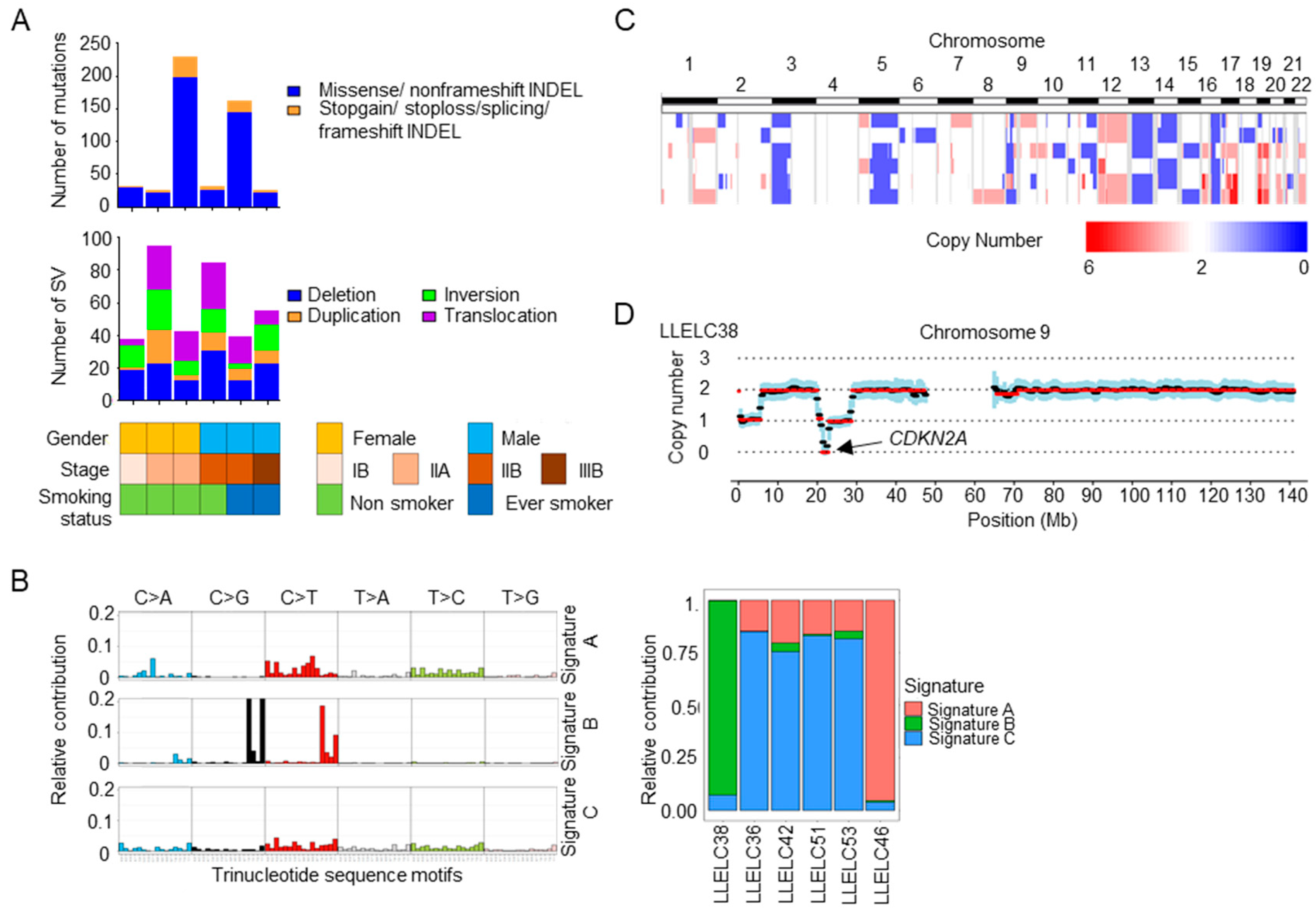
2.2. Mutational Signatures, Structural Variations and Copy Number Variations in Pulmonary LELC by Whole Genome Sequencing
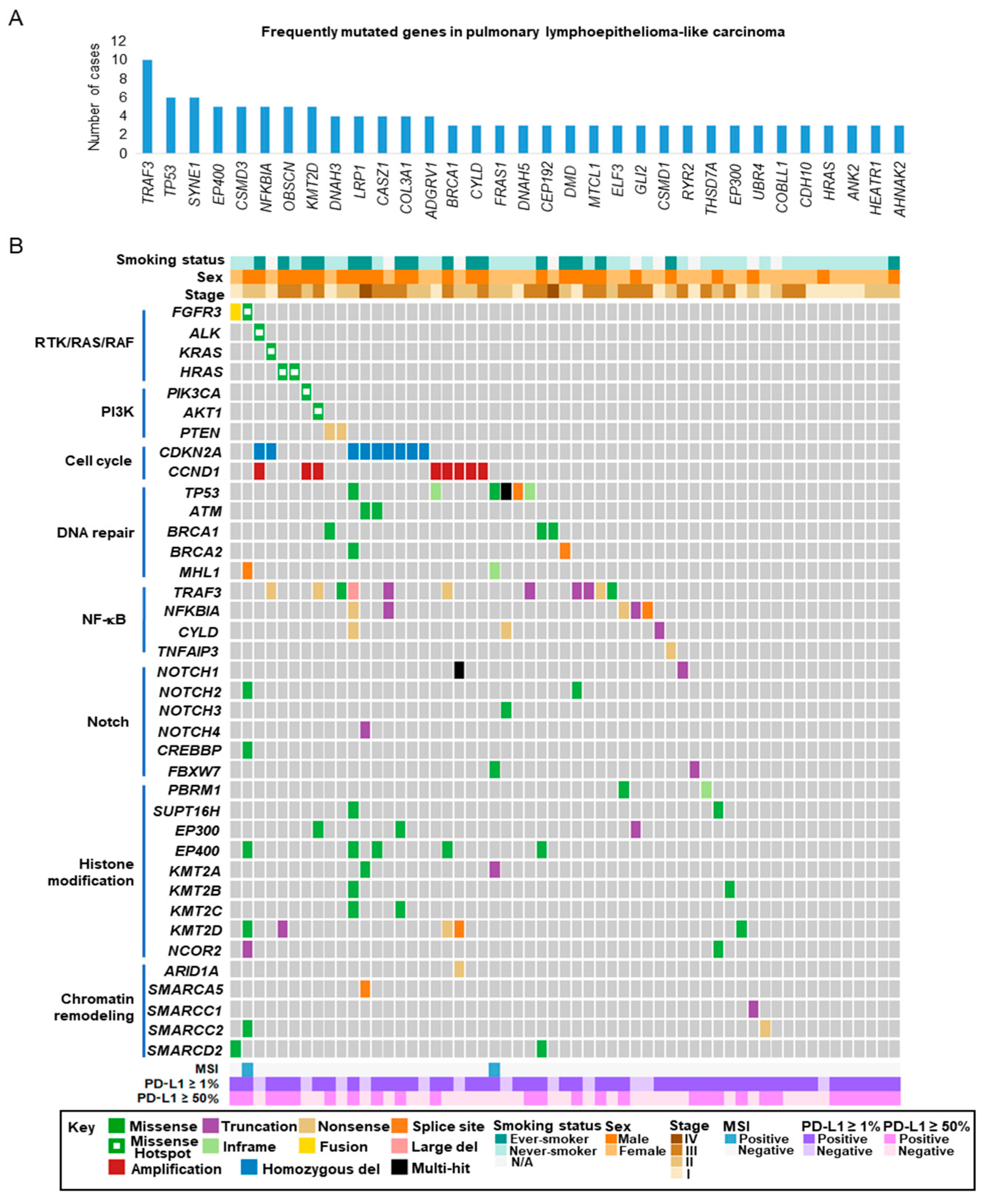
2.3. Targeted Sequencing of Pulmonary LELC
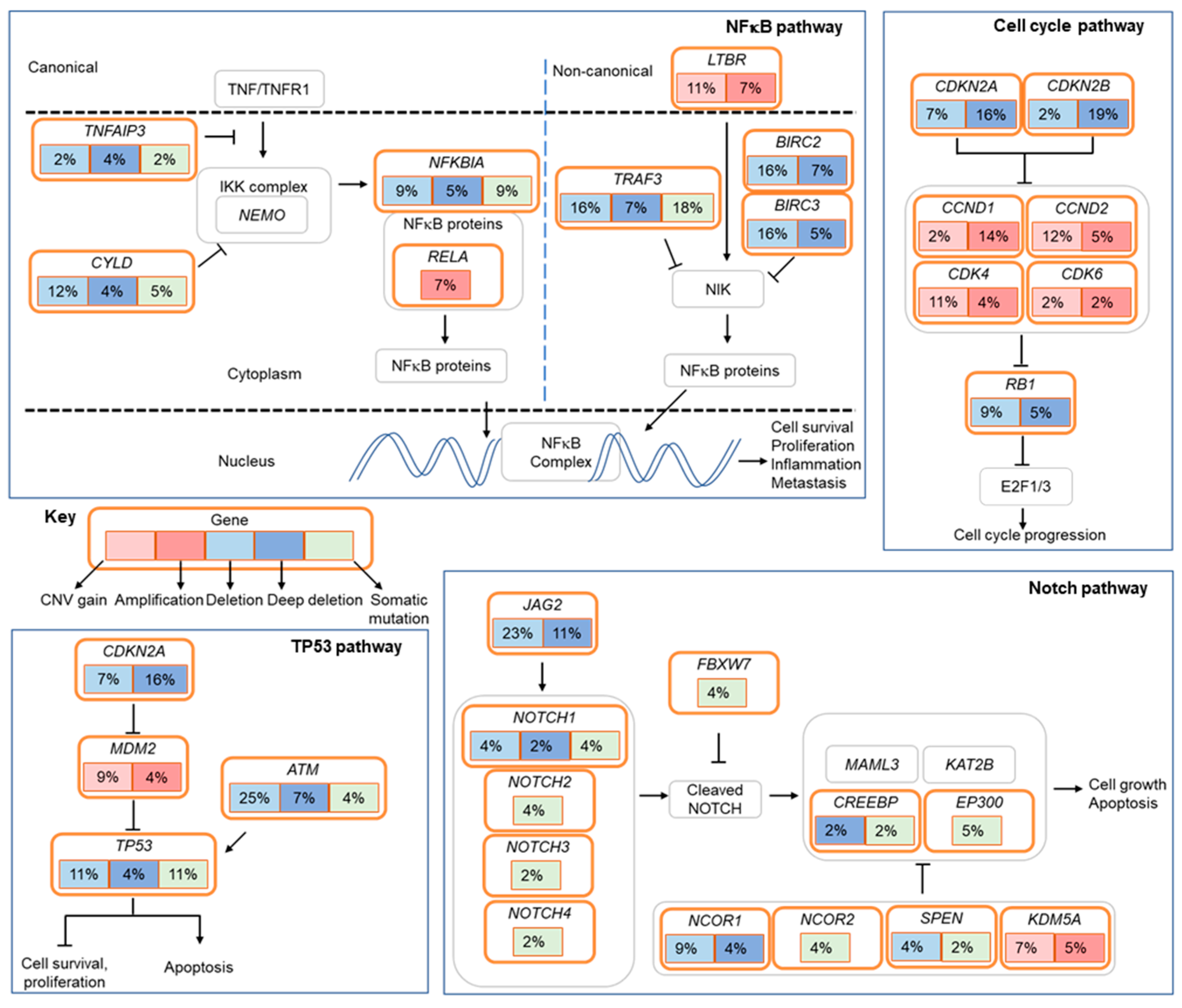
2.4. Actionable Alterations in the RTK/RAS/RAF and PI3K/AKT/mTOR Signaling Pathways
2.5. Alterations in Cell Cycle Regulatory Genes
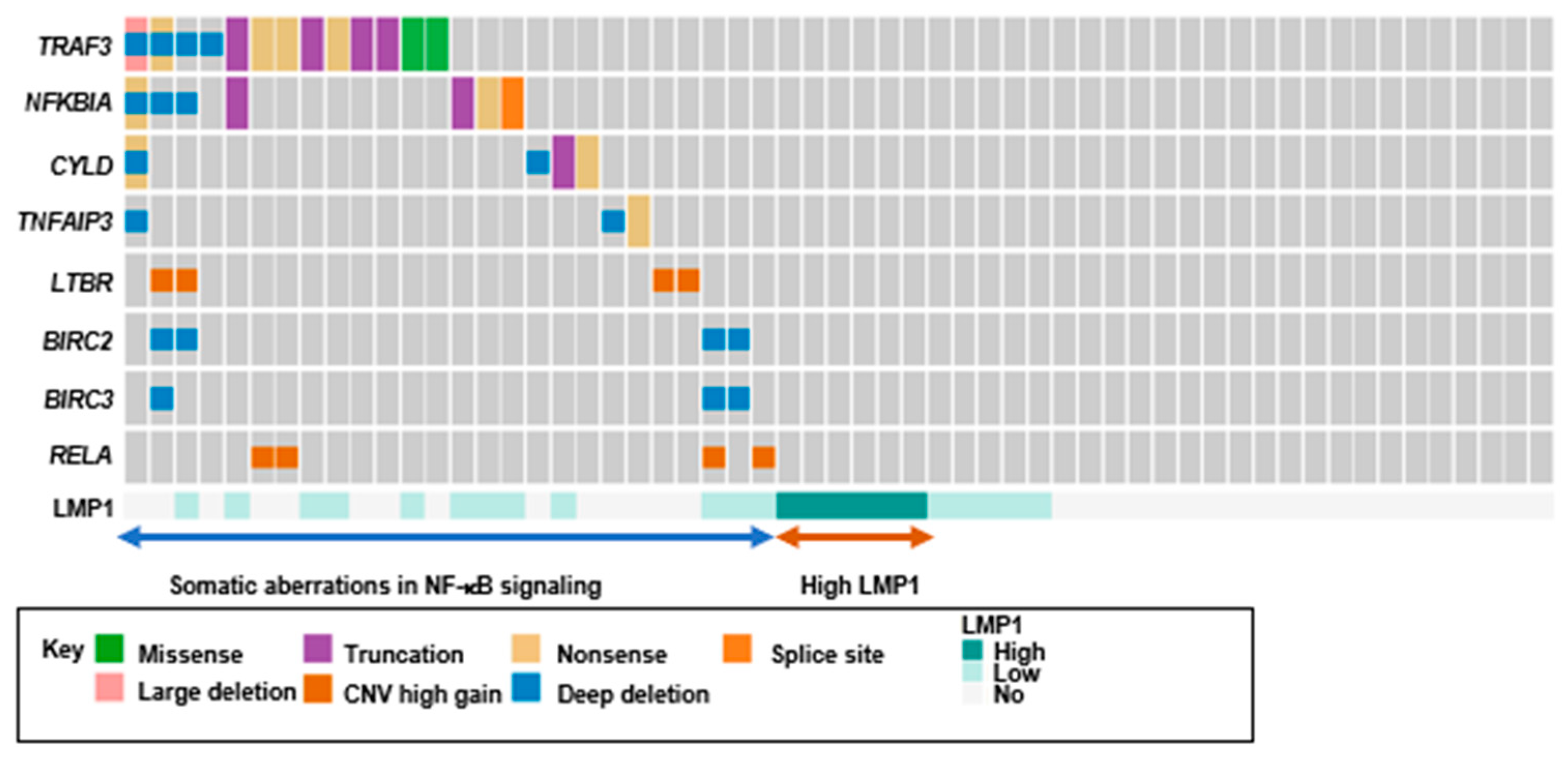
2.6. Loss-of-Function Mutations in Negative Regulators of The NF-κB Pathway
2.7. Alterations in Epigenetic Regulators
2.8. Alterations in the Notch and TP53 Pathways
2.9. Microsatellite Instability (MSI) in Pulmonary LELC
2.10. Programmed Cell Death Ligand-1 (PD-L1) Expression in Pulmonary LELC
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Whole Genome Sequencing (WGS)
4.3. Capture-Based Targeted Sequencing
4.4. Immunohistochemistry (IHC), Fluorescence in Situ Hybridization (FISH) and in Situ Hybridization (ISH)
4.5. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Han, A.J.; Xiong, M.; Zong, Y.S. Association of Epstein-Barr virus with lymphoepithelioma-like carcinoma of the lung in southern China. Am. J. Clin. Pathol. 2000, 114, 220–226. [Google Scholar] [CrossRef]
- Bégin, L.R.; Eskandari, J.; Joncas, J.; Panasci, L. Epstein-Barr-Virus Related Lymphoepithelioma-Like Carcinoma of Lung. J. Surg. Oncol. 1987, 36, 280–283. [Google Scholar]
