Identification of Novel microRNA Prognostic Markers Using Cascaded Wx, a Neural Network-Based Framework, in Lung Adenocarcinoma Patients

 , , , and
, , , and 
Abstract
1. Introduction
2. Results
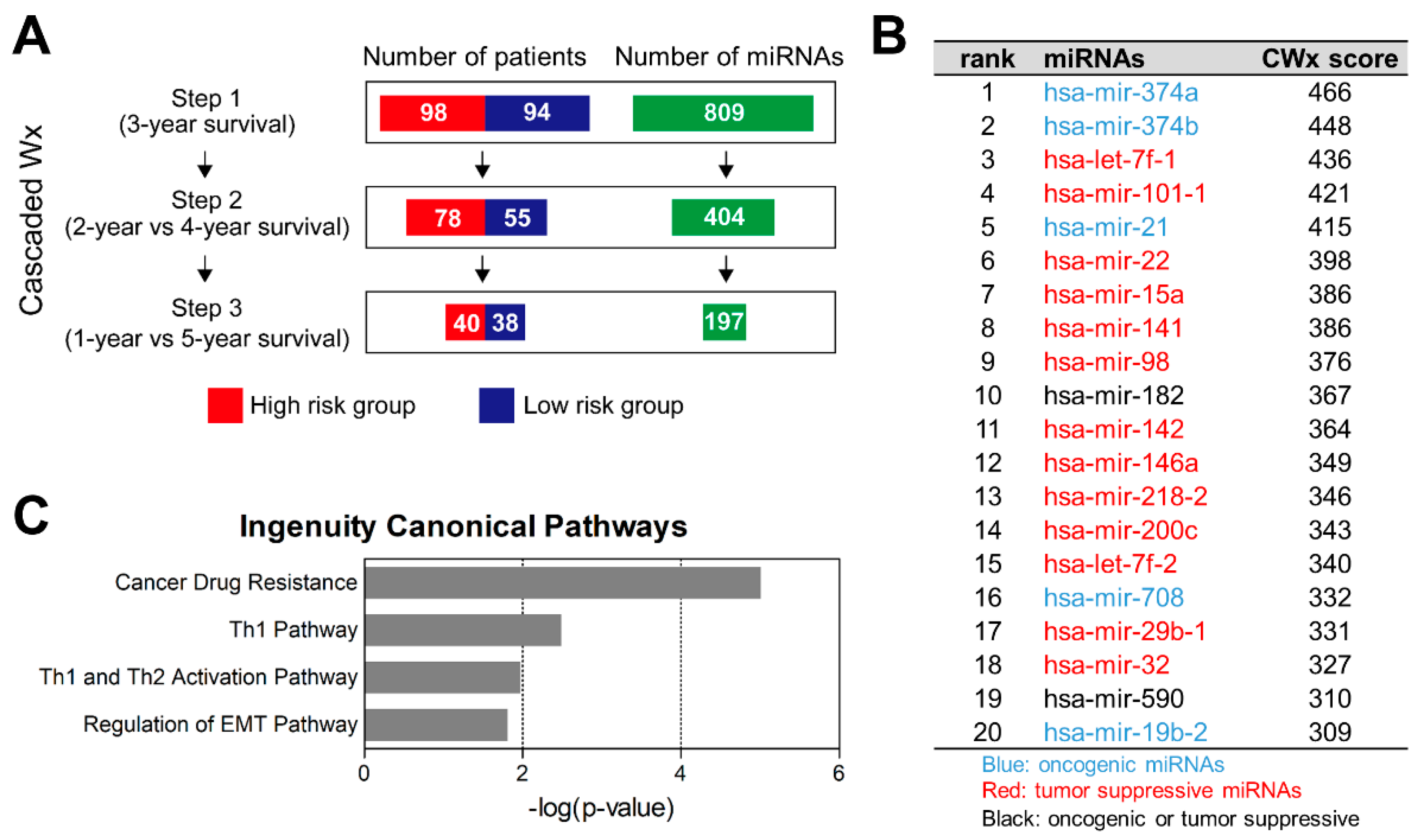
2.1. CWx Ranks miRNA Features Associated with Survival of LUAD Patients
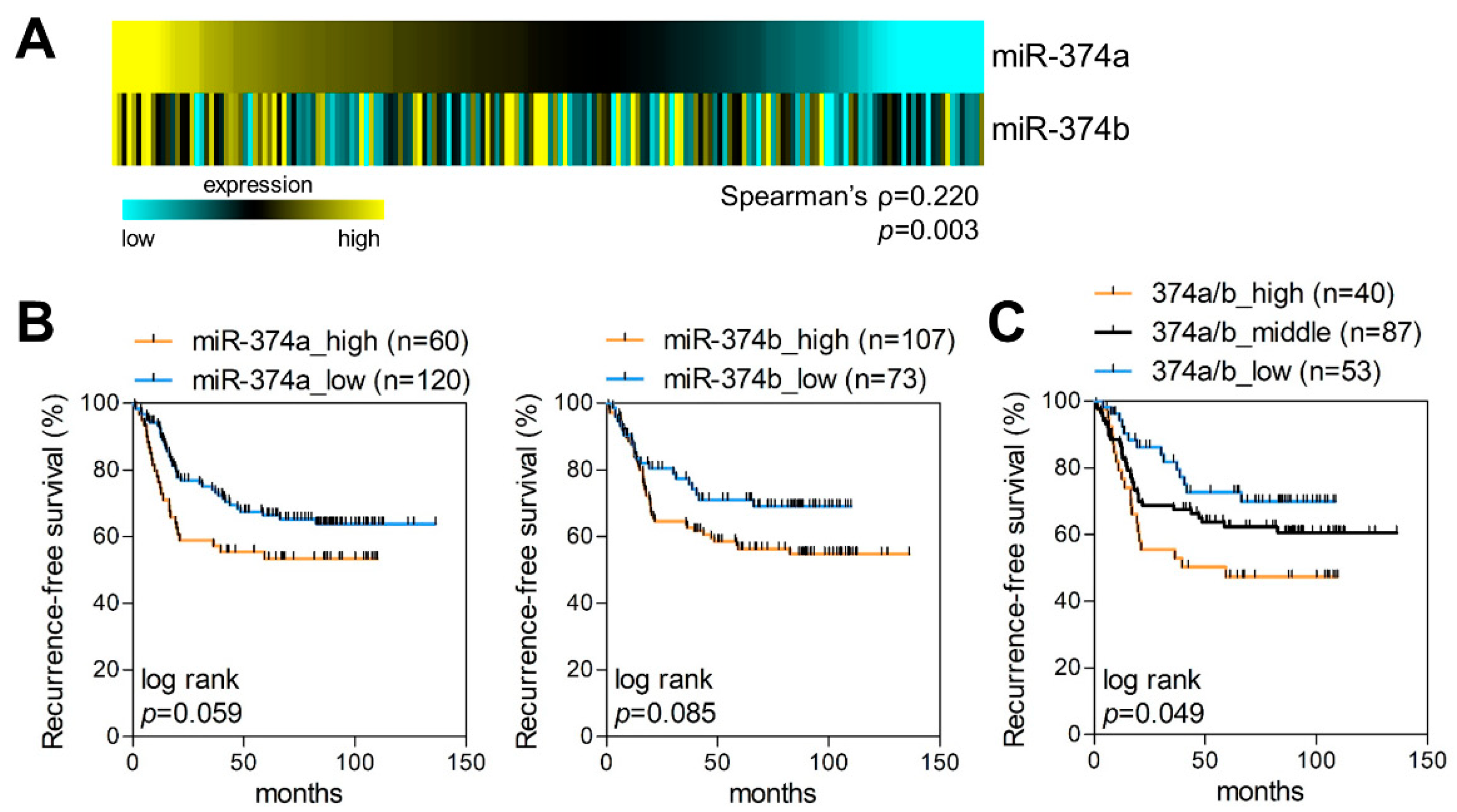
2.2. MiR-374a and MiR-374b Are Poor Prognostic Markers in LUAD
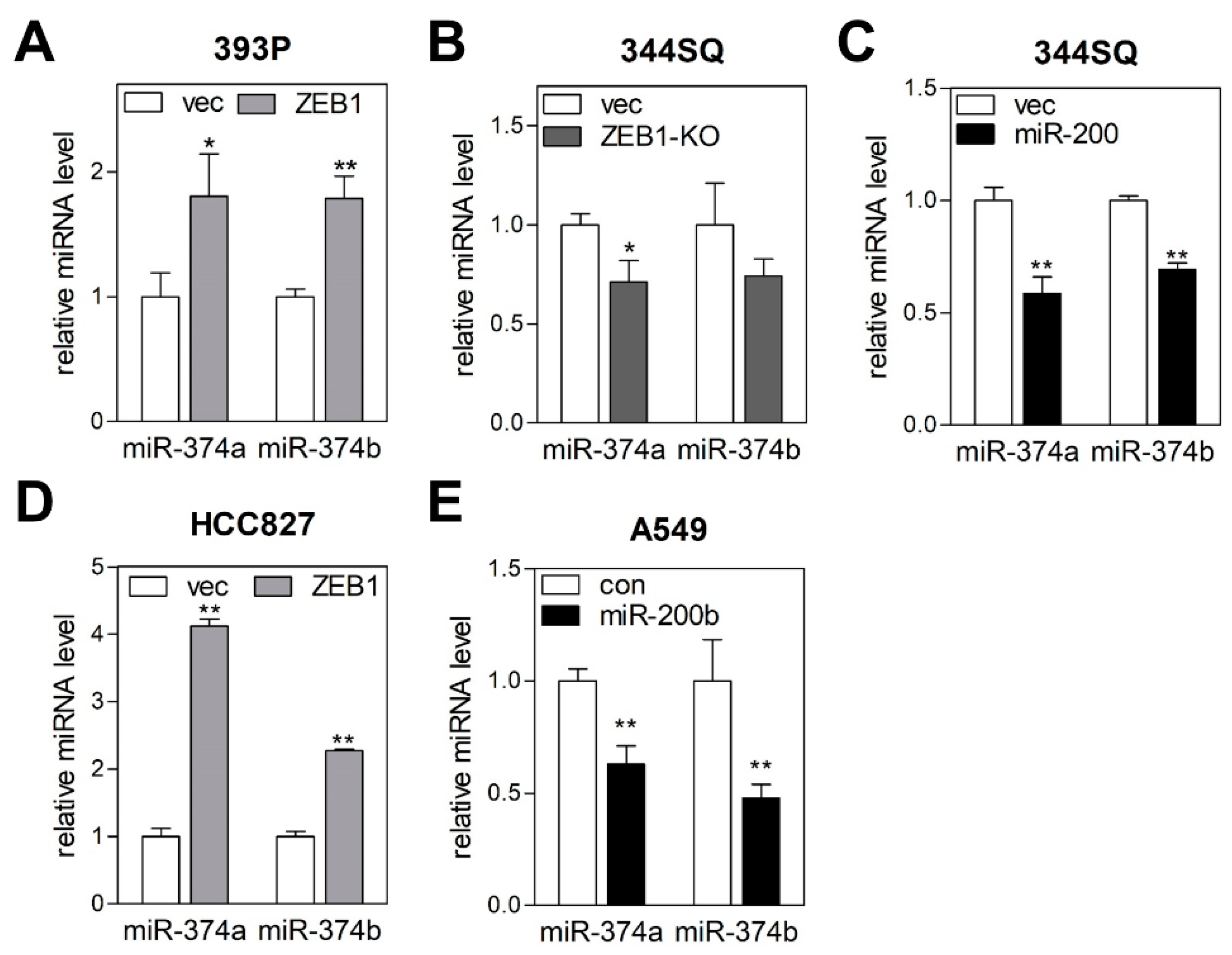
2.3. MiR-374a and MiR-374b Are Regulated by the ZEB1/miR-200 Feedback Loop
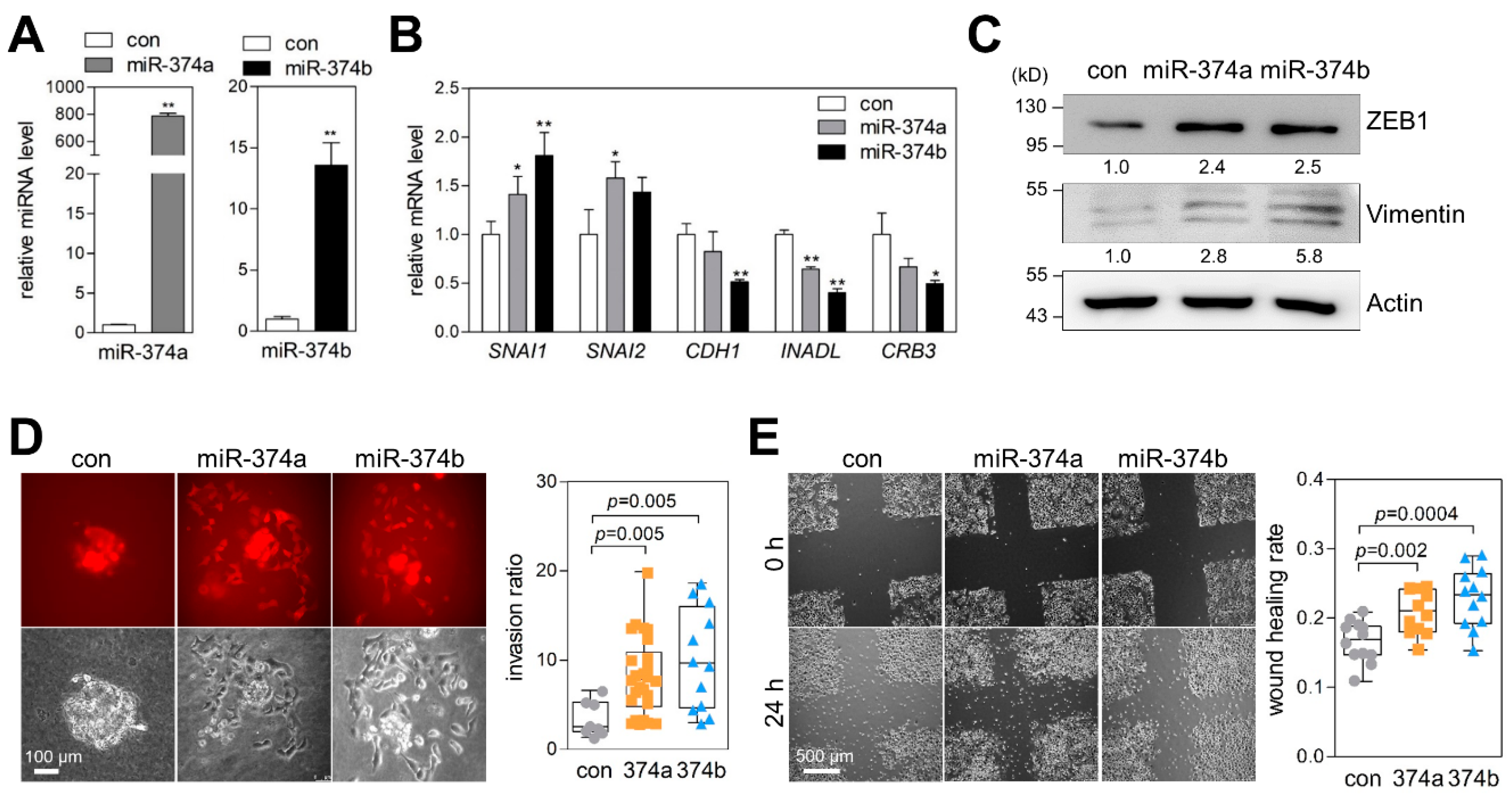
2.4. MiR-374a and MiR-374b Promote EMT, Migration, and Invasion of Lung Cancer Cells
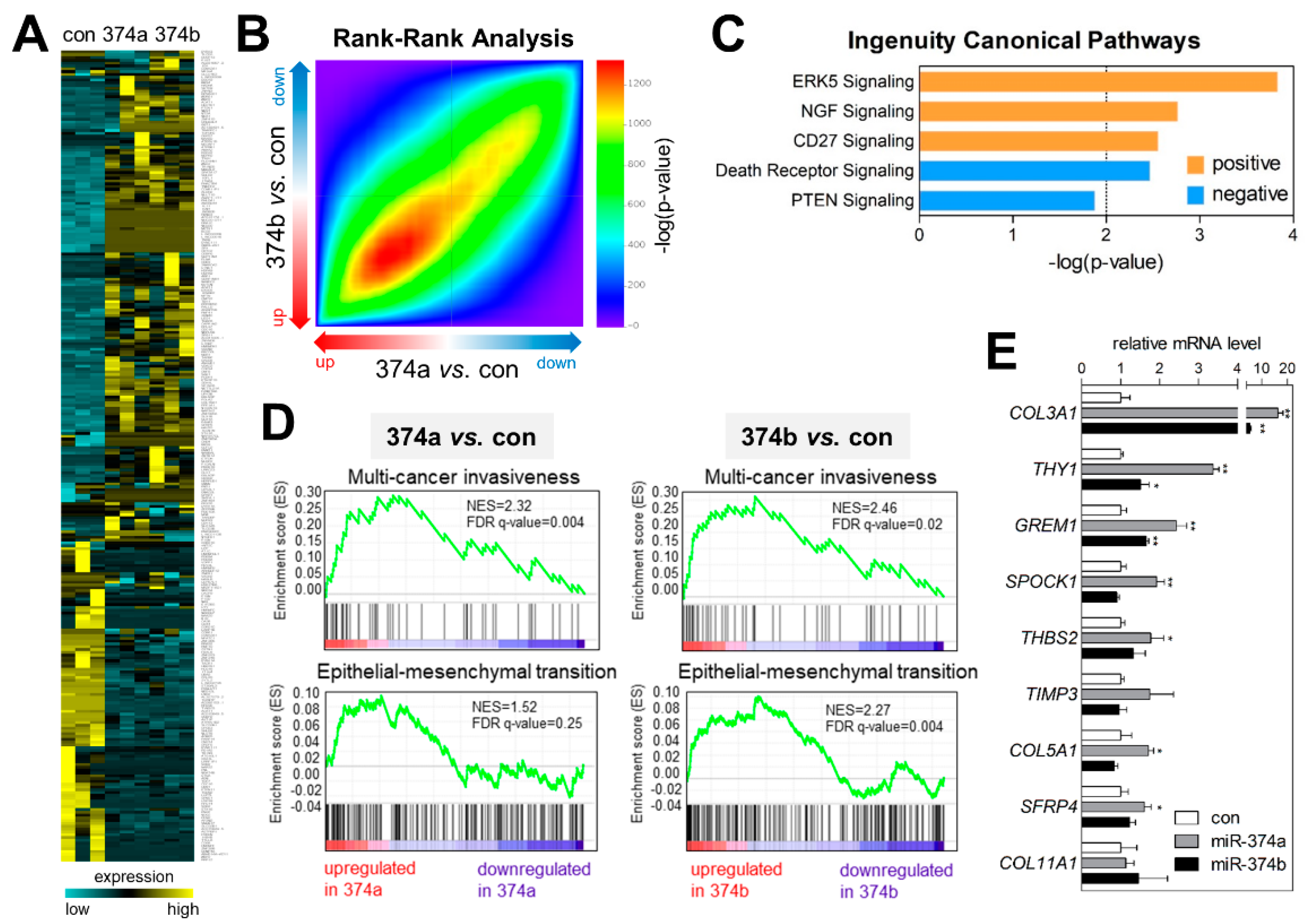
2.5. MiR-374a and MiR-374b Induce Gene-Expression Signatures Related to EMT and Invasiveness
3. Discussion
4. Materials and Methods
4.1. Data Acquisition
4.2. CWx Analysis
4.3. Cell Culture
4.4. Quantitative RT-PCR (qRT-PCR)
4.5. RNA Extraction from FFPE Tumors
4.6. MiRNA-Expression Profiling by NanoString
4.7. Western Blot
4.8. Spheroid Invasion Assay
4.9. RNA Sequencing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Boutros, P.C.; Lau, S.K.; Pintilie, M.; Liu, N.; Shepherd, F.A.; Der, S.D.; Tsao, M.S.; Penn, L.Z.; Jurisica, I. Prognostic gene signatures for non-small-cell lung cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 2824–2828. [Google Scholar] [CrossRef]
- Xie, Y.; Xiao, G.; Coombes, K.R.; Behrens, C.; Solis, L.M.; Raso, G.; Girard, L.; Erickson, H.S.; Roth, J.; Heymach, J.V.; et al. Robust gene expression signature from formalin-fixed paraffin-embedded samples predicts prognosis of non-small-cell lung cancer patients. Clin. Cancer Res. 2011, 17, 5705–5714. [Google Scholar] [CrossRef]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006, 9, 189–198. [Google Scholar] [CrossRef]
