Mechanisms of Androgen Receptor Agonist- and Antagonist-Mediated Cellular Senescence in Prostate Cancer



Abstract
1. Introduction
1.1. Prostate Cancer (PCa)
1.2. The Androgen Receptor (AR) and Adaptive Response of PCa
1.3. PCa Cell Response to Androgen Levels
1.4. Cellular Senescence in PCa
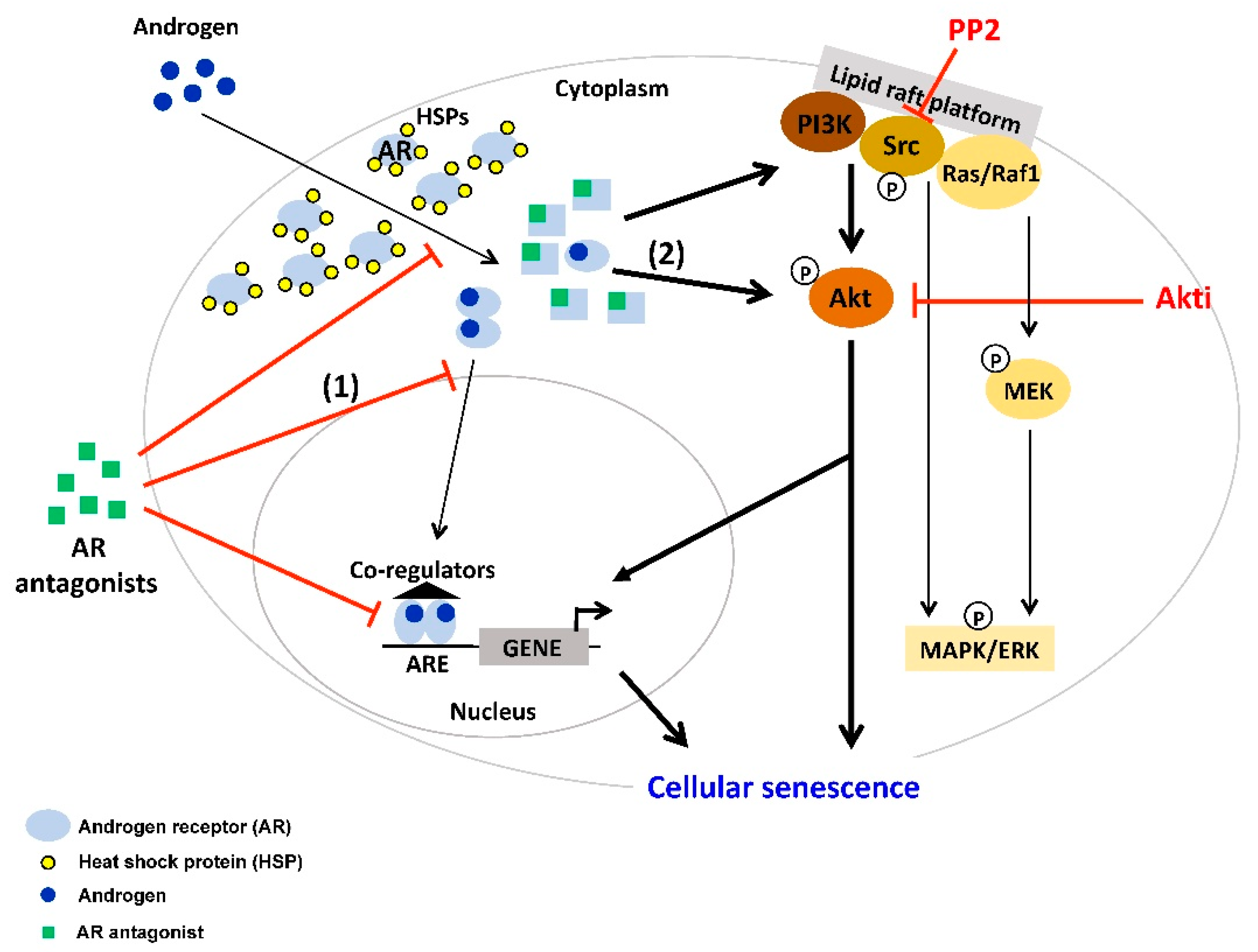
2. AR Antagonist-Induced Cellular Senescence
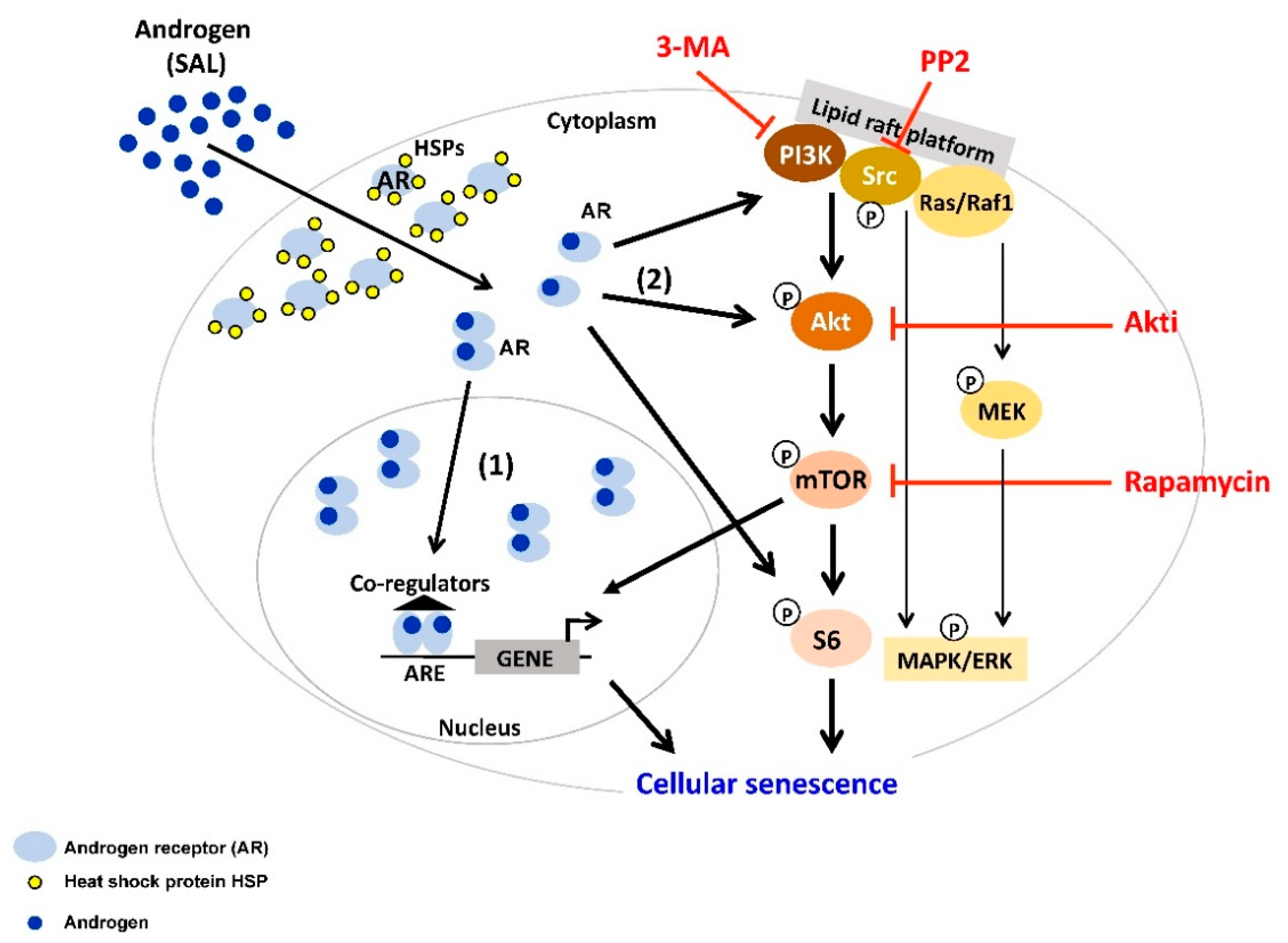
3. Supraphysiological Levels of Androgens Induce Cellular Senescence
4. Interplay between AR-Signaling and other Cellular Signaling Pathways in Senescent PCa
5. Targeting AR Ligand-Induced Cellular Senescent PCa Cells with Senolytic Compounds
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AA | atraric acid |
| ADT | androgen deprivation therapy |
| AR | androgen receptor |
| Apa | Apalutamide |
| BAT | bipolar androgen therapy |
| Bic | bicalutamide |
| CRPC | castration-resistant PCa |
| CSPC | castration-sensitive PCa |
| CDK | cyclin-dependent kinase |
| DHT | dihydrotestosterone |
| Enz | enzalutamide |
| LAL | low androgen level |
| LBD | ligand binding domain |
| nmCRPC | non-metastatic CRPC |
| mCRPC | metastatic CRPC |
| mTORC | mammalian target of rapamycin complex |
| PSA | prostate-specific antigen |
| PCa | prostate cancer |
| ROS | reactive oxygen species |
| SA-β-Gal | senescence-associated β-galactosidase |
| SAHF | senescence-associated heterochromatin foci |
| SAL | supraphysiological androgen level |
| SASP | senescence-associated secretory phenotype |
| TKs | tyrosine kinases |
| VDEC | vas deferens epithelial cells |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Mearini, L.; Zucchi, A.; Nunzi, E.; Villirillo, T.; Bini, V.; Porena, M. Low serum testosterone levels are predictive of prostate cancer. World J. Urol. 2013, 31, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Pastuszak, A.W.; Rodriguez, K.M.; Nguyen, T.M.; Khera, M. Testosterone therapy and prostate cancer. Transl. Androl. Urol. 2016, 5, 909–920. [Google Scholar] [CrossRef]
- Bousset, L.; Rambur, A.; Fouache, A.; Bunay, J.; Morel, L.; Lobaccaro, J.A.; Baron, S.; Trousson, A.; de Joussineau, C. New Insights in Prostate Cancer Development and Tumor Therapy: Modulation of Nuclear Receptors and Specific Role of Liver X Receptors. Int. J. Mol. Sci. 2018, 19, 2545. [Google Scholar] [CrossRef]
- Alpajaro, S.I.R.; Harris, J.A.K.; Evans, C.P. Non-metastatic castration resistant prostate cancer: A review of current and emerging medical therapies. Prostate Cancer Prostatic Dis. 2019, 22, 16–23. [Google Scholar] [CrossRef]
- Lin, T.T.; Chen, Y.H.; Wu, Y.P.; Chen, S.Z.; Li, X.D.; Lin, Y.Z.; Chen, S.H.; Zheng, Q.S.; Wei, Y.; Xu, N.; et al. Risk factors for progression to castration-resistant prostate cancer in metastatic prostate cancer patients. J. Cancer 2019, 10, 5608–5613. [Google Scholar] [CrossRef]
- Helsen, C.; Van den Broeck, T.; Voet, A.; Prekovic, S.; Van Poppel, H.; Joniau, S.; Claessens, F. Androgen receptor antagonists for prostate cancer therapy. Endocr. Relat. Cancer 2014, 21, T105–T118. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Chen, S.; Ng, P.; Bubley, G.J.; Nelson, P.S.; Mostaghel, E.A.; March, B.; Matsumoto, A.M.; Simon, N.I.; Wang, H.; et al. Intratumoral De Novo Steroid Synthesis Activates Androgen Receptor in Castration Resistant Prostate Cancer and is Upregulated by Treatment with CYP17A1 Inhibitors. Cancer Res. 2011, 71, 6503–6513. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Fang, H.; Chen, C.; Wu, Y.; Wang, Y.; Ge, H.; Wang, L.; Wan, Y.; He, H. Metastatic castration-resistant prostate cancer: Academic insights and perspectives through bibliometric analyses. Medicine 2020, 99, e19760. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.A.; Malhotra, S.V.; Stoyanova, T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front. Oncol. 2019, 9, 801. [Google Scholar] [CrossRef]
- Nguyen, M.M.; Dincer, Z.; Wade, J.R.; Alur, M.; Michalak, M.; Defranco, D.B.; Wang, Z. Cytoplasmic localization of the androgen receptor is independent of calreticulin. Mol. Cell. Endocrinol. 2009, 302, 65–72. [Google Scholar] [CrossRef]
- Lakshmana, G.; Baniahmad, A. Interference with the androgen receptor protein stability in therapy-resistant prostate cancer. Int. J. Cancer 2019, 144, 1775–1779. [Google Scholar] [CrossRef]