- Han, A.J.; Xiong, M.; Gu, Y.Y.; Lin, S.X.; Xiong, M. Lymphoepithelioma-like carcinoma of the lung with a better prognosis - A clinicopathologic study of 32 cases. Am. J. Clin. Pathol. 2001, 115, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.L.; Wu, C.T.; Shih, J.Y.; Lee, Y.C. New aspects in clinicopathologic and oncogene studies of 23 pulmonary lymphoepithelioma-like carcinomas. Am. J. Surg. Pathol. 2002, 26, 715–723. [Google Scholar] [CrossRef]
- Chang, Y.L.; Yang, C.Y.; Lin, M.W.; Wu, C.T.; Yang, P.C. PD-L1 is highly expressed in lung lymphoepithelioma-like carcinoma: A potential rationale for immunotherapy. Lung Cancer 2015, 88, 254–259. [Google Scholar] [CrossRef]
- Yin, K.; Feng, H.B.; Li, L.L.; Chen, Y.; Xie, Z.; Lv, Z.Y.; Guo, W.B.; Lu, D.X.; Yang, X.N.; Yan, W.Q.; et al. Low frequency of mutation of epidermal growth factor receptor (EGFR) and arrangement of anaplastic lymphoma kinase (ALK) in primary pulmonary lymphoepithelioma-like carcinoma. Thorac. Cancer 2020, 11, 346–352. [Google Scholar] [CrossRef]
- Yin, K.; Feng, H.B.; Li, L.L.; Chen, Y.; Xie, Z.; Lv, Z.Y.; Guo, W.B.; Lu, D.X.; Yang, X.N.; Yan, W.Q.; et al. Distinct epidermal growth factor receptor and KRAS mutation patterns in non-small cell lung cancer patients with different tobacco exposure and clinicopathologic features. Clin. Cancer Res. 2006, 12, 1647–1653. [Google Scholar]
- Xie, Z.; Liu, L.; Lin, X.; Xie, X.; Gu, Y.; Liu, M.; Zhang, J.; Ouyang, M.; Lizaso, A.; Zhang, H.; et al. A multicenter analysis of genomic profiles and PD-L1 expression of primary lymphoepithelioma-like carcinoma of the lung. Mod. Pathol. 2019, 33, 626–638. [Google Scholar] [CrossRef]