- Shin, B.; Park, S.; Hong, J.H.; An, H.J.; Chun, S.H.; Kang, K.; Ahn, Y.H.; Ko, Y.H.; Kang, K. Cascaded Wx: A novel prognosis-related feature selection framework in human lung adenocarcinoma transcriptomes. Front. Genet. 2019, 10, 662. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Wu, Y.; Song, Y.; Xiong, Y.; Wang, X.; Xu, K.; Han, B.; Bai, Y.; Li, L.; Zhang, Y.; Zhou, L. MicroRNA-21 (Mir-21) promotes cell growth and invasion by repressing tumor suppressor PTEN in colorectal cancer. Cell Physiol. Biochem. 2017, 43, 945–958. [Google Scholar] [CrossRef]
- Sarver, A.L.; Li, L.; Subramanian, S. MicroRNA miR-183 functions as an oncogene by targeting the transcription factor EGR1 and promoting tumor cell migration. Cancer Res. 2010, 70, 9570–9580. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Lin, W.; Creighton, C.J.; Rizvi, Z.H.; Gregory, P.A.; Goodall, G.J.; Thilaganathan, N.; Du, L.; Zhang, Y.; Pertsemlidis, A.; et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009, 23, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.; Peruzzi, P.P.; Lawler, S. MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 2014, 20, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Vosa, U.; Vooder, T.; Kolde, R.; Fischer, K.; Valk, K.; Tonisson, N.; Roosipuu, R.; Vilo, J.; Metspalu, A.; Annilo, T. Identification of miR-374a as a prognostic marker for survival in patients with early-stage nonsmall cell lung cancer. Genes Chromosomes Cancer 2011, 50, 812–822. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Creighton, C.J.; Gibbons, D.L.; Kurie, J.M. The role of epithelial-mesenchymal transition programming in invasion and metastasis: A clinical perspective. Cancer Manag. Res. 2013, 5, 187–195. [Google Scholar] [CrossRef]
- George, J.T.; Jolly, M.K.; Xu, S.; Somarelli, J.A.; Levine, H. Survival outcomes in cancer patients predicted by a partial EMT gene expression scoring metric. Cancer Res. 2017, 77, 6415–6428. [Google Scholar] [CrossRef]
