Cell-Mediated Release of Nanoparticles as a Preferential Option for Future Treatment of Melanoma



 ,
,  ,
, 
Abstract
1. Introduction
2. TIP and EPR, Two Opposite Physical Forces
2.1. TIP and Fluid Stress
2.2. TIP and Solid Stress
2.3. Consequences of TIP Increase on Circulation Times of Nanomedicines and EPR Effect
3. Cell-Mediated Delivery of Nanoparticles as a Means to Overcome TIP-Dependent Restraints of NP-Based Therapeutic Efficiency
3.1. Platelets
3.2. Neutrophils
3.3. Monocytes/Macrophages (Mϕs)
3.4. Lymphocytes
3.5. Red Blood Cells
3.6. Neural Stem Cells and Induced Pluripotent Stem Cells
3.7. Mesenchymal Stem Cells
3.8. Endothelial Colony Forming Cells (ECFCs)
4. Nanoparticles in the Preclinical Research and Clinic of Melanoma
5. Conclusions
“What the medicine does not cure, the scalpel heals, what the scalpel does not heal, the fire heals it, what fire does not heal must be considered incurable”Hippocrates (460–377 B.C)
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer Statistics 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Domingues, B.; Lopes, J.M.; Soares, P.; Pópulo, H. Melanoma treatment in review. Immunotargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, S.; Brancaccio, G.; Argenziano, G.; Martinelli, E.; Morgillo, F.; Ciardiello, F.; Troiani, T. It is finally time for adjuvant therapy in melanoma. Cancer Treat. Rev. 2018, 69, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.L.; Dudley, M.; Citrin, D.E.; Somerville, R.P.; Wunderlich, J.R.; Danforth, D.N.; Zlott, D.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; et al. A randomized prospective evaluation comparing intensity of lymphodepletion prior to adoptive transfer of tumor infiltrating lymphocytes for patients with metastatic melanoma. J. Clin. Oncol. 2016, 34, 2389–2397. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Restifo, N.P.; Royal, R.E.; Kammula, U.; White, D.E.; Mavroukakis, S.A.; et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol. 2005, 23, 2346–2357. [Google Scholar] [CrossRef]
- Bombelli, F.B.; Webster, C.A.; Moncrieff, M.; Sherwood, V. The scope of nanoparticle therapies for future metastatic melanoma treatment. Lancet Oncol. 2014, 15, e22–e32. [Google Scholar] [CrossRef]
- Mishra, H.; Mishra, P.K.; Ekielski, A.; Jaggi, M.; Iqbal, Z.; Talegaonkar, S. Melanoma treatment: From conventional to nanotechnology. J. Cancer Res. Clin. Oncol. 2018, 144, 2283–2302. [Google Scholar] [CrossRef]
- Bagheri, S.; Yasemi, M.; Safaie-Qamsari, E.; Rashidiani, J.; Abkar, M.; Hassani, M.; Mirhosseini, S.A.; Kooshki, H. Using gold nanoparticles in diagnosis and treatment of melanoma cancer. Artif. Cells Nanomed. Biotechnol. 2018, 46, 462–471. [Google Scholar] [CrossRef]
- Lombardo, D.; Kiselev, M.A.; Caccamo, M.T. Smart Nanoparticles for Drug Delivery Application: Development of Versatile Nanocarrier Platforms in Biotechnology and Nanomedicine. J. Nanomater. 2019, 12, 1–26. [Google Scholar] [CrossRef]
- Li, Z.; Tan, S.; Li, S.; Shen, Q.; Wang, K. Cancer drug delivery in the nano era: An overview and perspectives (Review). Oncol. Rep. 2017, 38, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Golombek, S.K.; May, J.N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef]
- Ding, S.; O’Banion, C.P.; Welfare, J.G.; Lawrence, D.S. Cellular Cyborgs: On the Precipice of a Drug Delivery Revolution. Cell Chem. Biol. 2018, 25, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. Cell-mediated delivery of nanoparticles: Taking advantage of circulatory cells to target nanoparticles. J. Control. Release 2014, 190, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Laurenzana, A.; Margheri, F.; Chillà, A.; Biagioni, A.; Margheri, G.; Calorini, L.; Fibbi, G.; Del Rosso, M. Endothelial Progenitor Cells as Shuttle of Anticancer Agents. Hum. Gene Ther. 2016, 27, 784–791. [Google Scholar] [CrossRef]