- Hong, S.; Liu, D.; Luo, S.; Fang, W.; Zhan, J.; Fu, S.; Zhang, Y.; Wu, X.; Zhou, H.; Chen, X.; et al. The genomic landscape of Epstein-Barr virus-associated pulmonary lymphoepithelioma-like carcinoma. Nat. Commun. 2019, 10, 3108. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstromet, E.N. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO. Precis. Oncol. 2017, 2017, 145–147. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Chung, G.T.; Lui, V.W.; To, K.F.; Ma, B.B.; Chow, C.; Woo, J.K.; Yip, K.Y.; Seo, J.; Hui, E.P.; et al. Exome and genome sequencing of nasopharynx cancer identifies NF-kappaB pathway activating mutations. Nat. Commun. 2017, 8, 14121. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.L.; Wu, C.T.; Shih, J.Y.; Lee, Y.C. Unique p53 and epidermal growth factor receptor gene mutation status in 46 pulmonary lymphoepithelioma-like carcinomas. Cancer Sci. 2011, 102, 282–287. [Google Scholar] [CrossRef]
- Fang, W.; Hong, S.; Chen, N.; He, X.; Zhan, J.; Qin, T.; Zhou, T.; Hu, Z.; Ma, Y.; Zhao, Y.; et al. PD-L1 is remarkably over-expressed in EBV-associated pulmonary lymphoepithelioma-like carcinoma and related to poor disease-free survival. Oncotarget. 2015, 6, 33019–33032. [Google Scholar] [CrossRef]
- Wu, Y.M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.F.; et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef]
- Yuan, L.; Liu, Z.H.; Lin, Z.R.; Xu, L.H.; Zhong, Q.; Zeng, M.S. Recurrent FGFR3-TACC3 fusion gene in nasopharyngeal carcinoma. Cancer Biol. Ther. 2014, 15, 1613–1621. [Google Scholar] [CrossRef]
- AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef]