- Cai, J.; Guan, H.; Fang, L.; Yang, Y.; Zhu, X.; Yuan, J.; Wu, J.; Li, M. MicroRNA-374a activates Wnt/beta-catenin signaling to promote breast cancer metastasis. J. Clin. Investig. 2013, 123, 566–579. [Google Scholar]
- Ma, L.; Shao, Z.; Zhao, Y. MicroRNA-374a promotes pancreatic cancer cell proliferation and epithelial to mesenchymal transition by targeting SRCIN1. Pathol. Res. Pract. 2019, 215, 152382. [Google Scholar] [CrossRef]
- Xu, X.; Wang, W.; Su, N.; Zhu, X.; Yao, J.; Gao, W.; Hu, Z.; Sun, Y. miR-374a promotes cell proliferation, migration and invasion by targeting SRCIN1 in gastric cancer. FEBS Lett. 2015, 589, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xia, H.; Zhuang, Z.; Miao, L.; Chen, X.; Cai, H. Axl-altered microRNAs regulate tumorigenicity and gefitinib resistance in lung cancer. Cell Death Dis. 2014, 5, e1227. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, G.; Zheng, W.; Xue, Q.; Wei, D.; Zheng, Y.; Yuan, J. MiR-454-3p and miR-374b-5p suppress migration and invasion of bladder cancer cells through targetting ZEB2. Biosci. Rep. 2018, 38, BSR20181436. [Google Scholar] [CrossRef]
- Li, H.; Liang, J.; Qin, F.; Zhai, Y. MiR-374b-5p-FOXP1 feedback loop regulates cell migration, epithelial-mesenchymal transition and chemosensitivity in ovarian cancer. Biochem. Biophys. Res. Commun. 2018, 505, 554–560. [Google Scholar] [CrossRef]
- Plaisier, S.B.; Taschereau, R.; Wong, J.A.; Graeber, T.G. Rank-rank hypergeometric overlap: Identification of statistically significant overlap between gene-expression signatures. Nucleic Acids Res. 2010, 38, e169. [Google Scholar] [CrossRef] [PubMed]