- Gil, D.; Zarzycka, M.; Dulinska-Litewka, J.; Ciolczyk-Wierzbicka, D.; Lekka, M.; Laidler, P. Dihydrotestosterone increases the risk of bladder cancer in men. Hum. Cell 2019, 32, 379–389. [Google Scholar] [CrossRef]
- Zboray, L.; Pluciennik, A.; Curtis, D.; Liu, Y.; Berman-Booty, L.D.; Orr, C.; Kesler, C.T.; Berger, T.; Gioeli, D.; Paschal, B.M.; et al. Preventing the Androgen Receptor N/C Interaction Delays Disease Onset in a Mouse Model of SBMA. Cell Rep. 2015, 13, 2312–2323. [Google Scholar] [CrossRef] [PubMed]
- Hankey, W.; Chen, Z.; Wang, Q. Shaping Chromatin States in Prostate Cancer by Pioneer Transcription Factors. Cancer Res. 2020, 80, 2427–2436. [Google Scholar] [CrossRef]
- De Silva, D.; Zhang, Z.; Liu, Y.; Parker, J.S.; Xu, C.; Cai, L.; Wang, G.G.; Earp, H.S.; Whang, Y.E. Interaction between androgen receptor and coregulator SLIRP is regulated by Ack1 tyrosine kinase and androgen. Sci. Rep. 2019, 9, 18637. [Google Scholar] [CrossRef]
- Regufe da Mota, S.; Bailey, S.; Strivens, R.A.; Hayden, A.L.; Douglas, L.R.; Duriez, P.J.; Borrello, M.T.; Benelkebir, H.; Ganesan, A.; Packham, G.; et al. LSD1 inhibition attenuates androgen receptor V7 splice variant activation in castration resistant prostate cancer models. Cancer Cell Int. 2018, 18, 71. [Google Scholar] [CrossRef]
- Van de Wijngaart, D.J.; Dubbink, H.J.; van Royen, M.E.; Trapman, J.; Jenster, G. Androgen receptor coregulators: Recruitment via the coactivator binding groove. Mol. Cell. Endocrinol. 2012, 352, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Heemers, H.V.; Tindall, D.J. Androgen Receptor (AR) Coregulators: A Diversity of Functions Converging on and Regulating the AR Transcriptional Complex. Endocr. Rev. 2007, 28, 778–808. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Zhang, Z.; Wu, Y.; Yu, W.Y.; Zhang, J.; Jiang, Z.M.; Zhang, Y.; Liang, H.; Gui, Y.T. Non-Genomic Action of Androgens is Mediated by Rapid Phosphorylation and Regulation of Androgen Receptor Trafficking. Cell. Physiol. Biochem. 2017, 43, 223–236. [Google Scholar] [CrossRef]
- Castoria, G.; Giovannelli., P.; Di Donato, M.; Ciociola, A.; Hayashi, R.; Bernal, F.; Appella, E.; Auricchio, F.; Migliaccio, A. Role of non-genomic androgen signaling in suppressing proliferation of fibroblasts and fibrosarcoma cells. Cell Death Dis. 2014, 5, e1548. [Google Scholar] [CrossRef]
- Decker, K.F.; Zheng, D.; Yuhong, H.; Bowman, T.; Edwards, J.R.; Jia, L. Persistent androgen receptor-mediated transcription in castration-resistant prostate cancer under androgen-deprived conditions. Nucleic Acids Res. 2012, 40, 10765–10779. [Google Scholar] [CrossRef] [PubMed]
- Mills, I.G. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nat. Rev. Cancer 2014, 14, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Perner, S.; Cronauer, M.V.; Schrader, A.J.; Klocker, H.; Culig, Z.; Baniahmad, A. Adaptive responses of androgen receptor signaling in castration-resistant prostate cancer. Oncotarget 2015, 6, 35542–35555. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, T.; Yang., J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar]
- Dutt, S.S.; Gao, A.C. Molecular mechanisms of castration-resistant prostate cancer progression. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef]
- Scott, L.J. Enzalutamide: A Review in Castration-Resistant Prostate Cancer. Drugs 2018, 78, 1913–1924. [Google Scholar] [CrossRef]
- Brave, M.; Weinstock, C.; Brewer, J.R.; Chi, D.C.; Suzman, D.L.; Cheng, J.; Zhang, L.; Sridhara, R.; Ibrahim, A.; Kluetz, P.G.; et al. An FDA Review of Drug Development in Non-Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Chi, K.N.; Agarwal, N.; Bjartell, A.; Chung, B.H.; Pereira de Santana Gomes, A.J.; Given, R.; Soto, A.J.; Merseburger, A.S.; Özgüroglu, M.; Uemura, H.; et al. Apalutamide for Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2019, 381, 13–24. [Google Scholar] [CrossRef]
- Sonnenschein, C.; Olea, N.; Pasanen, M.E.; Soto, A.M. Negative controls of cell proliferation: Human prostate cancer cells and androgens. Cancer Res. 1989, 49, 3474–3481. [Google Scholar]
- Mirochnik, Y.; Veliceasa, D.; Williams, L.; Maxwell, K.; Yemelyanov, A.; Budunova, I.; Volpert, O.V. Androgen receptor drives cellular senescence. PLoS ONE 2012, 7, e31052. [Google Scholar] [CrossRef] [PubMed]