- Scallan, J.; Huxley, V.H.; Korthius, R.J. Pathophisiology of Edema Formation. Capillary Fluid Exchange: Regulation, Functions, and Pathology; Morgan&Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Ariffin, A.B.; Forde, P.F.; Jahangeer, S.; Soden, D.M.; Hinchion, J. Releasing pressure in tumors: What do we know so far and where do we go from here? A review. Cancer Res. 2014, 74, 2655–2662. [Google Scholar] [CrossRef]
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef]
- Schmid-Schonbein, G.W. Microlymphatics and lymph flow. Physiol. Rev. 1990, 70, 987–1028. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 396–406. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, M.J.A.; Verweij, J. Renal Toxicities of Chemotherapy. Semin. Oncol. 2006, 33, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.; Catane, R.; Uziely, B.; Kaufman, B.; Safra, T.; Cohen, R.; Martin, F.; Huang, A.; Barenholz, Y. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994, 54, 987–992. [Google Scholar] [PubMed]
- Gabizon, A.A.; Barenholz, Y.; Bialer, M. Prolongation of the Circulation Time of Doxorubicin Encapsulated in Liposomes Containing a Polyethylene Glycol-Derivatized Phospholipid: Pharmacokinetic Studies in Rodents and Dogs. Pharm. Res. 1993, 10, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Jain, R.K.; Baxter, L.T. Mechanisms of heterogeneous distribution of monoclonal antibodies and other macromolecules in tumors: Significance of elevated interstitial pressure. Cancer Res. 1988, 48, 7022–7032. [Google Scholar]
- Padera, T.P.; Stoll, B.R.; Tooredman, J.B.; Capen, D.; Di Tomase, E.; Jain, R.K. Cancer cells compress intratumor vessels: Pressure from proliferating cells impedes transport of therapeutic drugs into tumors. Nature 2004, 247, 695. [Google Scholar] [CrossRef]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef]
- Harrington, K.J.; Mohammadtaghi, S.; Uster, P.S.; Glass, D.; Peters, A.M.; Vile, R.G.; Stewart, J.S.W. Effective Targeting of Solid Tumors in Patients With Locally Advanced Cancers by Radiolabeled Pegylated Liposomes. Clin. Cancer Res. 2001, 7, 243–254. [Google Scholar]
- Tanaka, N.; Kanatani, S.; Tomer, R.; Sahlgren, C.; Kronqvist, P.; Kaczynska, D.; Louhivuori, L.; Kis, L.; Lindh, C.; Mitura, P.; et al. Whole-tissue biopsy phenotyping of three-dimensional tumours reveals patterns of cancer heterogeneity. Nat. Biomed. Eng. 2017, 1, 796–806. [Google Scholar] [CrossRef]
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Natfji, A.A.; Ravishankar, D.; Osborn, H.M.I.; Greco, F. Parameters Affecting the Enhanced Permeability and Retention Effect: The Need for Patient Selection. J. Pharm. Sci. 2017, 106, 3179–3187. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Laginha, K.M.; Verwoert, S.; Charrois, G.J.R.; Allen, T.M. Determination of Doxorubicin Levels in Whole Tumor and Tumor Nuclei in Murine Breast Cancer Tumors. Clin. Cancer Res. 2005, 11, 6944–6949. [Google Scholar] [CrossRef] [PubMed]
- Van Vlerken, L.E.; Duan, Z.; Little, S.R.; Seiden, M.V.; Amiji, M.M. Biodistribution and pharmacokinetic analysis of Paclitaxel and ceramide administered in multifunctional polymer-blend nanoparticles in drug resistant breast cancer model. Mol. Pharm. 2008, 5, 516–526. [Google Scholar] [CrossRef][Green Version]
- Cui, Y.; Zhang, M.; Zeng, F.; Jin, H.; Xu, Q.; Huang, Y. Dual-Targeting Magnetic PLGA Nanoparticles for Codelivery of Paclitaxel and Curcumin for Brain Tumor Therapy. ACS Appl. Mater. Interfaces 2016, 8, 32159–32169. [Google Scholar] [CrossRef]