- de Miguel-Pérez, D.; Bayarri-Lara, C.I.; Ortega, F.G.; Russo, A.; Moyano Rodriguez, M.J.; Alvarez-Cubero, M.J.; Maza Serrano, E.; Lorente, J.A.; Rolfo, C.; Serrano, M.J. Post-surgery circulating tumor cells and AXL overexpression as new poor prognostic biomarkers in resected lung adenocarcinoma. Cancers 2019, 11, 1750. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Lin, C.J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 27. [Google Scholar] [CrossRef]
- Chou, H.L.; Yao, C.T.; Su, S.L.; Lee, C.Y.; Hu, K.Y.; Terng, H.J.; Shih, Y.W.; Chang, Y.T.; Lu, Y.F.; Chang, C.W.; et al. Gene expression profiling of breast cancer survivability by pooled cDNA microarray analysis using logistic regression, artificial neural networks and decision trees. BMC Bioinform. 2013, 14, 100. [Google Scholar] [CrossRef]
- Zhang, J.; Hadj-Moussa, H.; Storey, K.B. Current progress of high-throughput microRNA differential expression analysis and random forest gene selection for model and non-model systems: An R implementation. J. Integr. Bioinform. 2016, 13, 306. [Google Scholar] [CrossRef]
- Yao, D.; Yang, J.; Zhan, X.; Zhan, X.; Xie, Z. A novel random forests-based feature selection method for microarray expression data analysis. Int. J. Data Min. Bioinform. 2015, 13, 84–101. [Google Scholar] [CrossRef]
- Freres, P.; Wenric, S.; Boukerroucha, M.; Fasquelle, C.; Thiry, J.; Bovy, N.; Struman, I.; Geurts, P.; Collignon, J.; Schroeder, H.; et al. Circulating microRNA-based screening tool for breast cancer. Oncotarget 2016, 7, 5416–5428. [Google Scholar] [CrossRef]
- Wenric, S.; Shemirani, R. Using supervised learning methods for gene selection in RNA-seq case-control studies. Front. Genet. 2018, 9, 297. [Google Scholar] [CrossRef]
- Park, S.; Shin, B.; Sang Shim, W.; Choi, Y.; Kang, K.; Kang, K. Wx: A neural network-based feature selection algorithm for transcriptomic data. Sci. Rep. 2019, 9, 10500. [Google Scholar] [CrossRef] [PubMed]