- Qin, A.; Johnson, A.; Ross, J.S.; Miller, V.A.; Ali, S.M.; Schrock, A.B.; Gadgeel, S.M. Detection of Known and Novel FGFR Fusions in Non-Small Cell Lung Cancer by Comprehensive Genomic Profiling. J. Thorac. Oncol. 2019, 14, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Bahleda, R.; Dienstmann, R.; Infante, J.R.; Mita, A.; Italiano, A.; Calvo, E.; Moreno, V.; Adamo, B.; Gazzah, A.; et al. Phase I Dose-Escalation Study of JNJ-42756493, an Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2015, 33, 3401–3408. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Dai, W.; Cheung, A.K.; Ko, J.M.; Kan, R.; Wong, B.W.; Leong, M.M.; Deng, M.; Kwok, T.C.; Chan, J.Y.; et al. Whole-exome sequencing identifies multiple loss-of-function mutations of NF-kappaB pathway regulators in nasopharyngeal carcinoma. Proc. Natl. Acad. Sci. USA 2016, 113, 11283–11288. [Google Scholar] [CrossRef]
- Tsao, S.W.; Tsang, C.M.; To, K.F.; Lo, K.W. The role of Epstein-Barr virus in epithelial malignancies. J. Pathol. 2015, 235, 323–333. [Google Scholar] [CrossRef]
- Chung, G.T.; Lou, W.P.; Chow, C.; To, K.F.; Choy, K.W.; Leung, A.W.; Tong, C.Y.; Yuen, J.W.; Ko, C.W.; Yip, T.T.; et al. Constitutive activation of distinct NF-kappaB signals in EBV-associated nasopharyngeal carcinoma. J. Pathol. 2013, 231, 311–322. [Google Scholar] [CrossRef]
- Hau, P.M.; Lung, H.L.; Wu, M.; Tsang, C.M.; Wong, K.L.; Mak, N.K.; Lo, K.W. Targeting Epstein-Barr Virus in Nasopharyngeal Carcinoma. Frontiers in Oncology. Front. Oncol. 2020, 10, 35–38. [Google Scholar]
