Advances in Anti-Cancer Immunotherapy: Car-T Cell, Checkpoint Inhibitors, Dendritic Cell Vaccines, and Oncolytic Viruses, and Emerging Cellular and Molecular Targets

 ,
,  ,
, 
Abstract
1. Introduction
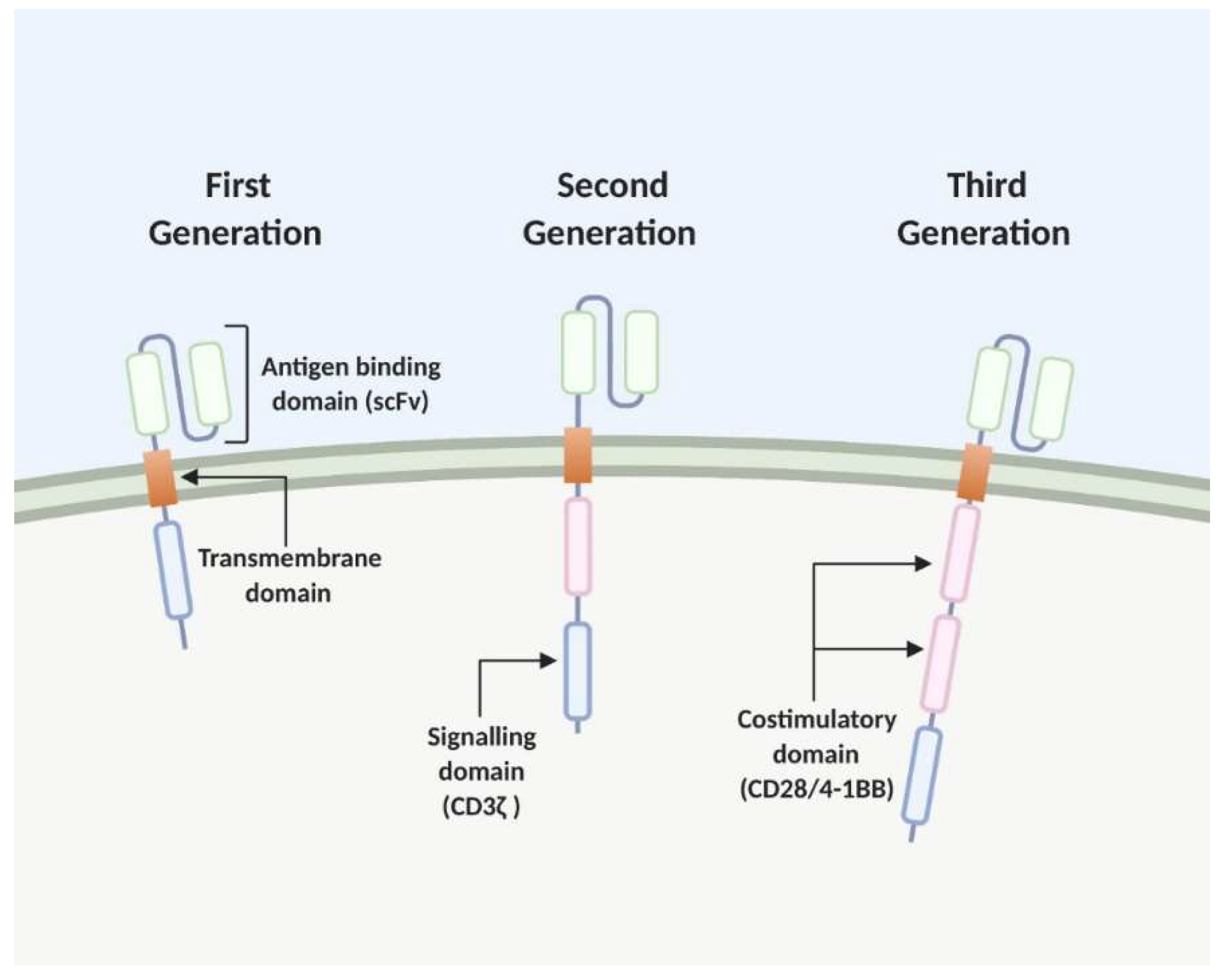
2. The Chimeric Antigen Receptor (CAR) T Cell
2.1. Development and Recent Advances CAR T Cell Therapy
2.1.1. Haematological Cancers
Limitations of CAR T Cell Therapy for Haematological Cancers
2.1.2. Solid Tumours—Challenges and Developments
2.1.3. Combination Treatment with CAR- T Cell Therapy
3. Immune Checkpoint Inhibitors
3.1. Development of Immune Checkpoint Inhibitors
3.1.1. Cytotoxic T-Lymphocyte-Associated Antigen 4—CTLA-4
3.1.2. Programmed Cell Death Protein 1—PD-1
3.1.3. Combination Therapy for CTLA-4 and PD-1
3.2. Beyond PD-1 and CTLA-4
3.2.1. Lymphocyte Activation Gene-3 – LAG-3
3.2.2. T Cell Immunoglobulin and Mucin-Domain Containing-3—TIM-3
3.2.3. T Cell Immunoglobulin and ITIM Domain—TIGIT
4. Dendritic Cell Vaccines
4.1. Development and Recent Advances of DC Vaccines
4.2. Main Challenges with DC Vaccine
4.2.1. Antigen Selection and Loading
4.2.2. Dendritic Cell Maturation
4.2.3. Administration of DC Vaccines
4.3. Dendritic Cell Vaccines in Combination Therapy
5. Oncolytic Viruses
5.1. Development and Recent Advances
5.1.1. Herpes Simplex Virus Type 1 (HSV-1)
5.1.2. Adenovirus
5.1.3. Combination Therapy- Pre-Clinical and Clinical Trials
6. Future Directions
6.1. Galectin-1 and Its Tumour-Immune Suppressing Role
6.2. Cathepsins in Cancer
6.3. OX40-Positive Regulatory T Cells and Plasmacytoid DCs
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cancer Research UK. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics-for-the-uk (accessed on 22 May 2020).