- Sozzi, G.; Boeri, M.; Rossi, M.; Verri, C.; Suatoni, P.; Bravi, F.; Roz, L.; Conte, D.; Grassi, M.; Sverzellati, N.; et al. Clinical utility of a plasma-based miRNA signature classifier within computed tomography lung cancer screening: A correlative MILD trial study. J. Clin. Oncol. 2014, 32, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Montani, F.; Marzi, M.J.; Dezi, F.; Dama, E.; Carletti, R.M.; Bonizzi, G.; Bertolotti, R.; Bellomi, M.; Rampinelli, C.; Maisonneuve, P.; et al. miR-Test: A blood test for lung cancer early detection. J. Natl. Cancer Inst. 2015, 107, 63. [Google Scholar] [CrossRef] [PubMed]
- Bian, H.; Zhou, Y.; Zhou, D.; Zhang, Y.; Shang, D.; Qi, J. The latest progress on miR-374 and its functional implications in physiological and pathological processes. J. Cell Mol. Med. 2019, 23, 3063–3076. [Google Scholar] [CrossRef]
- Son, D.; Kim, Y.; Lim, S.; Kang, H.G.; Kim, D.H.; Park, J.W.; Cheong, W.; Kong, H.K.; Han, W.; Park, W.Y.; et al. miR-374a-5p promotes tumor progression by targeting ARRB1 in triple negative breast cancer. Cancer Lett. 2019, 454, 224–233. [Google Scholar] [CrossRef]
- Long, Z.W.; Wu, J.H.; Cai, H.; Wang, Y.N.; Zhou, Y. MiR-374b promotes proliferation and inhibits apoptosis of human GIST cells by inhibiting PTEN through activation of the PI3K/Akt pathway. Mol. Cells 2018, 41, 532–544. [Google Scholar] [PubMed]
- Wu, H.; Liu, Y.; Shu, X.O.; Cai, Q. MiR-374a suppresses lung adenocarcinoma cell proliferation and invasion by targeting TGFA gene expression. Carcinogenesis 2016, 37, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Xu, P.; Liu, Z.; Zhen, Y.; Chen, Y.; Liu, Y.; Fu, Q.; Deng, X.; Liang, Z.; Li, Y.; et al. Dual roles of miR-374a by modulated c-Jun respectively targets CCND1-inducing PI3K/AKT signal and PTEN-suppressing Wnt/beta-catenin signaling in non-small-cell lung cancer. Cell Death Dis. 2018, 9, 78. [Google Scholar] [CrossRef]