- Messick, T.E.; Smith, G.R.; Soldan, S.S.; McDonnell, M.E.; Deakyne, J.S.; Malecka, K.A.; Tolvinski, L.; van den Heuvel, A.; Gu, B.W.; Cassel, J.A.; et al. Structure-based design of small-molecule inhibitors of EBNA1 DNA binding blocks Epstein-Barr virus latent infection and tumor growth. Sci. Transl. Med. 2019, 11, 101–105. [Google Scholar] [CrossRef]
- Chia, W.K.; Teo, M.; Wang, W.W.; Lee, B.; Ang, S.F.; Tai, W.M.; Chee, C.L.; Ng, J.; Kan, R.; Lim, W.T.; et al. Adoptive T-cell transfer and chemotherapy in the first-line treatment of metastatic and/or locally recurrent nasopharyngeal carcinoma. Mol. Ther. 2014, 22, 132–139. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar]
- Dacic, S.; Lomago, D.; Hunt, J.L.; Sepulveda, A.; Yousem, S.A. Microsatellite instability is uncommon in lymphoepithelioma-like carcinoma of the lung. Am. J. Clin. Pathol. 2007, 127, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.; Wu, Y.L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G.; Srimuninnimit, V., Jr.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Yu, X.Y.; Zhang, X.W.; Wang, F.; Lin, Y.B.; Wang, W.D.; Chen, Y.Q.; Zhang, L.J.; Cai, L. Correlation and prognostic significance of PD-L1 and P53 expression in resected primary pulmonary lymphoepithelioma-like carcinoma. J. Thorac. Dis. 2018, 10, 1891–1902. [Google Scholar] [CrossRef]
- Qiu, Z.X.; Zhou, P.; Wang, K. Primary Pulmonary Lymphoepithelioma-Like Carcinoma Response Favorably To Nivolumab: A Case Report. Onco. Targets. Ther. 2019, 12, 8595–8600. [Google Scholar] [CrossRef]
- Kumar, V.; Dave, V.; Harris, J.; Huang, Y. Response of advanced stage recurrent lymphoepithelioma-like carcinoma to nivolumab. Immunotherapy 2017, 9, 955–961. [Google Scholar] [CrossRef]