- Roediger, J.; Hessenkemper, W.; Bartsch, S.; Manvelyan, M.; Huettner, S.S.; Liehr, T.; Esmaeili, M.; Foller, S.; Petersen, I.; Grimm, M.O.; et al. Supraphysiological androgen levels induce cellular senescence in human prostate cancer through the Src-Akt pahtway. Mol. Cancer 2014, 13, 214. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.T.; D’Antonio, J.M.; Chen, S.; Antony, L.; Dalrymple, S.P.; Ndikuyeze, G.H.; Luo, J.; Denmeade, S.R. Adaptive auto-regulation of androgen receptor provides a paradigm shifting rationale for bipolar androgen therapy (BAT) for castrate resistant human prostate cancer. Prostate 2012, 72, 1491–1505. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Altuwaijri, S.; Wu, C.T.; Ricke, W.A.; Messing, E.M.; Yao, J.; Yeh, S.; Chang, C. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 12182–12187. [Google Scholar] [CrossRef]
- Teply, B.A.; Wang, H.; Luber, B.; Sullivan, R.; Rifkind, I.; Bruns, A.; Spitz, A.; DeCarli, M.; Sinibaldi, V.; Pratz, C.F.; et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: An open-label, phase 2, multicohort study. Lancet Oncol. 2018, 1, 76–86. [Google Scholar] [CrossRef]
- Denmeade, S.R. Bipolar Androgen Therapy in the Treatment of Prostate Cancer. Clin. Adv. Hematol. Oncol. 2018, 6, 408–411. [Google Scholar]
- Hessenkemper, W.; Roediger, J.; Bartscht, S.; Houtsmuller, A.B.; van Royen, M.E.; Petersen, I.; Grimm, M.O.; Baniahmad, A. A natural androgen receptor antagonist induces cellular senescence in prostate cancer cells. Mol. Endocrinol. 2014, 28, 1831–1840. [Google Scholar] [CrossRef]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguria, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef]
- Alimonti, A.; Nardella, C.; Chen, Z.; Clohessy, J.G.; Carracedo, A.; Trotman, L.C.; Cheng, K.; Varmeh, S.; Kozma, S.C.; Thomas, G.; et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J. Clin. Invest. 2010, 120, 681–693. [Google Scholar] [CrossRef]
- Qin, S.; Schulte, B.A.; Wang, G.W. Role of senescence induction in cancer treatment. World J. Clin. Oncol. 2018, 9, 180–187. [Google Scholar] [CrossRef]
- Fridlyanskaya, I.; Alekseenko, L.; Nikolsky, N. Senescence as a general cellular response to stress: A mini-review. Exp. Gerontol. 2015, 72, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, F.; Yang, P.; Xiong, F.; Yu, Q.; Li, J.; Zhou, Z.; Zhang, S.; Wang, C.Y. Aging and stress induced β cell senescence and its implication in diabetes development. Aging 2019, 11, 9947–9959. [Google Scholar] [CrossRef]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 99, 118. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Campisi, J.; Robert., L. Cell senescence: Role in aging and age-related diseases. Interdiscip. Top. Gerontol. 2014, 39, 45–61. [Google Scholar] [PubMed]
- Muñoz-Espin, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Indovina, P.; Marcelli, E.; Casini, N.; Rizzo, V.; Giordano, A. Emerging roles of RB family: New defense mechanisms against tumor progression. J. Cell. Physiol. 2013, 228, 525–535. [Google Scholar] [CrossRef]
- Kotolloshi, R.; Mirzakhani, K.; Ahlburg, J.; Kraft, F.; Pungsrinont, T.; Baniahmad, A. Thyroid hormone induces cellular senescence in prostate cancer cells through induction of DEC1. J. Steroid Biochem. Mol. Biol. 2020, 201, 105689. [Google Scholar] [CrossRef]
- Esmaeili, M.; Hennej, S.; Ludwig, S.; Klitzsch, A.; Kraft, F.; Melle, C.; Baniahmad, A. The tumor suppressor ING1b is a novel corepressor for the androgen receptor and induces cellular senescence in prostate cancer cells. J. Mol. Cell. Biol. 2016, 8, 207–220. [Google Scholar] [CrossRef]
- Esmaeili, M.; Pungsrinont, T.; Schaefer, A.; Baniahmad, A. A novel crosstalk between the tumor suppressors ING1 and ING2 regulates androgen receptor signaling. J. Mol. Med. 2016, 94, 1167–1179. [Google Scholar] [CrossRef]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Future Oncol. 2009, 5, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.Y.; Han, M.W.; Chang, H.W.; Kim, S.Y.; Kim, S.W. Prolonged autophagy by MTOR inhibitor leads radioresistant cancer cells into senescence. Autophagy 2013, 9, 1631–1632. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Kanno, A.; Ozawa, T.; Tao, H.; Umezawa, Y. Nongenomic Activity of Ligands in the Association of Androgen Receptor with Src. ACS Chem. Bio. 2007, 7, 484–492. [Google Scholar] [CrossRef]