- Fiorica, F.; Trovo, M.; Ottaiano, A.; Nasti, G.; Carandina, I.; Marzola, M.; De Paoli, P.; Berretta, M. Can the addition of radiotherapy postoperatively increase clinical outcome of patients with gastric cancer? A systematic review of the literature and meta-analysis. Oncotarget 2018, 9, 10734–10744. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression-implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H., Jr. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, S87–S97. [Google Scholar] [CrossRef]
- Ma, Q.; Gonzalo-Daganzo, R.M.; Junghans, R.P. Genetically engineered T cells as adoptive immunotherapy of cancer. Cancer Chemother Biol. Response Modif. 2002, 20, 315–341. [Google Scholar] [PubMed]
- Sadelain, M.; Riviere, I.; Brentjens, R. Targeting tumours with genetically enhanced T lymphocytes. Nat. Rev. Cancer 2003, 3, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Brocker, T.; Karjalainen, K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J. Exp. Med. 1995, 181, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef]
- Lamers, C.H.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef]
- Till, B.G.; Jensen, M.C.; Wang, J.; Chen, E.Y.; Wood, B.L.; Greisman, H.A.; Qian, X.; James, S.E.; Raubitschek, A.; Forman, S.J.; et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 2008, 112, 2261–2271. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Kalos, M.; Zheng, Z.; Levine, B.; June, C. Chimeric Antigen Receptor Therapy for B-cell Malignancies. J. Cancer 2011, 2, 331–332. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef]
- O’Leary, M.C.; Lu, X.; Huang, Y.; Lin, X.; Mahmood, I.; Przepiorka, D.; Gavin, D.; Lee, S.; Liu, K.; George, B.; et al. FDA Approval Summary: Tisagenlecleucel for Treatment of Patients with Relapsed or Refractory B-cell Precursor Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2019, 25, 1142–1146. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Zhao, J.; Song, Y.; Liu, D. Recent updates on CAR T clinical trials for multiple myeloma. Mol. Cancer 2019, 18, 154. [Google Scholar] [CrossRef] [PubMed]
- Yanez, L.; Sanchez-Escamilla, M.; Perales, M.A. CAR T Cell Toxicity: Current Management and Future Directions. Hemasphere 2019, 3, e186. [Google Scholar] [CrossRef] [PubMed]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Oncolytics. 2016, 3, 16011. [Google Scholar] [CrossRef]
- Gust, J.; Finney, O.C.; Li, D.; Brakke, H.M.; Hicks, R.M.; Futrell, R.B.; Gamble, D.N.; Rawlings-Rhea, S.D.; Khalatbari, H.K.; Ishak, G.E.; et al. Glial injury in neurotoxicity after pediatric CD19-directed chimeric antigen receptor T cell therapy. Ann. Neurol. 2019, 86, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy-assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; O’Brien, S.; Jorgensen, J.L.; Cortes, J.; Faderl, S.; Garcia-Manero, G.; Verstovsek, S.; Koller, C.; Pierce, S.; Huh, Y.; et al. Prognostic significance of CD20 expression in adults with de novo precursor B-lineage acute lymphoblastic leukemia. Blood 2009, 113, 6330–6337. [Google Scholar] [CrossRef]
- Fousek, K.; Watanabe, J.; Joseph, S.K.; George, A.; An, X.; Byrd, T.T.; Morris, J.S.; Luong, A.; Martinez-Paniagua, M.A.; Sanber, K.; et al. CAR T-cells that target acute B-lineage leukemia irrespective of CD19 expression. Leukemia 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, L.; Wang-Rodriguez, J.; Zhang, L.; Cui, B.; Frankel, W.; Wu, R.; Kipps, T.J. The onco-embryonic antigen ROR1 is expressed by a variety of human cancers. Am. J. Pathol. 2012, 181, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Salter, A.I.; Liggitt, D.; Yechan-Gunja, S.; Sarvothama, M.; Cooper, K.; Smythe, K.S.; Dudakov, J.A.; Pierce, R.H.; Rader, C.; et al. Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell 2019, 35, 489–503.e8. [Google Scholar] [CrossRef]
- Hudecek, M.; Schmitt, T.M.; Baskar, S.; Lupo-Stanghellini, M.T.; Nishida, T.; Yamamoto, T.N.; Bleakley, M.; Turtle, C.J.; Chang, W.C.; Greisman, H.A.; et al. The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood 2010, 116, 4532–4541. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, W.; Zhang, X.; Cai, Z.; Huang, W. A long way to the battlefront: CAR T cell therapy against solid cancers. J. Cancer 2019, 10, 3112–3123. [Google Scholar] [CrossRef]
- Van Elsas, M.J.; Van Hall, T.; Van Der Burg, S.H. Future Challenges in Cancer Resistance to Immunotherapy. Cancers (Basel) 2020, 12, 935. [Google Scholar] [CrossRef]
- Junttila, M.R.; De Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Eil, R.; Vodnala, S.K.; Clever, D.; Klebanoff, C.A.; Sukumar, M.; Pan, J.H.; Palmer, D.C.; Gros, A.; Yamamoto, T.N.; Patel, S.J.; et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 2016, 537, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W.; et al. CXCR1-or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 2019, 10, 4016. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Shou, P.; Smith, C.; Chen, Y.; Du, H.; Sun, C.; Porterfield Kren, N.; Michaud, D.; Ahn, S.; Vincent, B.; et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. Biotechnol. 2020, 38, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Rodgers, D.T.; Du, J.; Ahmad, I.; Hampton, E.N.; Ma, J.S.; Mazagova, M.; Choi, S.H.; Yun, H.Y.; Xiao, H.; et al. Design of Switchable Chimeric Antigen Receptor T Cells Targeting Breast Cancer. Angew. Chem. Int. Ed. Engl. 2016, 55, 7520–7524. [Google Scholar] [CrossRef]
- Rodgers, D.T.; Mazagova, M.; Hampton, E.N.; Cao, Y.; Ramadoss, N.S.; Hardy, I.R.; Schulman, A.; Du, J.; Wang, F.; Singer, O.; et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc. Natl. Acad. Sci. USA 2016, 113, E459–E468. [Google Scholar] [CrossRef]
- Raj, D.; Yang, M.H.; Rodgers, D.; Hampton, E.N.; Begum, J.; Mustafa, A.; Lorizio, D.; Garces, I.; Propper, D.; Kench, J.G.; et al. Switchable CAR-T cells mediate remission in metastatic pancreatic ductal adenocarcinoma. Gut 2019, 68, 1052–1064. [Google Scholar] [CrossRef]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdanifar, M.; Roy, L.D.; Whilding, L.M.; Gavrill, A.; Maher, J.; Mukherjee, P. CAR T Cells Targeting the Tumor MUC1 Glycoprotein Reduce Triple-Negative Breast Cancer Growth. Front. Immunol. 2019, 10, 1149. [Google Scholar] [CrossRef]
- Hegde, M.; Corder, A.; Chow, K.K.; Mukherjee, M.; Ashoori, A.; Kew, Y.; Zhang, Y.J.; Baskin, D.S.; Merchant, F.A.; Brawley, V.S.; et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther. 2013, 21, 2087–2101. [Google Scholar] [CrossRef]
- Li, X.; Ye, F.; Chen, H.; Lu, W.; Wan, X.; Xie, X. Human ovarian carcinoma cells generate CD4(+)CD25(+) regulatory T cells from peripheral CD4(+)CD25(−) T cells through secreting TGF-beta. Cancer Lett. 2007, 253, 144–153. [Google Scholar] [CrossRef]
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017, 7, 10541. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef] [PubMed]
- Traverso, I.; Fenoglio, D.; Negrini, S.; Parodi, A.; Battaglia, F.; Kalli, F.; Conteduca, G.; Tardito, S.; Traverso, P.; Indiveri, F.; et al. Cyclophosphamide inhibits the generation and function of CD8(+) regulatory T cells. Hum. Immunol. 2012, 73, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Bode, B.; Wenger, R.H.; Lehmann, K.; Sartori, A.A.; Moch, H.; Knuth, A.; Boehmer, L.; Broek, M. Gamma-Radiation promotes immunological recognition of cancer cells through increased expression of cancer-testis antigens in vitro and in vivo. PLoS ONE 2011, 6, e28217. [Google Scholar] [CrossRef]