- Zhou, N.; Lin, Y.; Peng, X.; Wang, Y.; Wang, Y. Thorough survey and analysis of pulmonary lymphoepithelioma-like carcinoma in Macau and multimodality treatment for advanced disease. Lung Cancer 2019, 138, 116–123. [Google Scholar] [CrossRef]
- Ju, Y.S.; Lee, W.C.; Shin, J.Y.; Lee, S.; Bleazard, T.; Won, J.K.; Kim, Y.T.; Kim, J.I.; Kang, J.H.; Seo, J.S. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res. 2012, 22, 436–445. [Google Scholar] [CrossRef]
- Raczy, C.; Petrovski, R.; Saunders, C.T.; Chorny, I.; Kruglyak, S.; Margulies, E.H.; Chuang, H.Y.; Källberg, M.; Kumar, S.A.; Liao, A.; et al. Isaac: Ultra-fast whole-genome secondary analysis on Illumina sequencing platforms. Bioinformatics 2013, 29, 2041–2043. [Google Scholar] [CrossRef]
- Saunders, C.T.; Wong, W.S.; Swamy, S.; Becq, J.; Murray, L.J.; Cheetham, R.K. Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012, 28, 1811–1817. [Google Scholar] [CrossRef]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef] [PubMed]
- Favero, F.; Joshi, T.; Marquard, A.M.; Birkbak, N.J.; Krzystanek, M.; Li, Q.; Szallasi, Z.; Eklund, A.C. Sequenza: Allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol. 2015, 26, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Chan, A.W.; Chau, S.L.; Tong, J.H.; Chow, C.; Kwan, J.; Chung, L.Y.; Lung, R.W.; Tong, C.Y.; Tin, E.K.; Law, P.P.; et al. The Landscape of Actionable Molecular Alterations in Immunomarker-Defined Large-Cell Carcinoma of the Lung. J. Thorac. Oncol. 