PVR and ICAM-1 on Blast Crisis CML Stem and Progenitor Cells with TKI Resistance Confer Susceptibility to NK Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
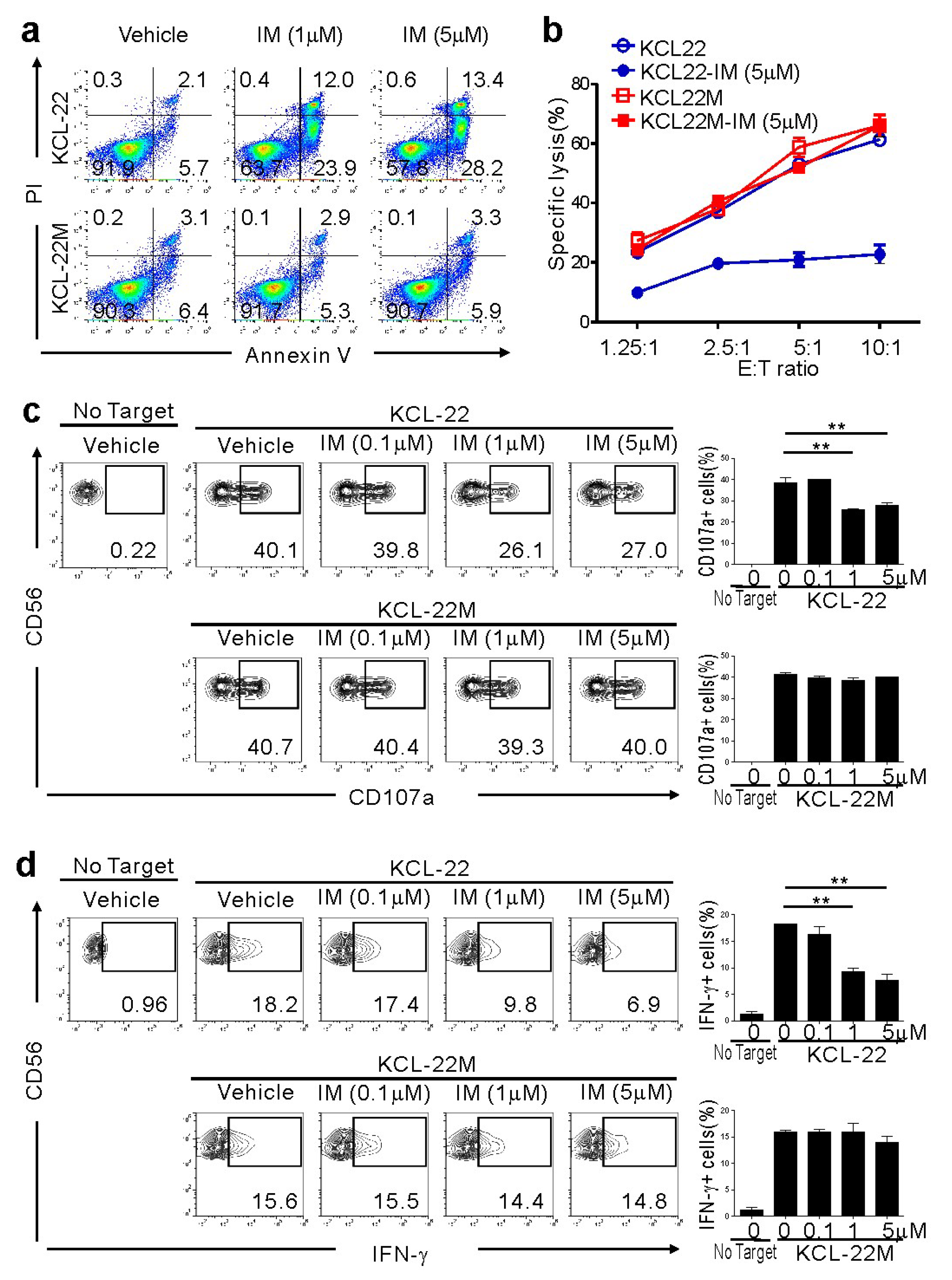
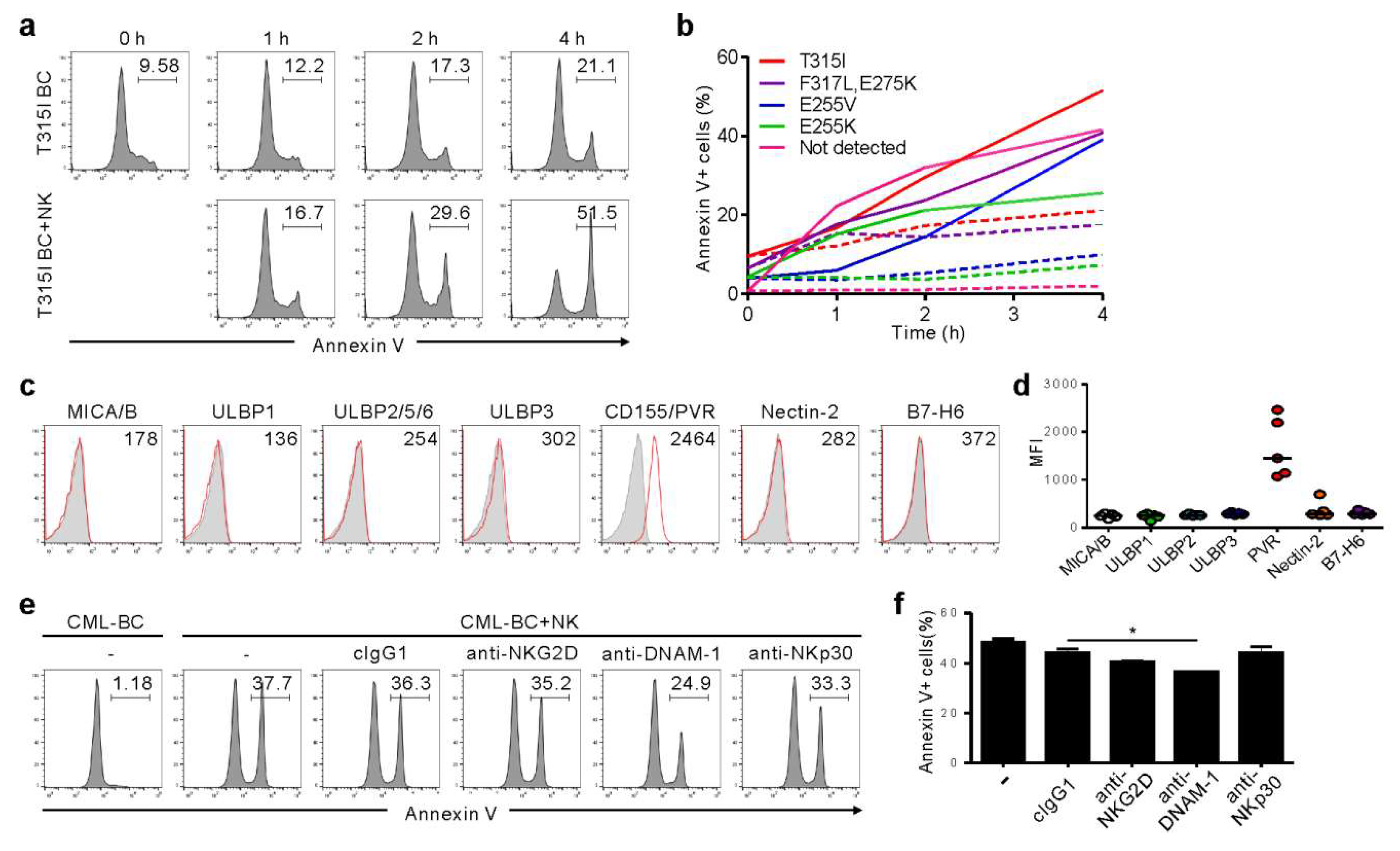
2.1. CML-BC Cells with BCR-ABL1 T315I Mutation are Susceptible to NK Cell-Mediated Killing
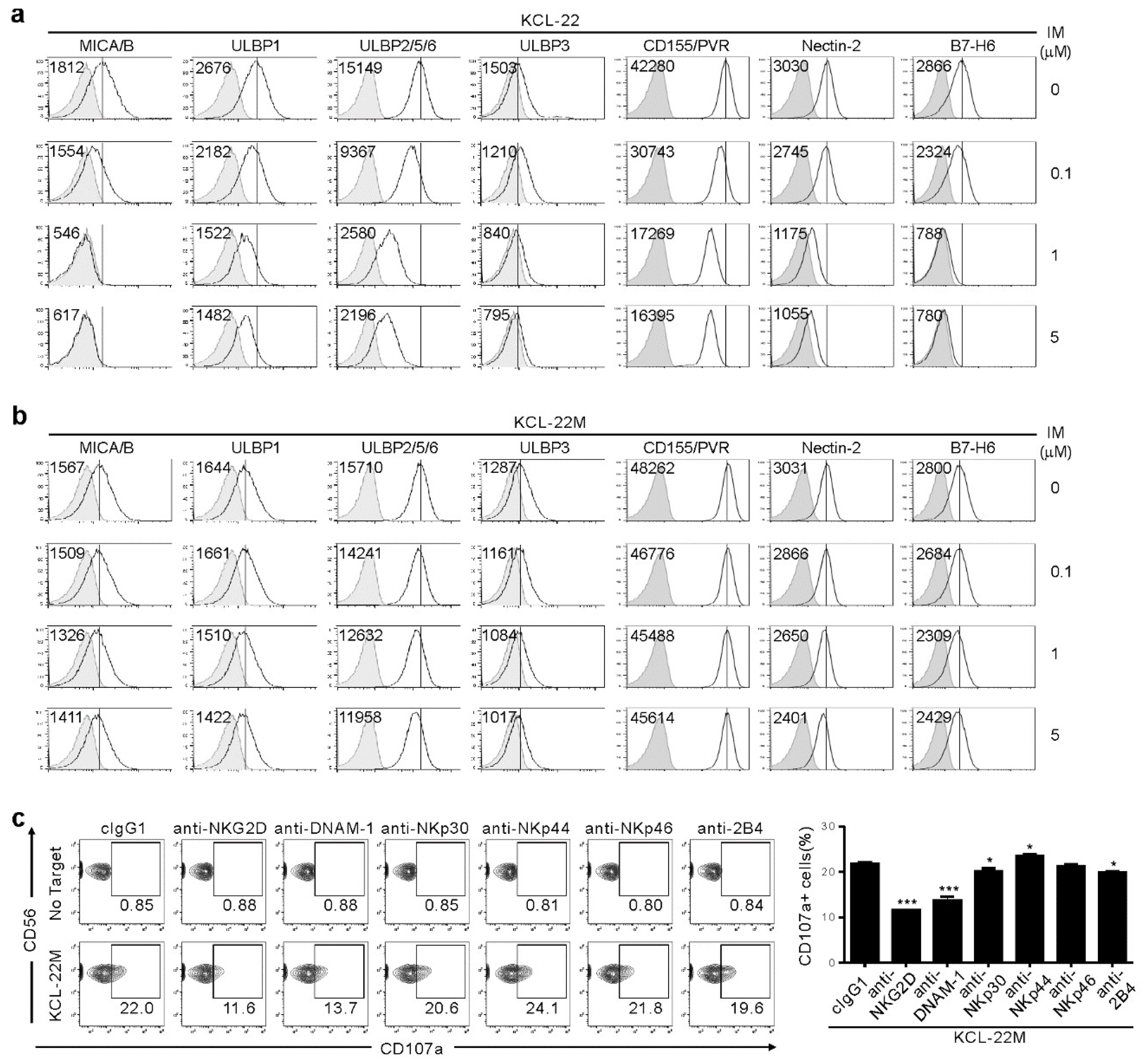
2.2. NK Cell Cytotoxicity Correlates with Activating Ligand Expression after IM Treatment
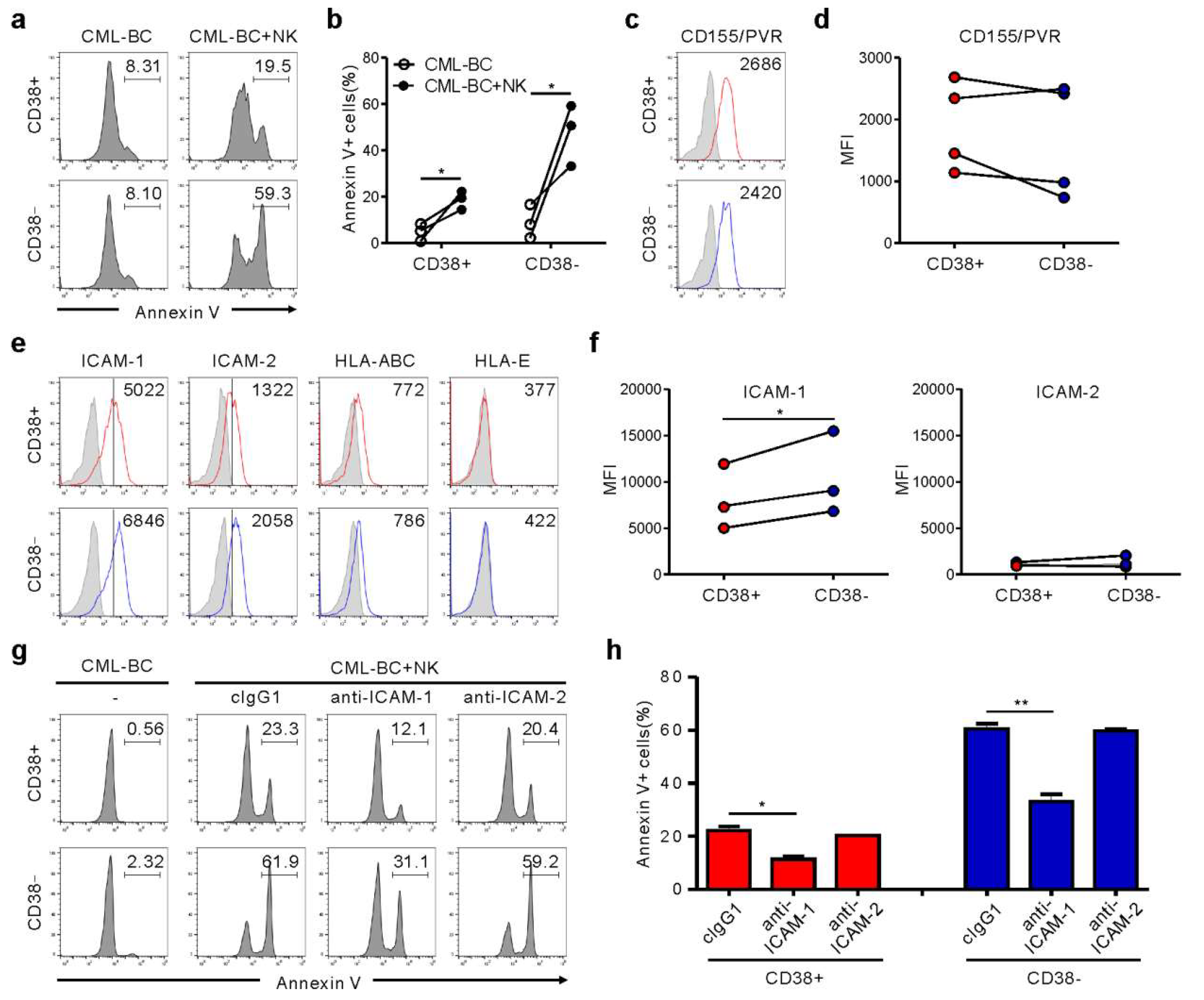
2.3. NK Cells Exert Cytotoxicity against Primary CD34+ CML-BC Cells with Different BCR-ABL Mutation
2.4. Association of ICAM-1 with Increased Susceptibility of CML-BC LSC to NK Cells
3. Materials and Methods
3.1. Cells and Reagents
3.2. NK Cell Expansion
3.3. Antibodies
3.4. NK Cell Degranulation Assay
3.5. NK Cell IFN-γ Production Assay
3.6. Assay of Target Cell Lysis by NK Cells
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- O’Hare, T.; Zabriskie, M.S.; Eiring, A.M.; Deininger, M.W. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat. Rev. Cancer 2012, 12, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; Mancini, M.; Bavaro, L.; Cavo, M.; Martinelli, G. Chronic myeloid leukemia: the paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 