- Yuan, X.; Cai, C.; Chen, S.; Chen, S.; Yu, Z.; Balk, S.P. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene 2014, 33, 2815–2825. [Google Scholar] [CrossRef]
- Sugawara, T.; Baumgart, S.J.; Nevedomskaya., E.; Reichert, K.; Streuber, H.; Lejeune, P.; Mumberg, D.; Haendler, B. Darolutamide is a potent androgen receptor antagonist with strong efficacy in prostate cancer models. Int. J. Cancer 2019, 145, 1382–1394. [Google Scholar] [CrossRef] [PubMed]
- Obst, J.K.; Wang, J.; Jian, K.; Williams, D.E.; Tien, A.H.; Mawji, N.; Tam, T.; Yang, Y.C.; Andersen, R.J.; Chi, K.N.; et al. Revealing Metabolic Liabilities of Ralaniten To Enhance Novel Androgen Receptor Targeted Therapies. ACS Pharm. Transl. Sci. 2019, 2, 453–467. [Google Scholar] [CrossRef]
- Burton, D.G.A.; Giribaldi, M.G.; Munoz, A.; Halvorsen, K.; Patel, A.; Jorda, M.; Perez-Stable, C.; Rai, P. Androgen Deprivation-Induced Senescence Promotes Outgrowth of Androgen-Refractory Prostate Cancer Cells. PLoS ONE 2013, 8, e68003. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.R.; Blute, M.L.; Yang, B.; Damaschke, N.; Jarrad, D.F. Synthetic lethal metabolic targeting of cellular senescence in prostate cancer with the repurposed drug metformin. J. Urol. 2018, 195, e673–e674. [Google Scholar] [CrossRef]
- Carpenter, V.J.; Saleh, T.; Gewirtz, D.A. The antiandrogen bicalutamide induces senescence in LNCaP cells and quiescence in Myc CaP cells. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Pungsrinont, T.; Sutter, M.F.; Ertingshausen, M.C.C.M.; Lakshmana, G.; Kokal, M.; Khan, A.S.; Baniahmad, A. Senolytic compounds control a distinct fate of androgen receptor agonist- and antagonist-induced cellular senescent LNCaP prostate cancer cells. Cell Biosci. 2020, 10, 59. [Google Scholar] [CrossRef]
- Gupta, S.; Pungsrinont, T.; Ženata, O.; Neubert, L.; Vrzal, R.; Baniahmad, A. Interleukin-23 represses the level of cell senescence induced by the androgen receptor antagonists enzalutamide and darolutamide in castration-resistant prostate cancer cells. Horm. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Fousteris, M.A.; Schubert, U.; Roell, D.; Roediger, J.; Bailis, N.; Nokolaropoulos, S.S.; Baniahmad, A.; Giannis, A. 20-Aminosteroids as a novel class of selective and complete androgen receptor antagonists and inhibitors of prostate cancer cell growth. Bioorg. Med. Chem. 2010, 18, 6960–6969. [Google Scholar] [CrossRef]
- Roell, D.; Rösler, T.W.; Hessenkemper, W.; Kraft, F.; Hauschild, M.; Bartsch, S.; Abraham, T.E.; Houtsmuller, M.; Matusch, R.; van Royen, M.E.; et al. Halogen-substituted anthranilic acid derivatives provide a novel chemical platform for androgen receptor antagonists. J. Steroid Biochem. Mol. Biol. 2019, 188, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, R.; Coss, C.C.; Yepuru, M.; Kearby, J.D.; Miller, D.D.; Dalton, J.T. Steroidal Androgens and Nonsteroidal, Tissue-Selective Androgen Receptor Modulator, S-22, Regulate Androgen Receptor Function through Distinct Genomic and Nongenomic Signaling Pathways. Mol. Endocrinol. 2008, 22, 2448–2465. [Google Scholar] [CrossRef]
- Dotzlaw, H.; Papaioannou, M.; Moehren, U.; Claessens, F.; Baniahmad, A. Agonist-antagonist induced coactivator and corepressor interplay on the human androgen receptor. Mol. Cell. Endocrinol. 2003, 213, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Bitzer, J.; Römer, T.; Lopes da Silva Filho, A. The use of cyproterone acetate/ethinyl estradiol in hyperandrogenic skin symptoms—A review. Eur. J. Contracep. Repr. 2017, 22, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, A.A.; Sarkar, F.H.; Yuhong, H. Overview on the complexity of androgen receptor-targeted therapy for prostate cancer. Cancer Cell Int. 2015, 15, 7. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Klokk, T.I.; Kurys, P.; Elbi, C.; Nagaich, A.K.; Hendarwanto, A.; Slagsvold, T.; Chang, C.Y.; Hager, G.L.; Saatcioglu, F. Ligand-specific dynamics of the androgen receptor at its response element in living cells. Mol. Cell. Biol. 2007, 27, 1823–1843. [Google Scholar] [CrossRef]
- Masiello, D.; Cheng, S.; Bubley, G.J.; Lu, M.L.; Balk, S.P. Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J. Biol. Chem. 2002, 277, 26321–26326. [Google Scholar] [CrossRef]