2019, 14, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Kelly, B.J.; Fitch, J.R.; Hu, Y.; Corsmeier, D.J.; Zhong, H.; Wetzel, A.N.; Nordquist, R.D.; Newsom, D.L.; White, P. Churchill: An ultra-fast, deterministic, highly scalable and balanced parallelization strategy for the discovery of human genetic variation in clinical and population-scale genomics. Genome Biol. 2015, 16, 6. [Google Scholar] [CrossRef]
- Lai, Z.; Markovets, A.; Ahdesmaki, M.; Chapman, B.; Hofmann, O.; McEwen, R.; Johnson, J.; Dougherty, B.; Barrett, J.C.; Dry, J.R. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic. Acids. Res. 2016, 44, e108. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R. KVarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS. Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.; Ye, K.; Zhang, Q.; Lu, C.; Xie, M.; McLellan, M.D.; Wendl, M.C.; Ding, L. MSIsensor: Microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 2014, 30, 1015–1016. [Google Scholar] [CrossRef]
- Chan, A.; Tong, J.; Kwan, J.; Chow, C.; Chung, L.Y.; Chau, S.L.; Lung, R.; Ng, C.; Wan, I.; Mok, T.; et al. Assessment of programmed cell death ligand-1 expression by 4 diagnostic assays and its clinicopathological correlation in a large cohort of surgical resected non-small cell lung carcinoma. Mod. Pathol. 2018, 31, 1381–1390. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Features | Total (n = 57) |
|---|---|
| Age | |
| Mean | 55 years |
| Median | 54 years |
| Range | 37–79 years |
| Tumor size | |
| Mean | 4.7 cm |
| Median | 4.5 cm |
| Range | 1–12 cm |
| Sex | |
| Female | 31(54) |
| Male | 26(46) |
| Smoking status | |
| Never-smoker | 31(65) |
| Ever-smoker | 17(35) |
| Stage | |
| I | 15(26) |
| II | 20(35) |
| III | 20(35) |
| IV | 2(4) |
| N status | |