- Lammers, T.; Kiessling, F.; Ashford, M.; Hennink, W.; Crommelin, D.; Storm, G. Cancer nanomedicine: Is targeting our target? Nat. Rev. Mater. 2016, 1, 16069. [Google Scholar] [CrossRef]
- Xu, P.; Zuo, H.; Chen, B.; Wang, R.; Ahmed, A.; Hu, Y.; Ouyang, J. Doxorubicin-loaded platelets as a smart drug delivery system: An improved therapy for lymphoma. Sci. Rep. 2017, 7, 42632. [Google Scholar] [CrossRef]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef]
- Sierko, E.; Wojtukiewicz, M.Z. Platelets and angiogenesis in malignancy. Semin. Thromb. Hemost. 2004, 30, 95–108. [Google Scholar] [PubMed]
- Prisco, D.; Paniccia, R.; Coppo, M.; Filippini, M.; Francalanci, I.; Brunelli, T.; Comeglio, P.; Abbate, R. Platelet activation and platelet lipid composition in pulmonary cancer Prostaglandins Leukot. Essent. Fatty Acids. 1995, 53, 65–68. [Google Scholar] [CrossRef]
- Blann, A.D.; Gurney, D.; Wadley, M.; Bareford, D.; Stonelake, P.; Lip, G.Y. Increased soluble P-selectin in patients with haematological and breast cancer: A comparison with fibrinogen, plasminogen activator inhibitor and von Willebrand factor. Blood Coagul. Fibrinolysis 2001, 12, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Verheul, H.M.W.; Hoekman, K.; Lupu, F.; Broxterman, H.J.; van der Valk, P.; Kakkar, A.K.; Pinedo, H.M. Platelet and coagulation activation with vascular endothelial growth factor generation in soft tissue sarcomas. Clin. Cancer Res. 2000, 6, 166–171. [Google Scholar]
- Honn, K.V.; Tang, D.G.; Chen, Y.Q. Platelets and cancer metastasis-more than an epiphenomenon. Semin. Thromb. Hemost. 1992, 18, 392–415. [Google Scholar] [CrossRef]
- Mehta, P. Potential role of platelets in the pathogenesis of tumor-metastasis. Blood 1984, 63, 55–63. [Google Scholar] [CrossRef]
- Jurasz, P.; Alonso-Escolano, D.; Radomski, M.W. Platelet-cancer interactions: Mechanisms and pharmacology of tumour cell-induced platelet aggregation. Br. J. Pharmacol. 2004, 143, 819–826. [Google Scholar] [CrossRef]
- Erpenbeck, L.; Schon, M.P. Deadly allies: The fatal interplay between platelets and metastasizing cancer cells. Blood 2010, 115, 3427–3436. [Google Scholar] [CrossRef]
- McCarty, O.J.T.; Mousa, S.A.; Bray, P.F.; Konstantopoulos, K. Immobilized platelets support human colon carcinoma cell tethering, rolling, and firm adhesion under dynamic flow conditions. Blood 2000, 96, 1789–1797. [Google Scholar] [CrossRef]
- Momi, S.; Falcinelli, E.; Giannini, S.; Gresele, P. Loss of matrix metalloproteinase 2 in platelets reduces arterial thrombosis in vivo. J. Exp. Med. 2009, 206, 2365–2379. [Google Scholar] [CrossRef] [PubMed]
- Movat, H.Z.; Weiser, W.J.; Glynn, M.F.; Mustard, J.F. Platelet phagocytosis and aggregation. J. Cell Biol. 1965, 27, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Rachidi, S.; Kaur, M.; Lautenschlaeger, T.; Li, Z. Platelet count correlates with stage and predicts survival in melanoma. Platelets 2019, 30, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Mannucci, P.M.; Cattaneo, M.; Canciani, M.T.; Maniezzo, M.; Vaglini, M.; Cascinelli, N. Early presence of activated (‘exhausted’) platelets in malignant tumors (breast adenocarcinoma and malignant melanoma). Eur. J. Cancer Clin. Oncol. 1989, 25, 1413–1417. [Google Scholar] [CrossRef]
- Li, N.; Diao, Z.; Huang, X.; Niu, Y.; Liu, T.; Liu, Z.P.; Wang, R.T.; Yu, K.J. Increased platelet distribution width predicts poor prognosis in melanoma patients. Sci. Rep. 2017, 7, 2970. [Google Scholar] [CrossRef]