- Farla, P.; Hersmus, R.; Trapman, J.; Houtsmuller, A.B. Antiandrogens prevent stable DNA-binding of the androgen receptor. J. Cell Sci. 2005, 118, 4187–4198. [Google Scholar] [CrossRef]
- McLeod, D.G.; Iversen, P.; See, W.A.; Morris, T.; Armstrong, J.; Wirth, M.P. Bicalutamide 150 mg plus standard care vs standard care alone for early prostate cancer. BJU Int. 2006, 97, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Wirth, M.; Tyrell, C.; Delaere, K.; Sánchez-Chapado, M.; Ramon, J.; Wallace, D.M.; Hetherington, J.; Pina, F.; Heyns, C.F.; Navani, S.; et al. Bicalutamide (Casodex) 150 mg plus standard care in early non-metastatic prostate cancer. Results from Early Prostate Cancer Trial 24 at a median 7 years’ follow-up. Prostate Cancer Prostatic Dis. 2007, 10, 87–93. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rodriguez-Vida, A.; Galazi, M.; Rudman, S.; Chowdhury, S.; Sternber, C.N. Enzalutamide for the treatment of metastatic castration-resistant prostate cancer. Drug Des. Dev. Ther. 2015, 9, 3325–3339. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.E.; Ouk, S.; Yoo, D.; Sawyers, C.L.; Chen, C.; Tran, C.; Wongvipat, J. Structure-activity relationship for thiohydantoin androgen receptor antagonists for castration-resistant prostate cancer (CRPC). J. Med. Chem. 2010, 53, 2779–2796. [Google Scholar] [CrossRef] [PubMed]
- Merseburger, A.S.; Haas, G.P.; Klot, C.A. An update on enzalutamide in the treatment of prostate cancer. Ther. Adv. Urol. 2015, 7, 9–21. [Google Scholar] [CrossRef]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef]
- Niraula, S.; Chi, K.; Joshua, A.M. Beyond castration-defining future directions in the hormonal treatment of prostate cancer. Horm. Cancer 2012, 3, 3–13. [Google Scholar] [CrossRef]
- Hoffmann-Censits, J.; Kelly, K.W. Enzalutamide. A novel antiandrogen for patients with castrate-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 1335–1339. [Google Scholar] [CrossRef]
- Ghashghaei, M.; Muanza, T.; Paliouras, M.; Niazi, T.M. Effect of enzalutamide on sensitivity in prostate cancer cells to radiation by inhibition of DNA double strand break repair. J. Clin. Oncol. 2017, 35, 208. [Google Scholar] [CrossRef]
- Al-Salama, Z.T. Apalutamide: A Review in Non-Metastatic Castration-Resistant Prostate Cancer. Drugs 2019, 79, 1591–1598. [Google Scholar] [CrossRef]
- Rathkopf, D.A.; Scher, H.I. Apalutamide for the treatment of prostate cancer. Expert Rev. Anticancer Ther. 2019, 18, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Clegg, N.J.; Wangvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. ARN-509: A Novel Antiandrogen for Prostate Cancer Treatment. Cancer Res. 2012, 72, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Borno, H.T.; Small, E.J. Apalutamide and its use in the treatment of prostate cancer. Future Oncol. 2019, 15, 591–599. [Google Scholar] [CrossRef]
- Fujiza, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Mens. Health 2019, 37, 288–295. [Google Scholar]
- Bastos, D.A.; Antonarakis, E.S. Darolutamide For Castration-Resistant Prostate Cancer. Onco. Targets Ther. 2019, 12, 8769–8777. [Google Scholar] [CrossRef]
- Fizazi, K.; Massard, C.; Bono, P.; Jones, R.; Kataja, V.; James, N.; Garcia, J.A.; Protheroe, A.; Tammela, T.L.; Elliott, T.; et al. Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES). An open-label 1 dose-escalation and randomized phase 2 dose expansion trial. Lancet Oncol. 2014, 15, 975–985. [Google Scholar] [CrossRef]
- Fizazi, K.; Albiges, L.; Loriot, J.; Massard, C. ODM-201: A new-generation androgen receptor inhibitor in castration-resistant prostate cancer. Expert Rev. Anticancer Ther. 2015, 15, 1007–1017. [Google Scholar] [CrossRef]
- Schleich, S.; Papaioannou, M.; Baniahmad, A.; Matusch, R. Extracts from Pygeum africanum and other ethnobotanical species with antiandrogenic activity. Planta Med. 2006, 72, 807–813. [Google Scholar] [CrossRef]
- Papaioannou, M.; Söderholm, A.A.; Hong, W.; Roediger, J.; Roell, D.; Thiele, M.; Nyrönen, T.H.; Baniahmad, A. Computational and functional analysis of the androgen receptor antagonist atraric acid and its derivatives. Anticancer Agents Med. Chem. 2013, 13, 801–810. [Google Scholar] [CrossRef]