- Balzeau, J.; Menezes, M.R.; Cao, S.; Hagan, J.P. The LIN28/let-7 pathway in cancer. Front. Genet. 2017, 8, 31. [Google Scholar] [CrossRef]
- Han, L.; Chen, W.; Xia, Y.; Song, Y.; Zhao, Z.; Cheng, H.; Jiang, T. MiR-101 inhibits the proliferation and metastasis of lung cancer by targeting zinc finger E-box binding homeobox 1. Am. J. Transl. Res. 2018, 10, 1172–1183. [Google Scholar] [PubMed]
- Feng, Y.H.; Tsao, C.J. Emerging role of microRNA-21 in cancer. Biomed. Rep. 2016, 5, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.H.; Gibbons, D.L.; Chakravarti, D.; Creighton, C.J.; Rizvi, Z.H.; Adams, H.P.; Pertsemlidis, A.; Gregory, P.A.; Wright, J.A.; Goodall, G.J.; et al. ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating miR-34a expression. J. Clin. Investig. 2012, 122, 3170–3183. [Google Scholar] [CrossRef]
- Kim, J.S.; Kim, E.J.; Lee, S.; Tan, X.; Liu, X.; Park, S.; Kang, K.; Yoon, J.S.; Ko, Y.H.; Kurie, J.M.; et al. MiR-34a and miR-34b/c have distinct effects on the suppression of lung adenocarcinomas. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Seo, H.D.; Hennighausen, L.; Lee, D.; Kang, K. Octopus-toolkit: A workflow to automate mining of public epigenomic and transcriptomic next-generation sequencing data. Nucleic Acids Res. 2018, 46, e53. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | All Patients (n = 180) | hsa-miR-374a-5p | hsa-miR-374b-5p | ||||
|---|---|---|---|---|---|---|---|
| Low (n = 120) | High (n = 60) | p | Low (n = 73) | High (n = 107) | p | ||
| Age, years | 63.78 (28–85) | 0.463 | 0.791 | ||||
| <65 | 94 (52.2%) | 65 (69.1%) | 29 (30.9%) | 39 (41.5%) | 55 (58.5%) | ||
| ≥65 | 86 (47.8%) | 55 (64.0%) | 31 (36.0%) | 34 (39.5%) | 52 (60.5%) | ||
| Gender | 0.916 | 0.312 | |||||
| Male | 97 (53.9%) | 65 (67.0%) | 32 (33.0%) | 36 (37.1%) | 61 (62.9%) | ||