2018, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, L.; Wang, Z.; Ho, Y.; McDonald, T.; Holyoake, T.L.; Chen, W.; Bhatia, R.; Li, L. Activation of p53 by SIRT1 Inhibition Enhances Elimination of CML Leukemia Stem Cells in Combination with Imatinib. Cancer Cell 2012, 21, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; McDonald, T.; Lin, A.; Chakraborty, S.; Huang, Q.; Snyder, D.S.; Bhatia, R. Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood 2011, 118, 5565–5572. [Google Scholar] [CrossRef] [PubMed]
- Long, E.O.; Kim, H.S.; Liu, N.; Peterson, M.E.; Rajagopalan, S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu. Rev. Immunol. 2013, 31, 227–258. [Google Scholar] [CrossRef]
- Carlsten, M.; Järås, M. Natural Killer Cells in Myeloid Malignancies: Immune Surveillance, NK Cell Dysfunction, and Pharmacological Opportunities to Bolster the Endogenous NK Cells. Front. Immunol. 2019, 10, 2357. [Google Scholar] [CrossRef]
- Yong, A.S.; Keyvanfar, K.; Hensel, N.; Eniafe, R.; Savani, B.N.; Berg, M.; Lundqvist, A.; Adams, S.; Sloand, E.M.; Goldman, J.M.; et al. Primitive quiescent CD34+ cells in chronic myeloid leukemia are targeted by in vitro expanded natural killer cells, which are functionally enhanced by bortezomib. Blood 2009, 113, 875–882. [Google Scholar] [CrossRef]