| N0 | 33(60) |
| N1-3 | 22(40) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chau, S.-L.; Tong, J.H.-M.; Chow, C.; Kwan, J.S.-H.; Lung, R.W.-M.; Chung, L.-Y.; Tin, E.K.-Y.; Wong, S.S.-Y.; Cheung, A.H.-K.; Lau, R.W.-H.; et al. Distinct Molecular Landscape of Epstein–Barr Virus Associated Pulmonary Lymphoepithelioma-Like Carcinoma Revealed by Genomic Sequencing. Cancers 2020, 12, 2065. https://doi.org/10.3390/cancers12082065
Chau S-L, Tong JH-M, Chow C, Kwan JS-H, Lung RW-M, Chung L-Y, Tin EK-Y, Wong SS-Y, Cheung AH-K, Lau RW-H, et al. Distinct Molecular Landscape of Epstein–Barr Virus Associated Pulmonary Lymphoepithelioma-Like Carcinoma Revealed by Genomic Sequencing. Cancers. 2020; 12(8):2065. https://doi.org/10.3390/cancers12082065
Chicago/Turabian StyleChau, Shuk-Ling, Joanna Hung-Man Tong, Chit Chow, Johnny Sheung-Him Kwan, Raymond Wai-Ming Lung, Lau-Ying Chung, Edith Ka-Yee Tin, Shela Shu-Yan Wong, Alvin Ho-Kwan Cheung, Rainbow Wing-Hung Lau, and et al. 2020. "Distinct Molecular Landscape of Epstein–Barr Virus Associated Pulmonary Lymphoepithelioma-Like Carcinoma Revealed by Genomic Sequencing" Cancers 12, no. 8: 2065. https://doi.org/10.3390/cancers12082065
APA StyleChau, S.-L., Tong, J. H.-M., Chow, C., Kwan, J. S.-H., Lung, R. W.-M., Chung, L.-Y., Tin, E. K.-Y., Wong, S. S.-Y., Cheung, A. H.-K., Lau, R. W.-H., Ng, C. S.-H., Mok, T. S.-K., Lo, K.-W., & To, K.-F. (2020). Distinct Molecular Landscape of Epstein–Barr Virus Associated Pulmonary Lymphoepithelioma-Like Carcinoma Revealed by Genomic Sequencing. Cancers, 12(8), 2065. https://doi.org/10.3390/cancers12082065