- Miao, H.; Choi, B.D.; Suryadevara, C.M.; Sanchez-Perez, L.; Yang, S.; De Leon, G.; Sayour, E.J.; McLendon, R.; Herndon, J.E., II; Healy, P.; et al. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLoS ONE 2014, 9, e94281. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Li, X.; Shao, C.; Shi, Y.; Han, W. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J. Hematol. Oncol. 2018, 11, 31. [Google Scholar] [CrossRef]
- Lafferty, K.J.; Cunningham, A.J. A new analysis of allogeneic interactions. Aust. J. Exp. Biol. Med. Sci. 1975, 53, 27–42. [Google Scholar] [CrossRef]
- Borcherding, N.; Kolb, R.; Gullicksrud, J.; Vikas, P.; Zhu, Y.; Zhang, W. Keeping Tumors in Check: A Mechanistic Review of Clinical Response and Resistance to Immune Checkpoint Blockade in Cancer. J. Mol. Biol. 2018, 430, 2014–2029. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Sun, H.; Sun, C. The Rise of NK Cell Checkpoints as Promising Therapeutic Targets in Cancer Immunotherapy. Front. Immunol. 2019, 10, 2354. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Freeman, G.J. The B7-CD28 superfamily. Nat. Rev. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef]
- Schwartz, R.H. Costimulation of T lymphocytes: The role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell 1992, 71, 1065–1068. [Google Scholar] [CrossRef]
- Linsley, P.S.; Greene, J.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef]
- Contardi, E.; Palmisano, G.L.; Tazzari, P.L.; Martelli, A.M.; Fala, F.; Fabbi, M.; Kato, T.; Lucarelli, E.; Donati, D.; Polito, L.; et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int. J. Cancer 2005, 117, 538–550. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Hodi, F.S.; Mihm, M.C.; Soiffer, R.J.; Haluska, F.G.; Butler, M.; Seiden, M.V.; Davis, T.; Henry-Spires, R.; MacRae, S.; Willman, A.; et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc. Natl. Acad. Sci. USA 2003, 100, 4712–4717. [Google Scholar] [CrossRef]
- Phan, G.Q.; Yang, J.C.; Sherry, R.M.; Hwu, P.; Topalian, S.L.; Schwartzentruber, D.J.; Restifo, N.P.; Haworth, L.R.; Seipp, C.A.; Freezer, L.J.; et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 2003, 100, 8372–8377. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Kim, H.T.; Bachireddy, P.; Costello, C.; Liguori, R.; Savell, A.; Lukez, A.P.; Avigan, D.; Chen, Y.B.; McSweeney, P.; et al. Ipilimumab for Patients with Relapse after Allogeneic Transplantation. N. Engl. J. Med. 2016, 375, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet. Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.N.; Pu, J.J. PD-1 pathway and its clinical application: A 20year journey after discovery of the complete human PD-1 gene. Gene 2018, 638, 20–25. [Google Scholar] [CrossRef]
- Zhu, B.; Tang, L.; Chen, S.; Yin, C.; Peng, S.; Li, X.; Liu, T.; Liu, W.; Han, C.; Stawski, L.; et al. Targeting the upstream transcriptional activator of PD-L1 as an alternative strategy in melanoma therapy. Oncogene 2018, 37, 4941–4954. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Zou, W.; Chen, L. Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef]
- Juneja, V.R.; McGuire, K.A.; Manguso, R.T.; LaFleur, M.W.; Collins, N.; Haining, W.N.; Freeman, G.J.; Sharpe, A.H. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med. 2017, 214, 895–904. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Antonia, S.J. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. Reply. N. Engl. J. Med. 2019, 380, 990. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Russell, J.S.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbe, C.; Milella, M.; Brownell, I.; et al. Updated efficacy of avelumab in patients with previously treated metastatic Merkel cell carcinoma after >/=1 year of follow-up: JAVELIN Merkel 200, a phase 2 clinical trial. J. Immunother. Cancer 2018, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhao, X.; Fu, J.; Wang, H. Progress and Challenges in Precise Treatment of Tumors with PD-1/PD-L1 Blockade. Front. Immunol. 2020, 11, 339. [Google Scholar] [CrossRef]
- Martins, F.; Sykiotis, G.P.; Maillard, M.; Fraga, M.; Ribi, C.; Kuntzer, T.; Michielin, O.; Peters, S.; Coukos, G.; Spertini, F.; et al. New therapeutic perspectives to manage refractory immune checkpoint-related toxicities. Lancet. Oncol. 2019, 20, e54–e64. [Google Scholar] [CrossRef]
- Puzanov, I.; Diab, A.; Abdallah, K.; Bingham, C.O., 3rd; Brogdon, C.; Dadu, R.; Hamad, L.; Kim, S.; Lacouture, M.E.; LeBoeuf, N.R.; et al. Managing toxicities associated with immune checkpoint inhibitors: Consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J. Immunother. Cancer 2017, 5, 95. [Google Scholar] [CrossRef]
- Haanen, J.; Carbonnel, F.; Robert, C.; Kerr, K.M.; Peters, S.; Larkin, J.; Jordan, K. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28, iv119–iv142. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Metro, G.; Signorelli, D. Immune checkpoints inhibitors rechallenge in non-small-cell lung cancer: Different scenarios with different solutions? Lung Cancer Manag. 2020, 8, LMT18. [Google Scholar] [CrossRef]
- Friedlaender, A.; Kim, C.; Addeo, A. Rethinking the Optimal Duration of Immune Checkpoint Inhibitors in Non-small Cell Lung Cancer Throughout the COVID-19 Pandemic. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet. Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Spain, L.; Diem, S.; Larkin, J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat. Rev. 2016, 44, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, F.; Montfort, A.; Marcheteau, E.; Imbert, C.; Gilhodes, J.; Filleron, T.; Rochaix, P.; Andrieu-Abadie, N.; Levade, T.; Meyer, N.; et al. TNFalpha blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat. Commun. 2017, 8, 2256. [Google Scholar] [CrossRef] [PubMed]
- Vetizou, M.; Pitt, J.M.; Daillere, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355. [Google Scholar] [CrossRef]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Andrews, L.P.; Yano, H.; Vignali, D.A.A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: Breakthroughs or backups. Nat. Immunol. 2019, 20, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef] [PubMed]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef]
- Huard, B.; Tournier, M.; Triebel, F. LAG-3 does not define a specific mode of natural killing in human. Immunol. Lett. 1998, 61, 109–112. [Google Scholar] [CrossRef]
- Macon-Lemaitre, L.; Triebel, F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology 2005, 115, 170–178. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Fougeray, S.; Brignone, C.; Triebel, F. A soluble LAG-3 protein as an immunopotentiator for therapeutic vaccines: Preclinical evaluation of IMP321. Vaccine 2006, 24, 5426–5433. [Google Scholar] [CrossRef]
- Yu, X.; Huang, X.; Chen, X.; Liu, J.; Wu, C.; Pu, Q.; Wang, Y.; Kang, X.; Zhou, L. Characterization of a novel anti-human lymphocyte activation gene 3 (LAG-3) antibody for cancer immunotherapy. MAbs 2019, 11, 1139–1148. [Google Scholar] [CrossRef]