- Bryceson, Y.T.; March, M.; Barber, D.F.; Ljunggren, H.-G.; Long, E.O. Cytolytic granule polarization and degranulation controlled by different receptors in resting NK cells. J. Exp. Med. 2005, 202, 1001–1012. [Google Scholar] [CrossRef]
- Pierson, B.; Miller, J.S. CD56+bright and CD56+dim natural killer cells in patients with chronic myelogenous leukemia progressively decrease in number, respond less to stimuli that recruit clonogenic natural killer cells, and exhibit decreased proliferation on a per cell basis. Blood 1996, 88, 2279–2287. [Google Scholar] [CrossRef]
- Ilander, M.; Olsson-Strömberg, U.; Schlums, H.; Guilhot, J.; Brück, O.; Lähteenmäki, H.; Kasanen, T.; Koskenvesa, P.; Söderlund, S.; Höglund, M.; et al. Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia. Leukemia 2016, 31, 1108–1116. [Google Scholar] [CrossRef]
- Yuan, H.; Wang, Z.; Gao, C.; Chen, W.; Huang, Q.; Yee, J.-K.; Bhatia, R.; Chen, W.Y. BCR-ABL gene expression is required for its mutations in a novel KCL-22 cell culture model for acquired resistance of chronic myelogenous leukemia. J. Biol. Chem. 2010, 285, 5085–5096. [Google Scholar] [CrossRef] [PubMed]
- Boissel, N.; Rea, D.; Tieng, V.; Dulphy, N.; Brun, M.; Cayuela, J.-M.; Rousselot, P.; Tamouza, R.; Le Bouteiller, P.; Mahon, F.-X.; et al. BCR/ABL Oncogene Directly Controls MHC Class I Chain-Related Molecule A Expression in Chronic Myelogenous Leukemia. J. Immunol. 2006, 176, 5108–5116. [Google Scholar] [CrossRef] [PubMed]