| Female | 83 (46.1%) | 55 (66.3%) | 28 (33.7%) | 37 (44.6%) | 46 (55.4%) | ||
| Smoking | 0.739 | <0.001 | |||||
| Never smoker | 101 (58.0%) | 66 (65.3%) | 35 (34.7%) | 51 (50.5%) | 50 (49.5%) | ||
| Current + Ex-smoker | 73 (21.3%) | 29 (54.7%) | 24 (45.3%) | 22 (30.1%) | 51 (69.9%) | ||
| Differentiation grade | 0.939 | 0.180 | |||||
| Well differentiated | 49 (27.2%) | 31 (63.3%) | 18 (36.7%) | 23 (46.9%) | 26 (53.1%) | ||
| Moderately | 102 (56.7%) | 70 (68.6%) | 32 (31.4%) | 41 (40.2%) | 61 (59.8%) | ||
| Poorly + Undifferentiated | 29 (16.1%) | 19 (65.5%) | 10 (34.5%) | 9 (31.0%) | 20 (69.0%) | ||
| Pathological stage | 0.059 | 0.135 | |||||
| I | 109 (61.2%) | 77 (70.6%) | 32 (29.4%) | 47 (43.1%) | 62 (56.9%) | ||
| II | 33 (18.6%) | 22 (66.7%) | 11 (33.3%) | 14 (42.4%) | 19 (57.6%) | ||
| III + IV | 36 (20.2%) | 19 (52.8%) | 17 (47.2%) | 10 (27.8%) | 26 (72.2%) | ||
| Tumor size | 0.134 | 0.510 | |||||
| <3 cm | 106 (58.9%) | 76 (71.7%) | 30 (28.3%) | 46 (43.4%) | 60 (56.6%) | ||
| 3≤ T <7 cm | 68 (37.8%) | 40 (58.8%) | 28 (41.2%) | 24 (35.3%) | 44 (64.7%) | ||
| ≥7 cm | 6 (3.3%) | 4 (66.7%) | 2 (33.3%) | 3 (50.0%) | 3 (50.0%) | ||
| Vascular invasion | 0.907 | 0.852 | |||||
| No | 152 (84.4%) | 105 (69.1%) | 47 (30.9%) | 63 (41.4%) | 89 (58.6%) | ||
| Yes or unknown | 28 (15.6%) | 15 (53.6%) | 13 (46.4%) | 10 (35.7%) | 18 (64.3%) | ||
| Lymphatic invasion | 0.758 | 0.889 | |||||
| No | 114 (63.3%) | 76 (66.7%) | 38 (33.3%) | 48 (42.1%) | 66 (57.9%) | ||
| Yes or unknown | 66 (36.7%) | 44 (66.7%) | 22 (33.3%) | 25 (37.9%) | 41 (62.1%) | ||
| Perineural invasion | 0.969 | 0.949 | |||||
| No | 169 (93.9%) | 115 (68.0%) | 54 (32.0%) | 71 (42.0%) | 98 (58.0%) | ||
| Yes or unknown | 11 (6.1%) | 5 (45.5%) | 6 (54.5%) | 2 (18.2%) | 9 (81.8%) | ||
| EGFR mutation | 0.104 | 0.737 | |||||