- Huang, R.Y.; Francois, A.; McGray, A.R.; Miliotto, A.; Odunsi, K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology 2017, 6, e1249561. [Google Scholar] [CrossRef]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar] [CrossRef]
- Anderson, A.C. Tim-3, a negative regulator of anti-tumor immunity. Curr. Opin. Immunol. 2012, 24, 213–216. [Google Scholar] [CrossRef]
- Liu, F.; Liu, Y.; Chen, Z. Tim-3 expression and its role in hepatocellular carcinoma. J. Hematol. Oncol. 2018, 11, 126. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, A.; Laken, H.; Correia, J.D.S.; McNeeley, P.; Altobell, L.; Zhang, J.; Vancutsem, P.; Wilcoxen, K.; Jenkins, D. Discovery of TSR-022, a novel, potent anti-human TIM-3 therapeutic antibody. Eur. J. Cancer 2016, 69, S102. [Google Scholar] [CrossRef]
- Chen, X.; Song, X.; Li, K.; Zhang, T. FcgammaR-Binding Is an Important Functional Attribute for Immune Checkpoint Antibodies in Cancer Immunotherapy. Front. Immunol. 2019, 10, 292. [Google Scholar] [CrossRef]
- Levin, S.D.; Taft, D.W.; Brandt, C.S.; Bucher, C.; Howard, E.D.; Chadwick, E.M.; Johnston, J.; Hammond, A.; Bontadelli, K.; Ardourel, D.; et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur. J. Immunol. 2011, 41, 902–915. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.L.; Garrido-Laguna, I. TIGIT: A novel immunotherapy target moving from bench to bedside. Cancer Immunol. Immunother. 2018, 67, 1659–1667. [Google Scholar] [CrossRef]
- Saxena, M.; Bhardwaj, N. Turbocharging Vaccines: Emerging Adjuvants for Dendritic Cell Based Therapeutic Cancer Vaccines; Elsevier Ltd.: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Tel, J.; Erik, H.J.G.A.; Baba, T.; Schreibelt, G.; Schulte, B.M.; Benitez-Ribas, D.; Boerman, O.C.; Croockewit, S.; Wim, J.G.O.; Van Rossum, M.; et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res. 2013, 73, 1063–1075. [Google Scholar] [CrossRef]
- Van Tendeloo, V.F.; Van De Veldea, A.; Van Driesschea, A.; Coolsa, N.; Anguille, S.; Ladell, K.; Gostick, E.; Vermeulen, K.; Pieters, K.; Nijs, G.; et al. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms’ tumor 1 antigen-targeted dendritic cell vaccination. Proc. Natl. Acad. Sci. USA 2010, 107, 13824–13829. [Google Scholar] [CrossRef]
- Rosenblatt, J.; Avivi, I.; Vasir, B.; Uhl, L.; Munshi, N.C.; Katz, T.; Dey, B.R.; Somaiya, P.; Mills, H.; Campigotto, F.; et al. Vaccination with dendritic cell/tumor fusions following autologous stem cell transplant induces immunologic and clinical responses in multiple myeloma patients. Clin. Cancer Res. 2013, 19, 3640–3648. [Google Scholar] [CrossRef]
- Ogasawara, M.; Miyashita, M.; Yamagishi, Y.; Ota, S. Phase I/II Pilot Study of Wilms’ Tumor 1 Peptide-Pulsed Dendritic Cell Vaccination Combined With Conventional Chemotherapy in Patients With Head and Neck Cancer. Ther. Apher. Dial. 2019, 23, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Tanyi, J.L.; Bobisse, S.; Ophir, E.; Tuyaerts, S.; Roberti, A.; Genolet, R.; Baumgartner, P.; Stevenson, B.J.; Iseli, C.; Dangaj, D.; et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Van Willigen, W.W.; Bloemendal, M.; Gerritsen, W.R.; Schreibelt, G.; De Vries, I.J.M.; Bol, K.F. Dendritic cell cancer therapy: Vaccinating the right patient at the right time. Front. Media 2018, 9, 2265. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. New Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Lissoni, P.; Malugani, F.; Bonfanti, A.; Bucovec, R.; Secondino, S.; Brivio, F.; Ferrari-Bravo, A.; Ferrante, R.; Vigore, L.; Rovelli, F.; et al. Abnormally enhanced blood concentrations of vascular endothelial growth factor (VEGF) in metastatic cancer patients and their relation to circulating dendritic cells, IL-12 and endothelin-1. J. Biol. Regul. Homeost. Agents 2001, 15, 140–144. [Google Scholar] [PubMed]
- Chen, W.; Ten Dijke, P. Immunoregulation by members of the TGFβ superfamily. Nature 2016, 16, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.T.; Murray, G.I. Current mechanistic insights into the roles of matrix metalloproteinases in tumour invasion and metastasis. J. Pathol. 2015, 237, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Mastelic-Gavillet, B.; Balint, K.; Boudousquie, C.; Gannon, P.O.; Kandalaft, L.E. Personalized dendritic cell vaccines-recent breakthroughs and encouraging clinical results. Front. Media 2019, 10, 766. [Google Scholar] [CrossRef] [PubMed]
- Baldin, A.V.; Savvateeva, L.V.; Bazhin, A.V.; Zamyatnin, A.A. Dendritic cells in anticancer vaccination: Rationale for ex vivo loading or in vivo targeting. MDPI AG 2020, 12, 590. [Google Scholar] [CrossRef]
- Sprinzl, G.M.; Kacani, L.; Schrott-Fischer, A.; Romani, N.; Thumfart, W.F. Dendritic cell vaccines for cancer therapy. Cancer Treat. Rev. 2001, 27, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Touzot, M.; Bohineust, A.; Cappuccio, A.; Chiocchia, G.; Hosmalin, A.; Dalod, M.; Soumelis, V.; Amigorena, S. Human Inflammatory Dendritic Cells Induce Th17 Cell Differentiation. Immunity 2013, 38, 336–348. [Google Scholar] [CrossRef]
- Sallusto, F.; Lanzavecchi, A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus iuterleukin 4 and downregulated by tumor necrosis factor α. J. Exp. Med. 1994, 179, 1109–1118. [Google Scholar] [CrossRef]
- Breckpot, K.; Corthals, J.; Bonehill, A.; Michiels, A.; Tuyaerts, S.; Aerts, C.; Heirman, C.; Thielemans, K. Dendritic cells differentiated in the presence of IFN-{beta} and IL-3 are potent inducers of an antigen-specific CD8+ T cell response. J. Leukoc. Biol. 2005, 78, 898–908. [Google Scholar] [CrossRef]
- Bol, K.F.; Schreibelt, G.; Rabold, K.; Wculek, S.K.; Schwarze, J.K.; Dzionek, A.; Teijeira, A.; Kandalaft, L.E.; Romero, P.; Coukos, G.; et al. The clinical application of cancer immunotherapy based on naturally circulating dendritic cells. BioMed Central. 2019, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Trinchieri, G.; Liu, Y.-J. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 2004, 5, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.C.; Tullett, K.M.; Lee, Y.S.; Idris, A.; Ding, Y.; McDonald, K.J.; Kassianos, A.; Leal Rojas, I.M.; Jeet, V.; Lahoud, M.H.; et al. Differential uptake and cross-presentation of soluble and necrotic cell antigen by human DC subsets. Eur. J. Immunol. 2016, 46, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Sittig, S.P.; Bakdash, G.; Weiden, J.; Sköld, A.E.; Tel, J.; Figdor, C.G.; Vries, I.J.M.d.; Schreibelt, G. A Comparative Study of the T Cell Stimulatory and Polarizing Capacity of Human Primary Blood Dendritic Cell Subsets. Mediat. Inflamm. 2016, 2016. [Google Scholar] [CrossRef]
- Theisen, D.J.; Davidson, J.T.; Briseño, C.G.; Gargaro, M.; Lauron, E.J.; Wang, Q.; Desai, P.; Durai, V.; Bagadia, P.; Brickner, J.R.; et al. WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science 2018, 362, 694–699. [Google Scholar] [CrossRef]
- Schreibelt, G.; Bol, K.F.; Westdorp, H.; Wimmers, F.; Aarntzen, E.H.J.G.; Duiveman-De Boer, T.; Van De Rakt, M.W.M.M.; Scharenborg, N.M.; De Boer, A.J.; Pots, J.M.; et al. Effective clinical responses in metastatic melanoma patients after vaccination with primary myeloid dendritic cells. Clin. Cancer Res. 2016, 22, 2155–2166. [Google Scholar] [CrossRef]