- Roell, D.; Baniahmad, A. The natural compounds atraric acid and N-butylbenzene-sulfonamide as antagonists of the human androgen receptor and inhibitors of prostate cancer cell growth. Mol. Cell. Endocrinol. 2011, 322, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, M.; Schleich, S.; Prade, I.; Degen, S.; Roell, D.; Schubert, U.; Tanner, T.; Claessens, F.; Matusch, R.; Baniahmad, A. The natural compound atraric acid is an antagonist of the human androgen receptor inhibiting cellular invasiveness and prostate cancer cell growth. J. Cell. Mol. Med. 2009, 13, 2210–2223. [Google Scholar] [CrossRef] [PubMed]
- Fenner, A. A new class of AR antagonists? Nat. Rev. Endocrinol. 2019, 15, 128. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Antonarakis, E.S.; Wang, H.; Ajiboye, A.S.; Spitz, A.; Cao, H.; Luo, J.; Haffner, M.C.; Yegnasubramanian, S.; Carducci, M.A.; et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: Results from a pilot clinical study. Sci. Transl. Med. 2015, 7, 269ra2. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.T.; Brennen, W.N.; Denmeade, S.R. Rationale for bipolar androgen therapy (BAT) for metastatic prostate cancer. Cell Cycle 2017, 16, 1639–1640. [Google Scholar] [CrossRef][Green Version]
- Lam, H.M.; Corey, E. Supraphysiological Testosterone Therapy as Treatment for Castration-Resistant Prostate Cancer. Front. Oncol. 2018, 8, 167. [Google Scholar] [CrossRef]
- Kokontis, J.M.; Lin, H.P.; Jiang, S.S.; Lin, C.Y.; Fukuchi, J.; Hiipakka, R.A.; Chung, C.J.; Chan, T.M.; Liao, S.; Chang, C.H.; et al. Androgen suppresses the proliferation of androgen receptor-positive castration-resistant prostate cancer cells via inhibition of Cdk2, CyclinA, and Skp2. PLoS ONE 2014, 9, e109170. [Google Scholar] [CrossRef]
- Liao, R.S.; Shihong, M.; Lu, M.; Li, R.; Yin, Y.; Raj, G.V. Androgen receptor-mediated non-genomic regulation of prostate cancer cell proliferation. Transl. Androl. Urol. 2013, 2, 187–196. [Google Scholar]
- Cinar, B.; Mukhopadhyay, N.K.; Meng, G.; Freeman, M.R. Phosphoinositide 3-kinase-independent non-genomic signals transit from the androgen receptor to Akt1 in membrane raft microdomains. J. Biol. Chem. 2007, 282, 29584–29593. [Google Scholar] [CrossRef]
- Peterziel, H.; Mink, S.; Schonert, A.; Becker, M.; Klocker, H.; Cato, A.C. Rapid signaling by androgen receptor in prostate cancer cells. Oncogene 1999, 18, 6322–6329. [Google Scholar] [CrossRef]
- Vlaeminck-Guillem, V.; Gillet, G.; Rimokh, R. SRC: Marker or actor in prostate cancer aggressiveness. Front. Oncol. 2014, 4, 222. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.K.; Sadar, M.D. Non-Genomic Actions of the Androgen Receptor in Prostate Cancer. Front. Endocrinol. 2017, 15, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Mandel, A.; Larsson, P.; Sarwar, M.; Semenas, J.; Syed Khaja, A.S.; Persson, J.L. The interplay between AR, EGF receptor and MMP-9 signaling pathways in invasive prostate cancer. Mol. Med. 2018, 24, 34. [Google Scholar] [CrossRef] [PubMed]
- Saranyutanon, S.; Srivastava, S.K.; Pai, S.; Singh, S.; Singh, A.P. Therapies targeted to androgen receptor signaling axis in prostate cancer: Progress, challenges, and hope. Cancers 2019, 12, 51. [Google Scholar] [CrossRef]
- Tatarov, O.; Mitchell, T.J.; Seywright, M.; Leung, H.Y.; Brunton, V.G.; Edwards, J. SRC family kinase activity is up-regulated in hormone-refractory prostate cancer. Clin. Cancer Res. 2009, 15, 3540–3549. [Google Scholar] [CrossRef]
- Migliaccio, A.; Castoria, G.; Di Domenico, M.; de Falco, A.; Bilancio, A.; Lombardi, M.; Barone, M.V.; Ametrano, D.; Zannini, M.S.; Abbondanza, C.; et al. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000, 19, 5406–5417. [Google Scholar] [CrossRef]
- Zarif, J.C.; Lamb, L.E.; Schulz, V.V.; Nollet, E.A.; Miranti, C.K. Androgen receptor non-nuclear regulation of prostate cancer cell invasion mediated by Src and matriptase. Oncotarget 2015, 6, 6862–6876. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Peter, H.G.; Jie, N.; Hao, J.; Bucci, J.; Cozzi, P.J.; Li, Y. Targeting PI3K/Akt/mTOR signaling pathway in the treatment of prostate cancer radioresistance. Crit. Rev. Oncol. Hematol. 2015, 96, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Lorente, D.; De Bono, J.S. Molecular alterations and emerging targets in castration resistant prostate cancer. Eur. J. Cancer 2014, 50, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Edlind, M.P.; Hsieh, A.C. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J. Androl. 2014, 16, 378–386. [Google Scholar] [PubMed]