- Sconocchia, G.; Lau, M.; Provenzano, M.; Rezvani, K.; Wongsena, W.; Fujiwara, H.; Hensel, N.; Melenhorst, J.; Li, J.; Ferrone, S.; et al. The antileukemia effect of HLA-matched NK and NK-T cells in chronic myelogenous leukemia involves NKG2D–target-cell interactions. Blood 2005, 106, 3666–3672. [Google Scholar] [CrossRef] [PubMed]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Invest. 2011, 121, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, Z.; Wu, X.; Chu, S.; Wang, J.; Yuan, H.; Roth, M.; Yuan, Y.-C.; Bhatia, R.; Chen, W. ATRA-Induced Cellular Differentiation and CD38 Expression Inhibits Acquisition of BCR-ABL Mutations for CML Acquired Resistance. PLoS Genet. 2014, 10, e1004414. [Google Scholar] [CrossRef] [PubMed]
- Maio, M.; Pinto, A.; Carbone, A.; Zagonel, V.; Gloghini, A.; Marotta, G.; Cirillo, D.; Colombatti, A.; Ferrara, F.; Del Vecchio, L. Differential expression of CD54/intercellular adhesion molecule-1 in myeloid leukemias and in lymphoproliferative disorders. Blood 1990, 76, 783–790. [Google Scholar] [CrossRef]
- Chowdhury, S.; Bandyopadhyay, S.; Chandra, S.; Mandal, C. Comparative analysis of differential expression of sialic acids and adhesion molecules on mononuclear cells of bone marrow and peripheral blood in childhood acute lymphoblastic leukaemia at diagnosis and clinical remission. Indian J. Biochem. Biophys. 2007, 44, 357–365. [Google Scholar]