- Perez, C.R.; De Palma, M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat. Res. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Aspord, C.; Leccia, M.T.; Salameire, D.; Laurin, D.; Chaperot, L.; Charles, J.; Plumas, J. HLA-A 0201 plasmacytoid dendritic cells provide a cell-based immunotherapy for melanoma patients. J. Investig. Dermatol. 2012, 132, 2395–2406. [Google Scholar] [CrossRef]
- De Paula Peres, L.; Da Luz, F.A.C.; Dos Anjos Pultz, B.; Brígido, P.C.; De Araújo, R.A.; Goulart, L.R.; Silva, M.J.B. Peptide vaccines in breast cancer: The immunological basis for clinical response. Biotechnol. Adv. 2015, 33, 1868–1877. [Google Scholar] [CrossRef]
- Capietto, A.H.; Jhunjhunwala, S.; Delamarre, L. Characterizing neoantigens for personalized cancer immunotherapy. Curr. Opin. Immunol. 2017, 46, 58–65. [Google Scholar] [CrossRef]
- Sahin, U.; Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef]
- Aldous, A.R.; Dong, J.Z. Personalized neoantigen vaccines: A new approach to cancer immunotherapy. Bioorganic. Med. Chem. 2018, 26, 2842–2849. [Google Scholar] [CrossRef] [PubMed]
- Vandenberk, L.; Belmans, J.; Van Woensel, M.; Riva, M.; Van Gool, S.W. Exploiting the immunogenic potential of cancer cells for improved dendritic cell vaccines. Front. Media 2016, 6, 663. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Liang, S.; Jiang, F.; Xu, J.; Zhu, W.; Qian, W.; Hu, Y.; Zhou, Z.; Chen, J.; Niu, L.; et al. 2003–2013, a valuable study: Autologous tumor lysate-pulsed dendritic cell immunotherapy with cytokine-induced killer cells improves survival in stage IV breast cancer. Immunol. Lett. 2017, 183, 37–43. [Google Scholar] [CrossRef]
- Saxena, M.; Bhardwaj, N. Re-Emergence of Dendritic Cell Vaccines for Cancer Treatment. Cell Press 2018, 4, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Scandella, E.; Men, Y.; Gillessen, S.; Förster, R.; Groettrup, M. Prostaglandin E2 is a key factor for CCR7 surface expression and migration of monocyte-derived dendritic cells. Blood 2002, 100, 1354–1361. [Google Scholar] [CrossRef] [PubMed]
- Jongmans, W.; Tiemessen, D.M.; Van Vlodrop, I.J.H.; Mulders, P.F.A.; Oosterwijk, E. Th1-polarizing capacity of clinical-grade dendritic cells is triggered by ribomunyl but is compromised by PGE2: The importance of maturation cocktails. J. Immunother. 2005, 28, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis, E.S.C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022.e14–1037.e14. [Google Scholar] [CrossRef]
- Kalinski, P.; Okada, H. Polarized dendritic cells as cancer vaccines: Directing effector-type T cells to tumors. Semin. Immunol. 2010, 22, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Bonehill, A.; Van Nuffel, A.M.T.; Corthals, J.; Tuyaerts, S.; Heirman, C.; François, V.; Colau, D.; Van Der Bruggen, P.; Neyns, B.; Thielemans, K. Single-step antigen loading and activation of dendritic cells by mRNA electroporation for the purpose of therapeutic vaccination in melanoma patients. Clin. Cancer Res. 2009, 15, 3366–3375. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; Van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) plus ipilimumab in patientswith pretreated advanced melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef]
- Mullins, D.W.; Sheasley, S.L.; Ream, R.M.; Bullock, T.N.J.; Fu, Y.X.; Engelhard, V.H. Route of immunization with peptide-pulsed dendritic cells controls the distribution of memory and effector T cells in lymphoid tissues and determines the pattern of regional tumor control. J. Exp. Med. 2003, 198, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Lesterhuis, W.J.; De Vries, I.J.M.; Schreibelt, G.; Lambeck, A.J.A.; Aarntzen, E.H.J.G.; Jacobs, J.F.M.; Scharenborg, N.M.; Van De Rakt, M.W.M.M.; De Boer, A.J.; Croockewit, S.; et al. Route of administration modulates the induction of dendritic cell vaccine-induced antigen-specific T cells in advanced melanoma patients. Clin. Cancer Res. 2011, 17, 5725–5735. [Google Scholar] [CrossRef] [PubMed]
- Hochnadel, I.; Kossatz-Boehlert, U.; Jedicke, N.; Lenzen, H.; Manns, M.P.; Yevsa, T. Cancer vaccines and immunotherapeutic approaches in hepatobiliary and pancreatic cancers. Hum. Vaccines Immunother. 2017, 13, 2931–2952. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Uchida, H.; Marzulli, M.; Nakano, K.; Goins, W.F.; Chan, J.; Hong, C.S.; Mazzacurati, L.; Yoo, J.Y.; Haseley, A.; Nakashima, H.; et al. Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol. Ther. 2013, 21, 561–569. [Google Scholar] [CrossRef]
- Vile, R.G.; Hart, I.R. Targeting of cytokine gene expression to malignant melanoma cells using tissue specific promoter sequences. Ann. Oncol. 1994, 5, 59–65. [Google Scholar] [CrossRef]
- Xia, T.; Konno, H.; Ahn, J.; Barber, G.N. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates with Tumorigenesis. Cell Rep. 2016, 14, 282–297. [Google Scholar] [CrossRef]
- Xia, T.; Konno, H.; Barber, G.N. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 2016, 76, 6747–6759. [Google Scholar] [CrossRef]
- Bhattacharya, P.; Budnick, I.; Singh, M.; Thiruppathi, M.; Alharshawi, K.; Elshabrawy, H.; Holterman, M.J.; Prabhakar, B.S. Dual Role of GM-CSF as a Pro-Inflammatory and a Regulatory Cytokine: Implications for Immune Therapy. J. Interferon Cytokine Res. 2015, 35, 585–599. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Foreman, P.M.; Friedman, G.K.; Cassady, K.A.; Markert, J.M. Oncolytic Virotherapy for the Treatment of Malignant Glioma. Neurotherapeutics 2017, 14, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef]
- Parker, J.N.; Bauer, D.F.; Cody, J.J.; Markert, J.M. Oncolytic viral therapy of malignant glioma. Neurotherapeutics 2009, 6, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef]
- Harrow, S.; Papanastassiou, V.; Harland, J.; Mabbs, R.; Petty, R.; Fraser, M.; Hadley, D.; Patterson, J.; Brown, S.M.; Rampling, R. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: Safety data and long-term survival. Gene Ther. 2004, 11, 1648–1658. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.N.; Gillespie, G.Y.; Love, C.E.; Randall, S.; Whitley, R.J.; Markert, J.M. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 2208–2213. [Google Scholar] [CrossRef]
- Rehman, H.; Silk, A.W.; Kane, M.P.; Kaufman, H.L. Into the clinic: Talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J. Immunother. Cancer 2016, 4, 53. [Google Scholar] [CrossRef]
- Russell, L.; Peng, K.W.; Russell, S.J.; Diaz, R.M. Oncolytic Viruses: Priming Time for Cancer Immunotherapy. BioDrugs 2019, 33, 485–501. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kim, D.W.; DeRaffele, G.; Mitcham, J.; Coffin, R.S.; Kim-Schulze, S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 2010, 17, 718–730. [Google Scholar] [CrossRef]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Cheng, P.H.; Wechman, S.L.; McMasters, K.M.; Zhou, H.S. Oncolytic Replication of E1b-Deleted Adenoviruses. Viruses 2015, 7, 5767–5779. [Google Scholar] [CrossRef]