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.G.; et al. Cell Autonomous Role of PTEN in Regulating Castration-Resistant Prostate Cancer Growth. Cancer Cell 2011, 19, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabernero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143–153. [Google Scholar] [CrossRef]
- Statz, C.M.; Patterson, S.E.; Mockus, S.M. mTOR Inhibitors in Castration-Resistant Prostate Cancer: A Systematic Review. Target. Oncol. 2017, 12, 47–59. [Google Scholar] [CrossRef]
- Majumder, P.K.; Yeh, J.J.; George, D.J.; Febbo, P.G.; Kum, J.; Xue, Q.; Bikoff, R.; Ma, H.; Kantoff, P.W.; Golub, T.R.; et al. Prostate intraepithelial neoplasia induced by prostate restricted Akt activation: The MPAKT model. Proc. Natl. Acad. Sci. USA 2003, 100, 7841–7846. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- McCall, P.; Gemmell, L.K.; Mukherjee, R.; Bartlett, J.M.; Edwards, J. Phosphorylation of the androgen receptor is associated with reduced survival in hormone-refractory prostate cancer patients. Br. J. Cancer 2008, 98, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xie, S.; Jamaluddin, M.S.; Altuwaijri, S.; Ni, J.; Kim, E.; Chen, Y.T.; Hu, Y.C.; Wang, L.; Chuang, K.H.; et al. Induction of androgen receptor expression by phosphatidylinositol 3-kinase/Akt downstream substrate, FOXO3a, and their roles in apoptosis of LNCaP prostate cancer cells. J. Biol. Chem. 2005, 280, 33558–33565. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Li, S.; Gan, L.; Kao, T.P.; Huang, H. A Transcription-Independent Function of FOXO1 in Inhibition of Androgen-Independent Activation of the Androgen Receptor in Prostate Cancer Cells. Cancer Res. 2008, 68, 10290–10299. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Hu, M.C.; Makino, K.; Spohn, B.; Bartholomeusz, G.; Yan, D.H.; Hung, M.C. HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res. 2000, 60, 6841–6845. [Google Scholar]
- Singh, R.; Dhanyamraju, P.K.; Lauth, M. DYRK1B blocks canonical and promotes non-canonical Hedgehog signaling through activation of the mTOR/AKT pathway. Oncotarget 2017, 8, 833–845. [Google Scholar] [CrossRef]
- Malik, S.N.; Brattain, M.; Ghosh, P.M.; Toyer, D.A.; Prihoda, T.; Bedolla, R.; Kreisberg, J.I. Immunohistochemical demonstration of phospho-Akt in high Gleason grade prostate cancer. Clin. Cancer Res. 2002, 8, 1168–1171. [Google Scholar]
- Axanova, L.S.; Chen, Y.Q.; McCoy, T.; Sui, G.; Cramer, S.D. 1,25-dihydroxyvitamin D(3) and PI3K/AKT inhibitors synergistically inhibit growth and induce senescence in prostate cancer cells. Prostate 2010, 70, 1658–1671. [Google Scholar] [CrossRef]
- Nowicki, T.S.; Zhao, H.; Darzynkiewicz, Z.; Moscatello, A.; Shin, E.; Schantz, S.; Tiwari, R.K.; Geliebter, J. Downregulation of uPAR inhibits migration, invasion, proliferation, FAK/PI3K/Akt signaling and induces senescence in papillary thyroid carcinoma cells. Cell Cycle 2011, 10, 100–107. [Google Scholar] [CrossRef]
- Van der Steen, T.; Tindall, D.J.; Huang, H. Posttranslational Modification of the Androgen Receptor in Prostate Cancer. Int. J. Mol. Sci. 2013, 14, 14833–14859. [Google Scholar] [CrossRef]
- Elia, U.; Flescher, E. PI3K/Akt Pathway Activation Attenuates the Cytotoxic Effect of Methyl Jasmonate Toward Sarcoma Cells. Neoplasia 2008, 11, 1303–1313. [Google Scholar] [CrossRef]
- Hu, Y.; Qiao, L.; Wang, S.; Rong, S.B.; Meuillet, E.J.; Berggren, M.; Gallegos, A.; Powis, G.; Kozikowski, A.P. 3-(Hydroxymethyl)-bearing Phosphatidylinositol Ether Lipid Analogues and Carbonate Surrogates Block PI3-K, Akt, and Cancer Cell Growth. J. Med. Chem. 2000, 43, 3045–3051. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crossby, K.; Sheen, J.H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. The mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 2009, 15, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Baron, S.M.; Manin, C.; Beaudoin, L.; Leotoing, L.; Communai, Y.; Veyssiere, G.; Morel, L. Androgen receptor mediates non-genomic activation of phosphatidylinositol 3-OH kinase in androgen-sensitive epithelial cells. J. Biol. Chem. 2004, 279, 14579–14586. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Birch, J.; Fielder, E.; Rahmatika, D.; Taylor, J.; Chapman, J.; Lagnado, A.; Carroll, B.M.; Miwa, S.; Richardson, G.; et al. Rapamycin improves healthspan but not inflammaging in nfκb1−/− mice. Aging Cell 2019, 18, e12882. [Google Scholar] [CrossRef] [PubMed]