- Sovalat, H.; Racadot, E.; Ojeda, M.; Lewandowski, H.; Chabouté, V.; Henon, P. CD34+Cells and CD34+CD38–Subset from Mobilized Blood Show Different Patterns of Adhesion Molecules Compared to Those from Steady-State Blood, Bone Marrow, and Cord Blood. J. Hematotherapy 2003, 12, 473–489. [Google Scholar] [CrossRef]
- Windisch, R.; Pirschtat, N.; Kellner, C.; Chen-Wichmann, L.; Lausen, J.; Humpe, A.; Krause, D.S.; Wichmann, C. Oncogenic Deregulation of Cell Adhesion Molecules in Leukemia. Cancers 2019, 11, 311. [Google Scholar] [CrossRef]
- Kwon, H.J.; Choi, G.E.; Ryu, S.; Kwon, S.J.; Kim, S.C.; Booth, C.; Nichols, K.E.; Kim, H.S. Stepwise phosphorylation of p65 promotes NF-kappaB activation and NK cell responses during target cell recognition. Nat. Commun. 2016, 7, e11686. [Google Scholar] [CrossRef]
- Nievergall, E.; Ramshaw, H.; Yong, A.S.; Biondo, M.; Busfield, S.J.; Vairo, G.; López, A.F.; Hughes, T.P.; White, D.L.; Hiwase, D.K. Monoclonal antibody targeting of IL-3 receptor α with CSL362 effectively depletes CML progenitor and stem cells. Blood 2014, 123, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Landberg, N.; Hansen, N.; Askmyr, M.; Ågerstam, H.; Lassen, C.; Rissler, M.; Hjorth-Hansen, H.; Mustjoki, S.; Järås, M.; Richter, J.; et