- O’Shea, C.C.; Johnson, L.; Bagus, B.; Choi, S.; Nicholas, C.; Shen, A.; Boyle, L.; Pandey, K.; Soria, C.; Kunich, J.; et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell 2004, 6, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Abbed, K.M.; Tatter, S.; Louis, D.N.; Hochberg, F.H.; Barker, F.; Kracher, J.; Grossman, S.A.; Fisher, J.D.; Carson, K.; et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-Attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol. Ther. 2004, 10, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Liang, M. Oncorine, the World First Oncolytic Virus Medicine and its Update in China. Curr. Cancer Drug Targets 2018, 18, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Fueyo, J.; Alemany, R.; Gomez-Manzano, C.; Fuller, G.N.; Khan, A.; Conrad, C.A.; Liu, T.J.; Jiang, H.; Lemoine, M.G.; Suzuki, K.; et al. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway. J. Natl. Cancer Inst. 2003, 95, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, J.K.; Chiocca, E.A. Glioma virus therapies between bench and bedside. Neuro Oncol. 2014, 16, 334–351. [Google Scholar] [CrossRef]
- Cervera-Carrascon, V.; Quixabeira, D.C.A.; Havunen, R.; Santos, J.M.; Kutvonen, E.; Clubb, J.H.A.; Siurala, M.; Heinio, C.; Zafar, S.; Koivula, T.; et al. Comparison of Clinically Relevant Oncolytic Virus Platforms for Enhancing T Cell Therapy of Solid Tumors. Mol. Ther. Oncolytics. 2020, 17, 47–60. [Google Scholar] [CrossRef]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef]
- Chesney, J.; Puzanov, I.; Collichio, F.; Milhem, M.M.; Hauschild, A.; Chen, L.; Sharma, A.; Garbe, C.; Singh, P.; Mehnert, J.M. Patterns of response with talimogene laherparepvec in combination with ipilimumab or ipilimumab alone in metastatic unresectable melanoma. Br. J. Cancer 2019, 121, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma. J. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Woller, N.; Gurlevik, E.; Fleischmann-Mundt, B.; Schumacher, A.; Knocke, S.; Kloos, A.M.; Saborowski, M.; Geffers, R.; Manns, M.P.; Wirth, T.C.; et al. Viral Infection of Tumors Overcomes Resistance to PD-1-immunotherapy by Broadening Neoantigenome-directed T-cell Responses. Mol. Ther. 2015, 23, 1630–1640. [Google Scholar] [CrossRef] [PubMed]
- Sivanandam, V.; LaRocca, C.J.; Chen, N.G.; Fong, Y.; Warner, S.G. Oncolytic Viruses and Immune Checkpoint Inhibition: The Best of Both Worlds. Mol. Ther. Oncolytics 2019, 13, 93–106. [Google Scholar] [CrossRef]
- Shen, W.; Patnaik, M.M.; Ruiz, A.; Russell, S.J.; Peng, K.W. Immunovirotherapy with vesicular stomatitis virus and PD-L1 blockade enhances therapeutic outcome in murine acute myeloid leukemia. Blood 2016, 127, 1449–1458. [Google Scholar] [CrossRef]
- Aiken, R.; Chen, C.; Cloughesy, T.; Colman, H.; Daras, M.; Groves, M.; Khagi, S.; Kumthekar, P.; Lang, F.; Nassiri, F.; et al. Atim-33. Interim Results of a Phase Ii Multi-Center Study of Oncolytic Adenovirus Dnx-2401 with Pembrolizumab for Recurrent Glioblastoma; Captive Study (Keynote-192). Neuro Oncol. 2019, 21, vi8–vi9. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Jiang, H.; Gomez-Manzano, C.; Lang, F.F.; Alemany, R.; Fueyo, J. Oncolytic adenovirus: Preclinical and clinical studies in patients with human malignant gliomas. Curr. Gene Ther. 2009, 9, 422–427. [Google Scholar] [CrossRef]
- Cousin, J.M.; Cloninger, M.J. The Role of Galectin-1 in Cancer Progression, and Synthetic Multivalent Systems for the Study of Galectin-1. Int. J. Mol. Sci. 2016, 17, 1566. [Google Scholar] [CrossRef]
- Thijssen, V.L.; Postel, R.; Brandwijk, R.J.; Dings, R.P.; Nesmelova, I.; Satijn, S.; Verhofstad, N.; Nakabeppu, Y.; Baum, L.G.; Bakkers, J.; et al. Galectin-1 is essential in tumor angiogenesis and is a target for antiangiogenesis therapy. Proc. Natl. Acad. Sci. USA 2006, 103, 15975–15980. [Google Scholar] [CrossRef]
- Thijssen, V.L.; Barkan, B.; Shoji, H.; Aries, I.M.; Mathieu, V.; Deltour, L.; Hackeng, T.M.; Kiss, R.; Kloog, Y.; Poirier, F.; et al. Tumor cells secrete galectin-1 to enhance endothelial cell activity. Cancer Res. 2010, 70, 6216–6224. [Google Scholar] [CrossRef] [PubMed]
- Croci, D.O.; Salatino, M.; Rubinstein, N.; Cerliani, J.P.; Cavallin, L.E.; Leung, H.J.; Ouyang, J.; Ilarregui, J.M.; Toscano, M.A.; Domaica, C.I.; et al. Disrupting galectin-1 interactions with N-glycans suppresses hypoxia-driven angiogenesis and tumorigenesis in Kaposi’s sarcoma. J. Exp. Med. 2012, 209, 1985–2000. [Google Scholar] [CrossRef]
- D’Haene, N.; Sauvage, S.; Maris, C.; Adanja, I.; Le Mercier, M.; Decaestecker, C.; Baum, L.; Salmon, I. VEGFR1 and VEGFR2 involvement in extracellular galectin-1- and galectin-3-induced angiogenesis. PLoS ONE 2013, 8, e67029. [Google Scholar] [CrossRef] [PubMed]
- Zinovkin, D.A.; Achinovich, S.L.; Zubritskiy, M.G.; Whatmore, J.L.; Pranjol, M.Z.I. High Expression of Galectin-1, VEGF and Increased Microvessel Density Are Associated with MELF Pattern in Stage I-III Endometrioid Endometrial Adenocarcinoma. J. Pathol. Transl. Med. 2019, 53, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Pranjol, M.Z.I.; Zinovkin, D.A.; Maskell, A.R.T.; Stephens, L.J.; Achinovich, S.L.; Los, D.M.; Nadyrov, E.A.; Hannemann, M.; Gutowski, N.J.; Whatmore, J.L. Cathepsin L-induced galectin-1 may act as a proangiogenic factor in the metastasis of high-grade serous carcinoma. J. Transl. Med. 2019, 17, 216. [Google Scholar] [CrossRef] [PubMed]
- Camby, I.; Le Mercier, M.; Lefranc, F.; Kiss, R. Galectin-1: A small protein with major functions. Glycobiology 2006, 16, 137R–157R. [Google Scholar] [CrossRef]
- Pace, K.E.; Lee, C.; Stewart, P.L.; Baum, L.G. Restricted receptor segregation into membrane microdomains occurs on human T cells during apoptosis induced by galectin-1. J. Immunol. 1999, 163, 3801–3811. [Google Scholar]
- Hernandez, J.D.; Nguyen, J.T.; He, J.; Wang, W.; Ardman, B.; Green, J.M.; Fukuda, M.; Baum, L.G. Galectin-1 binds different CD43 glycoforms to cluster CD43 and regulate T cell death. J. Immunol. 2006, 177, 5328–5336. [Google Scholar] [CrossRef]
- Perone, M.J.; Bertera, S.; Shufesky, W.J.; Divito, S.J.; Montecalvo, A.; Mathers, A.R.; Larregina, A.T.; Pang, M.; Seth, N.; Wucherpfennig, K.W.; et al. Suppression of autoimmune diabetes by soluble galectin-1. J. Immunol. 2009, 182, 2641–2653. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; Daly, G.; Dreja, H.; Tailor, H.; Riera, C.M.; Hirabayashi, J.; Chernajovsky, Y. Recombinant galectin-1 and its genetic delivery suppress collagen-induced arthritis via T cell apoptosis. J. Exp. Med. 1999, 190, 385–398. [Google Scholar] [CrossRef]
- Santucci, L.; Fiorucci, S.; Cammilleri, F.; Servillo, G.; Federici, B.; Morelli, A. Galectin-1 exerts immunomodulatory and protective effects on concanavalin A-induced hepatitis in mice. Hepatology 2000, 31, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Santucci, L.; Fiorucci, S.; Rubinstein, N.; Mencarelli, A.; Palazzetti, B.; Federici, B.; Rabinovich, G.A.; Morelli, A. Galectin-1 suppresses experimental colitis in mice. Gastroenterology 2003, 124, 1381–1394. [Google Scholar] [CrossRef]
- Motran, C.C.; Molinder, K.M.; Liu, S.D.; Poirier, F.; Miceli, M.C. Galectin-1 functions as a Th2 cytokine that selectively induces Th1 apoptosis and promotes Th2 function. Eur. J. Immunol. 2008, 38, 3015–3027. [Google Scholar] [CrossRef] [PubMed]
- Verschuere, T.; Toelen, J.; Maes, W.; Poirier, F.; Boon, L.; Tousseyn, T.; Mathivet, T.; Gerhardt, H.; Mathieu, V.; Kiss, R.; et al. Glioma-derived galectin-1 regulates innate and adaptive antitumor immunity. Int. J. Cancer 2014, 134, 873–884. [Google Scholar] [CrossRef]