- Carroll, B.G.; Nelson, Y.; Rabanal-Ruiz, O.; Kucheryavenko, O.; Dunhill-Turner, N.A.; Chesterman, C.C.; Zahari, Q.; Zhang, T.; Conduit, S.E.; Mitchell, C.A.; et al. Persistent mTORC1 signaling in cell senescence results from defects in amino acid and growth factor sensing. J. Cell. Biol. 2017, 216, 1949–1957. [Google Scholar] [CrossRef]
- Carroll, B.V.; Korolchuk, I.; Sarkar, S. Amino acids and autophagy: Cross-talk and co-operation to control cellular homeostasis. Amino Acids 2015, 47, 2065–2088. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Young, A.R.; Arakawa, S.; Samarajiwa, S.A.; Nakashima, T.; Yoshida, S.; Hong, S.; Berry, L.S.; Reichelt, S.; Ferreira, M.; et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 2011, 332, 966–970. [Google Scholar] [CrossRef]
- Wong, J.; Qudrat, A.; Al Mosabbir, A.; Truong, K. Advances in Senotherapies. In Molecular Basis and Emerging Strategies for Anti-Aging Interventions; Rizvi, S.I., Çakatay, U., Eds.; Springer: Singapore, 2018; pp. 67–82. [Google Scholar]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Denmaria, M. Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol. 2017, 27, 2652–2660. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Kim, I.K.; Hong, S.H.; Nan, H.; Kim, H.J.; Lee, H.J.; Masuda, E.S.; Meyuhas, O.; Oh, B.H.; Jung, Y.K. Ribosomal protein S6 is a selective mediator of TRAIL-apoptotic signaling. Oncogene 2008, 27, 4344–4352. [Google Scholar] [CrossRef]
- Meyuhas, O. Ribosomal protein S6 phosphorylation: Four decades of research. Int. Rev. Cell Mol. Biol. 2015, 320, 41–73. [Google Scholar]
- Wittenberg, A.D.; Azar, S.; Klochendler, A.; Stolovich-Rain, M.; Avraham, S.; Birnbaum, L.; Binder Gallimidi, A.; Katz, M.; Dor, Y.; Meyuhas, O. Phosphorylated ribosomal protein S6 is required for Akt-driven hyperplasia and malignant transformation, but not for hypertrophy, aneuploidy and hyperfunction of pancreatic β-cells. PLoS ONE 2016, 11, e0149995. [Google Scholar] [CrossRef] [PubMed][Green Version]



{kind=link}
{kind=link}
| AR Ligands | Detection of Cellular Senescence and Molecular Pathways | Cell Lines and PCa Tissues | References |
|---|---|---|---|
| AR antagonists | |||
| Bicalutamide | SA-β-gal, p16INK4A, p27KIP1 | LNCaP, PC3 AR, CWR22PC | [50,58,59,60] |
| Enzalutamide | SA-β-gal, p16INK4A | LNCaP, C4-2 | [61,62] |
| Darolutamide | SA-β-gal, p16INK4A | LNCaP, C4-2 | [62] |
| Atraric acid | SA-β-gal, p16INK4A, pRb, Src, Akt | LNCaP | [37] |
| Novel 20-aminosteroid (Compound 18) | SA-β-gal | LNCaP | [63] |
| Halogen-substituted anthranilic acid esters | SA-β-gal | LNCaP | [64] |
| AR agonists | |||
| Dihydrotestosterone | SA-β-gal, SAHF, p14ARF, p16INK4A, p21CIP1, Cyclin D1, pRb, p63, mTOR, ROS, PML | PC3 AR, LNCaP, C4-2, RWPE AR, PCa tissue ex vivo | [31,32,50] |
| Methyltrienolone | SA-β-gal, SAHF, p14ARF, p16INK4A, p21CIP1, p27KIP1, Cyclin D1, E2F1, pRb, Src, Akt | PC3 AR, LNCaP, C4-2, PCa tissue ex vivo | [32,50] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kokal, M.; Mirzakhani, K.; Pungsrinont, T.; Baniahmad, A. Mechanisms of Androgen Receptor Agonist- and Antagonist-Mediated Cellular Senescence in Prostate Cancer. Cancers 2020, 12, 1833. https://doi.org/10.3390/cancers12071833
Kokal M, Mirzakhani K, Pungsrinont T, Baniahmad A. Mechanisms of Androgen Receptor Agonist- and Antagonist-Mediated Cellular Senescence in Prostate Cancer. Cancers. 2020; 12(7):1833. https://doi.org/10.3390/cancers12071833
Chicago/Turabian StyleKokal, Miriam, Kimia Mirzakhani, Thanakorn Pungsrinont, and Aria Baniahmad. 2020. "Mechanisms of Androgen Receptor Agonist- and Antagonist-Mediated Cellular Senescence in Prostate Cancer" Cancers 12, no. 7: 1833. https://doi.org/10.3390/cancers12071833
APA StyleKokal, M., Mirzakhani, K., Pungsrinont, T., & Baniahmad, A. (2020). Mechanisms of Androgen Receptor Agonist- and Antagonist-Mediated Cellular Senescence in Prostate Cancer. Cancers, 12(7), 1833. https://doi.org/10.3390/cancers12071833