al. IL1RAP expression as a measure of leukemic stem cell burden at diagnosis of chronic myeloid leukemia predicts therapy outcome. Leukemia 2015, 30, 255–258. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, N.; Kim, M.-Y.; Cho, Y.-U.; Chen, W.; Lee, K.-H.; Kim, H.S. PVR and ICAM-1 on Blast Crisis CML Stem and Progenitor Cells with TKI Resistance Confer Susceptibility to NK Cells. Cancers 2020, 12, 1923. https://doi.org/10.3390/cancers12071923
Kim N, Kim M-Y, Cho Y-U, Chen W, Lee K-H, Kim HS. PVR and ICAM-1 on Blast Crisis CML Stem and Progenitor Cells with TKI Resistance Confer Susceptibility to NK Cells. Cancers. 2020; 12(7):1923. https://doi.org/10.3390/cancers12071923
Chicago/Turabian StyleKim, Nayoung, Mi-Yeon Kim, Young-Uk Cho, WenYong Chen, Kyoo-Hyung Lee, and Hun Sik Kim. 2020. "PVR and ICAM-1 on Blast Crisis CML Stem and Progenitor Cells with TKI Resistance Confer Susceptibility to NK Cells" Cancers 12, no. 7: 1923. https://doi.org/10.3390/cancers12071923
APA StyleKim, N., Kim, M.-Y., Cho, Y.-U., Chen, W., Lee, K.-H., & Kim, H. S. (2020). PVR and ICAM-1 on Blast Crisis CML Stem and Progenitor Cells with TKI Resistance Confer Susceptibility to NK Cells. Cancers, 12(7), 1923. https://doi.org/10.3390/cancers12071923