- Nambiar, D.K.; Aguilera, T.; Cao, H.; Kwok, S.; Kong, C.; Bloomstein, J.; Wang, Z.; Rangan, V.S.; Jiang, D.; von Eyben, R.; et al. Galectin-1-driven T cell exclusion in the tumor endothelium promotes immunotherapy resistance. J. Clin. Invest. 2019, 129, 5553–5567. [Google Scholar] [CrossRef] [PubMed]
- Bozorgmehr, F.; Hommertgen, A.; Krisam, J.; Lasitschka, F.; Kuon, J.; Maenz, M.; Huber, P.E.; Konig, L.; Kieser, M.; Debus, J.; et al. Fostering efficacy of anti-PD-1-treatment: Nivolumab plus radiotherapy in advanced non-small cell lung cancer-study protocol of the FORCE trial. BMC Cancer 2019, 19, 1074. [Google Scholar] [CrossRef]
- Kuo, P.; Bratman, S.V.; Shultz, D.B.; Von Eyben, R.; Chan, C.; Wang, Z.; Say, C.; Gupta, A.; Loo, B.W., Jr.; Giaccia, A.J.; et al. Galectin-1 mediates radiation-related lymphopenia and attenuates NSCLC radiation response. Clin. Cancer Res. 2014, 20, 5558–5569. [Google Scholar] [CrossRef]
- Mereiter, S.; Balmana, M.; Campos, D.; Gomes, J.; Reis, C.A. Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell 2019, 36, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Pranjol, M.Z.; Gutowski, N.; Hannemann, M.; Whatmore, J. The Potential Role of the Proteases Cathepsin D and Cathepsin L in the Progression and Metastasis of Epithelial Ovarian Cancer. Biomolecules 2015, 5, 3260–3279. [Google Scholar] [CrossRef] [PubMed]
- Dana, D.; Pathak, S.K. A Review of Small Molecule Inhibitors and Functional Probes of Human Cathepsin L. Molecules 2020, 25, 698. [Google Scholar] [CrossRef] [PubMed]
- Pranjol, M.Z.I.; Gutowski, N.J.; Hannemann, M.; Whatmore, J.L. Cathepsin L Induces Proangiogenic Changes in Human Omental Microvascular Endothelial Cells via Activation of the ERK1/2 Pathway. Curr. Cancer Drug Targets 2019, 19, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Pranjol, Z.I.; Whatmore, J.L. Cathepsin D in the Tumor Microenvironment of Breast and Ovarian Cancers. In Tumor Microenvironment; Birbrair, A., Ed.; Springer International Publishing: Cham, Switzerland, 2020; Volume 1259, pp. 1–16. [Google Scholar]
- Pranjol, M.Z.I.; Gutowski, N.J.; Hannemann, M.; Whatmore, J.L. Cathepsin D non-proteolytically induces proliferation and migration in human omental microvascular endothelial cells via activation of the ERK1/2 and PI3K/AKT pathways. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 25–33. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, S.A.; Beith, J.M.; Millar, E.K.; West, R.; McLean, A.; Cazet, A.; Swarbrick, A.; Oakes, S.R. Therapeutic targets in triple negative breast cancer. J. Clin. Pathol. 2013, 66, 530–542. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- van Roozendaal, L.M.; Smit, L.H.M.; Duijsens, G.; de Vries, B.; Siesling, S.; Lobbes, M.B.I.; de Boer, M.; De Wilt, J.H.W.; Smidt, M.L. Risk of regional recurrence in triple-negative breast cancer patients: A Dutch cohort study. Breast Cancer Res. Treat. 2016, 156, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Liu, Z.; Chen, S.; Liu, Y.; Shao, Z. A prognostic model for triple-negative breast cancer patients based on node status, cathepsin-D and Ki-67 index. PLoS ONE 2013, 8, e83081. [Google Scholar] [CrossRef]
- Ashraf, Y.; Mansouri, H.; Laurent-Matha, V.; Alcaraz, L.B.; Roger, P.; Guiu, S.; Derocq, D.; Robin, G.; Michaud, H.A.; Delpech, H.; et al. Immunotherapy of triple-negative breast cancer with cathepsin D-targeting antibodies. J. Immunother. Cancer 2019, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Yull, F.; Khabele, D. Bipolar Tumor-Associated Macrophages in Ovarian Cancer as Targets for Therapy. Cancers (Basel) 2018, 10, 366. [Google Scholar] [CrossRef]
- Mansouri, H.; Alcaraz, L.B.; Mollevi, C.; Mallavialle, A.; Jacot, W.; Boissière-Michot, F.; Simony-Lafontaine, J.; Laurent-Matha, V.; Roger, P.; Liaudet-Coopman, E.; et al. Co-Expression of Androgen Receptor and Cathepsin D Defines a Triple-Negative Breast Cancer Subgroup with Poorer Overall Survival. Cancers (Basel) 2020, 12, 1244. [Google Scholar] [CrossRef]
- Tabish, T.A.; Pranjol, M.Z.I.; Horsell, D.W.; Rahat, A.A.M.; Whatmore, J.L.; Winyard, P.G.; Zhang, S. Graphene Oxide-Based Targeting of Extracellular Cathepsin D and Cathepsin L as A Novel Anti-Metastatic Enzyme Cancer Therapy. Cancers (Basel) 2019, 11, 319. [Google Scholar] [CrossRef]
- Irvine, D.J.; Dane, E.L. Enhancing cancer immunotherapy with nanomedicine. Nat. Rev. Immunol. 2020, 20, 321–334. [Google Scholar] [CrossRef]
- Tabish, T.A.; Pranjol, M.Z.I.; Whatmore, J.L.; Zhang, S. Status and future directions of anti-metastatic cancer nanomedicines for the inhibition of cathepsin L. Front. Nanotechnol. 2020. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Shang, B.; Liu, Y.; Jiang, S.J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef]
- Sasada, T.; Kimura, M.; Yoshida, Y.; Kanai, M.; Takabayashi, A. CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies: Possible involvement of regulatory T cells in disease progression. Cancer 2003, 98, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef]
- Griseri, T.; Asquith, M.; Thompson, C.; Powrie, F. OX40 is required for regulatory T cell-mediated control of colitis. J. Exp. Med. 2010, 207, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Abu-Eid, R.; Shrimali, R.; Webb, M.; Verma, V.; Doroodchi, A.; Berrong, Z.; Samara, R.; Rodriguez, P.C.; Mkrtichyan, M.; et al. Differential PI3Kdelta Signaling in CD4(+) T-cell Subsets Enables Selective Targeting of T Regulatory Cells to Enhance Cancer Immunotherapy. Cancer Res. 2017, 77, 1892–1904. [Google Scholar] [CrossRef]
- Linch, S.N.; McNamara, M.J.; Redmond, W.L. OX40 Agonists and Combination Immunotherapy: Putting the Pedal to the Metal. Front. Oncol. 2015, 5, 34. [Google Scholar] [CrossRef]
- Schreiber, T.H. Parallel Costimulation of Effector and Regulatory T Cells by OX40, GITR, TNFRSF25, CD27, and CD137: Implications for Cancer Immunotherapy. In Novel Immunotherapeutic Approaches to the Treatment of Cancer: Drug Development and Clinical Application; Rennert, P.D., Ed.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 59–78. [Google Scholar]
- Curti, B.D.; Kovacsovics-Bankowski, M.; Morris, N.; Walker, E.; Chisholm, L.; Floyd, K.; Walker, J.; Gonzalez, I.; Meeuwsen, T.; Fox, B.A.; et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013, 73, 7189–7198. [Google Scholar] [CrossRef]
- Hamid, O.; Thompson, J.A.; Diab, A.; Ros, W.; Eskens, F.; Bermingham, C.; Konto, C.; Long, H.; Liao, K.; Ganguly, B.J.; et al. First in human (FIH) study of an OX40 agonist monoclonal antibody (mAb) PF-04518600 (PF-8600) in adult patients (pts) with select advanced solid tumors: Preliminary safety and pharmacokinetic (PK)/pharmacodynamic results. J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Zippelius, A.; Schreiner, J.; Herzig, P.; Muller, P. Induced PD-L1 expression mediates acquired resistance to agonistic anti-CD40 treatment. Cancer Immunol. Res. 2015, 3, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Polesso, F.; Weinberg, A.D.; Moran, A.E. Late-Stage Tumor Regression after PD-L1 Blockade Plus a Concurrent OX40 Agonist. Cancer Immunol. Res. 2019, 7, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Poropatich, K.O.; Dominguez, D.; Chan, W.-C.; Andrade, J.; Zha, Y.; Wray, B.D.; Miska, J.; Qin, L.; Cole, L.E.; Coates, S.; et al. OX40+ plasmacytoid dendritic cells in the tumor microenvironment promote antitumor immunity. J. Clin. Investig. 2020. [Google Scholar] [CrossRef]