| No or unknown | 116 (64.4%) | 77 (66.4%) | 39 (33.6%) | 45 (18.9%) | 71 (45.6%) | ||
| Yes | 63 (35.6%) | 42 (66.7%) | 21 (33.3%) | 28 (33.3%) | 35 (2.2%) | ||
| Disease recurrence | 0.128 | 0.053 | |||||
| No | 113 (62.8%) | 80 (38.8%) | 33 (61.2%) | 52 (46.0%) | 61 (54.0%) | ||
| Yes | 67 (37.2%) | 40 (44.4%) | 27 (55.6%) | 21 (31.3%) | 46 (68.7%) | ||
| Disease survival | 0.155 | 0.407 | |||||
| Survival | 115(63.9%) | 81(70.4%) | 34(29.6%) | 44(38.3%) | 71(61.7%) | ||
| Death | 65(36.1%) | 39(60.0%) | 26(40.0%) | 29(44.6%) | 36(55.4%) | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.S.; Chun, S.H.; Park, S.; Lee, S.; Kim, S.E.; Hong, J.H.; Kang, K.; Ko, Y.H.; Ahn, Y.-H. Identification of Novel microRNA Prognostic Markers Using Cascaded Wx, a Neural Network-Based Framework, in Lung Adenocarcinoma Patients. Cancers 2020, 12, 1890. https://doi.org/10.3390/cancers12071890
Kim JS, Chun SH, Park S, Lee S, Kim SE, Hong JH, Kang K, Ko YH, Ahn Y-H. Identification of Novel microRNA Prognostic Markers Using Cascaded Wx, a Neural Network-Based Framework, in Lung Adenocarcinoma Patients. Cancers. 2020; 12(7):1890. https://doi.org/10.3390/cancers12071890
Chicago/Turabian StyleKim, Jeong Seon, Sang Hoon Chun, Sungsoo Park, Sieun Lee, Sae Eun Kim, Ji Hyung Hong, Keunsoo Kang, Yoon Ho Ko, and Young-Ho Ahn. 2020. "Identification of Novel microRNA Prognostic Markers Using Cascaded Wx, a Neural Network-Based Framework, in Lung Adenocarcinoma Patients" Cancers 12, no. 7: 1890. https://doi.org/10.3390/cancers12071890
APA StyleKim, J. S., Chun, S. H., Park, S., Lee, S., Kim, S. E., Hong, J. H., Kang, K., Ko, Y. H., & Ahn, Y.-H. (2020). Identification of Novel microRNA Prognostic Markers Using Cascaded Wx, a Neural Network-Based Framework, in Lung Adenocarcinoma Patients. Cancers, 12(7), 1890. https://doi.org/10.3390/cancers12071890