- Lu, C.; Klement, J.D.; Ibrahim, M.L.; Xiao, W.; Redd, P.S.; Nayak-Kapoor, A.; Zhou, G.; Liu, K. Type I interferon suppresses tumor growth through activating the STAT3-granzyme B pathway in tumor-infiltrating cytotoxic T lymphocytes. J. Immunother. Cancer 2019, 7, 157. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | CAR Construct | Malignancy | Phase | Reference Clinicaltrials.gov |
|---|---|---|---|---|
| CD70 | CD3ζ, CD28 | Pancreatic, renal cell, breast, melanoma and ovarian | I and II | NCT02830724 |
| Mesothelin | CD3ζ, 4-1BB | Pancreatic, ovarian and mesothelioma | I | NCT02159716 |
| Muc16 (CA125) | CD3ζ, CD28 Armoured with IL-12 secretion | Ovarian | I | NCT02498912 |
| HER2 | CD3ζ, CD28 | Glioblastoma | I | NCT02442297 |
| Glypican-3 | CD3ζ, CD28 and 4-1BB | Hepatocellular carcinoma | I | NCT02395250 |
| CEA | CD3ζ, CD28 | Peritoneal carcinomatosis, colorectal, gastric, breast and pancreatic cancer | I | NCT03682744 |
| EGRFvIII | CD3ζ, 4-1BB | Glioblastoma | I | NCT03726515 |
| PSMA | CD3ζ, CD28 | Prostate Cancer | I | NCT01140373 |
| Drug | First FDA Approval Date | Cancer Type | Ref |
|---|---|---|---|
| Pembrolizumab (Anti-PD-1) | 2014 | Melanoma; non-small cell lung cancer; head and neck squamous cell cancer; classical Hodgkin lymphoma; primary mediastinal large b-cell lymphoma; urothelial carcinoma; microsatellite instability-high cancer; gastric cancer; cervical cancer; hepatocellular carcinoma; merkel cell carcinoma | [73] |
| Nivolumab (Anti-PD-1) | 2014 | Unresectable or metastatic melanoma; adjuvant treatment of melanoma; metastatic non-small cell lung cancer; small cell lung cancer; advanced renal cell carcinoma; classical Hodgkin lymphoma; squamous cell carcinoma of the head and neck; urothelial carcinoma; microsatellite instability-high (MSI-H) or mismatch repair; deficient (dMMR) metastatic colorectal cancer; hepatocellular carcinoma | [74] |
| Cemiplimab (Anti-PD-1) | 2018 | Metastatic cutaneous squamous cell carcinoma or locally advanced cutaneous squamous cell carcinoma who are not candidates for curative surgery or curative radiation | [75] |
| Atezolizumab (Anti-PD-L1) | 2016 | Locally advanced or metastatic urothelial carcinoma; metastatic non-small cell lung cancer; locally advanced or metastatic triple-negative breast cancer | [76] |
| Durvalumab (Anti-PD-L1) | 2017 | Locally advanced or metastatic urothelial carcinoma | [77] |
| Avelumab (Anti-PD-L1) | 2017 | Metastatic Merkel cell carcinoma (>12 yo); Locally advanced or metastatic urothelial carcinoma; advanced renal cell carcinoma | [78] |
| Start Date | Title | Conditions | NCT Number | Intervention | Phase | Current Status |
|---|---|---|---|---|---|---|
| 2012 | Natural Dendritic Cell Vaccines in Metastatic Melanoma Patients | Melanoma | NCT01690377 | Biological: PDC or myDC | I | Complete |
| 2013 | Safety Study of a Dendritic Cell-based Cancer Vaccine in Melanoma (GeniusVac-Mel4) | Melanoma | NCT01863108 | Biological: GeniusVac-Mel4 | I | Complete |
| 2015 | myDC/pDC in Stage III Melanoma Patients | Melanoma | NCT02574377 | Drug A: myDC vaccination Drug B: pDC vaccination Drug C: combined myDC/pDC vaccination | I & II | Unknown |
| 2016 | Melanoma Patients Immunized with Natural Dendritic Cells (MIND-DC) | Melanoma | NCT02993315 | Biological: nDC vaccination Biological: Placebo injection | III | Active, not recruiting |
| 2016 | Natural Dendritic Cells for Immunotherapy of Chemo-naive Metastatic Castration-resistant Prostate Cancer Patients | Prostatic Neoplasms | NCT02692976 | Biological:mDC vaccination Biological: pDC vaccination Biological: mDC and pDC vaccination | II | Complete |
| 2017 | Dendritic Cell Therapy, Cryosurgery, and Pembrolizumab in Treating Patients With Non-Hodgkin Lymphoma | Non-Hodgkins Lymphoma | NCT03035331 | Procedure: Cryosurgery Biological: Dendritic Cell Therapy Biological: Pembrolizumab Biological: Pneumococcal 13-valent Conjugate Vaccine Other: Quality-of-Life Assessment | I & II | Recruiting |
| 2018 | Intratumoral Injection of Autologous CD1c (BDCA-1)+ Myeloid Dendritic Cells Plus Talimogene Laherparepvec (T-VEC) (myDCTV) | Melanoma | NCT03747744 | Other: CD1c (BDCA-1)+ myDC | I | Recruiting |
| 2018 | Intratumoral Injection of Autologous CD1c (BDCA-1)+ myDC, Avelumab, and Ipilimumab Plus Systemic Nivolumab (myDAvIpNi) | Solid Tumours Metastases of Soft Tissue | NCT03707808 | Drug: intratumoral injection of autologous CD1c (BDCA-1)+ myDC | I | Recruiting |
| 2019 | Treatment of Recurrent Bladder Cancer With Dendritic Cells | Bladder cancer | NCT04184232 | Biological: Dendritic cells Other: Standard treatment according to the Clinical protocols | II | Recruiting |
| 2019 | Dendritic Cells for Immunotherapy of Metastatic Endometrial Cancer Patients (DECENDO) | Endometrical Cancer | NCT04212377 | Biological: Dendritic Cells for endometrial cancer | II | Recruiting |
| 2019 | Arm 1: Infusion of Autologous Monocyte-derived Lysate Pulsed Dendritic Cells (PV-001-DC) in Patients With Advanced Melanoma | Metastatic Melanoma | NCT03803397 | Biological: PV-001-DC | I | Not yet recruiting |
| 2019 | DCVAC/OvCa and Standard of Care (SoC) in Relapsed Ovarian, Fallopian Tube, and Primary Peritoneal Carcinoma (VITALIA) | Ovarian Cancer Fallopian Tube Cancer Peritoneal Carcinoma | NCT03905902 | Biological: DCVAC/OvCa Bioloigcal: DCVAC/OvCa placebo | III | Not yet recruiting |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alard, E.; Butnariu, A.-B.; Grillo, M.; Kirkham, C.; Zinovkin, D.A.; Newnham, L.; Macciochi, J.; Pranjol, M.Z.I. Advances in Anti-Cancer Immunotherapy: Car-T Cell, Checkpoint Inhibitors, Dendritic Cell Vaccines, and Oncolytic Viruses, and Emerging Cellular and Molecular Targets. Cancers 2020, 12, 1826. https://doi.org/10.3390/cancers12071826
Alard E, Butnariu A-B, Grillo M, Kirkham C, Zinovkin DA, Newnham L, Macciochi J, Pranjol MZI. Advances in Anti-Cancer Immunotherapy: Car-T Cell, Checkpoint Inhibitors, Dendritic Cell Vaccines, and Oncolytic Viruses, and Emerging Cellular and Molecular Targets. Cancers. 2020; 12(7):1826. https://doi.org/10.3390/cancers12071826
Chicago/Turabian StyleAlard, Emilie, Aura-Bianca Butnariu, Marta Grillo, Charlotte Kirkham, Dmitry Aleksandrovich Zinovkin, Louise Newnham, Jenna Macciochi, and Md Zahidul Islam Pranjol. 2020. "Advances in Anti-Cancer Immunotherapy: Car-T Cell, Checkpoint Inhibitors, Dendritic Cell Vaccines, and Oncolytic Viruses, and Emerging Cellular and Molecular Targets" Cancers 12, no. 7: 1826. https://doi.org/10.3390/cancers12071826
APA StyleAlard, E., Butnariu, A.-B., Grillo, M., Kirkham, C., Zinovkin, D. A., Newnham, L., Macciochi, J., & Pranjol, M. Z. I. (2020). Advances in Anti-Cancer Immunotherapy: Car-T Cell, Checkpoint Inhibitors, Dendritic Cell Vaccines, and Oncolytic Viruses, and Emerging Cellular and Molecular Targets. Cancers, 12(7), 1826. https://doi.org/10.3390/cancers12071826