- Echtler, K.; Konrad, I.; Lorenz, M.; Schneider, S.; Hofmaier, S.; Plenagl, F.; Stark, K.; Czermak, T.; Tirniceriu, A.; Eichhorn, M.; et al. Platelet GPIIb supports initial pulmonary retention but inhibits subsequent proliferation of melanoma cells during hematogenic metastasis. PLoS ONE 2017, 12, e0172788. [Google Scholar] [CrossRef]
- Kim, M.W.; Lee, G.; Niidome, T.; Komohara, Y.; Lee, R.; Park, Y.I. Platelet-Like Gold Nanostars for Cancer Therapy: The Ability to Treat Cancer and Evade Immune Reactions. Front. Bioeng. Biotechnol. 2020, 8, 133. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef]
- Grecian, R.; Whyte, M.K.B.; Walmsley, S.R. The role of neutrophils in cancer. Br. Med. Bull. 2018, 128, 5–14. [Google Scholar] [CrossRef]
- Galdiero, M.R.; Varricchi, G.; Loffredo, S.; Mantovani, A.; Marone, G. Roles of neutrophils in cancer growth and progression. J. Leukoc. Biol. 2018, 103, 457–464. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Wu, L.; Saxena, S.; Awaji, M.; Rakesh, K.; Singh, R.K. Tumor-Associated Neutrophils in Cancer: Going Pro. Cancers 2019, 11, 564. [Google Scholar] [CrossRef] [PubMed]
- Forsthuber, A.; Lipp, K.; Andersen, L.; Ebersberger, S.; Graña-Castro, I.; Ellmeier, W.; Petzelbauer, P.; Lichtenberger, B.M.; Loewe, R. CXCL5 as Regulator of Neutrophil Function in Cutaneous Melanoma. J. Investig. Dermatol. 2019, 139, 186–194. [Google Scholar] [CrossRef]
- Soler-Cardona, A.; Forsthuber, A.; Lipp, K.; Ebersberger, S.; Heinz, M.; Schossleitner, K.; Buchberger, E.; Gröger, M.; Petzelbauer, P.; Hoeller, C.; et al. CXCL5 Facilitates Melanoma Cell-Neutrophil Interaction and Lymph Node Metastasis. J. Investig. Dermatol. 2018, 138, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Hölzel, M.; Tüting, T. Inflammation-Induced Plasticity in Melanoma Therapy and Metastasis. Trends Immunol. 2016, 37, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zhang, S.; Qiao, J. Prognostic value of neutrophil-to-lymphocyte ratio in melanoma. Evidence from a PRISMA-compliant meta-analysis. Medicine (Baltimore) 2018, 97, e11446. [Google Scholar] [CrossRef]
- Martin, C.; Burdon, P.C.; Bridger, G.; Gutierrez-Ramos, J.C.; Williams, T.J.; Rankin, S.M. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 2003, 19, 583–593. [Google Scholar] [CrossRef]
- Furze, R.C.; Rankin, S.M. Neutrophil mobilization and clearance in the bone marrow. Immunology 2008, 125, 281–288. [Google Scholar] [CrossRef]
- Eash, K.J.; Greenbaum, A.M.; Gopalan, P.K.; Link, D.C. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Investig. 2010, 120, 2423–2431. [Google Scholar] [CrossRef]
- Chow, M.T.; Luster, A.D. Chemokines in cancer. Cancer Immunol. Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef]
- Hong, C.W. Current Understanding in Neutrophil Differentiation and Heterogeneity. Immune Netw. 2017, 17, 298–306. [Google Scholar] [CrossRef]
- Belperio, J.A.; Keane, M.P.; Burdick, M.D.; Londhe, V.; Xue, Y.Y.; Li, K.; Phillips, R.J.; Strieter, R.M. Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. J. Clin. Investig. 2002, 110, 1703–1716. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Nawandar, D.M.; Nannuru, K.C.; Varney, M.L.; Singh, R.K. Targeting CXCR2 enhances chemotherapeutic response, inhibits mammary tumor growth, angiogenesis, and lung metastasis. Mol. Cancer Ther. 2013, 12, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Albelda, S.M. Tumor-associated neutrophils: Friend or foe? Carcinogenesis 2012, 33, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liu, W.; Zheng, Y.; Wang, S.; Yang, B.; Li, M.; Song, J.; Zhang, F.; Zhang, X.; Wang, Q.; et al. CXCL1 derived from tumor-associated macrophages promotes breast cancer metastasis via activating NF-kappaB/SOX4 signaling. Cell Death Dis. 2018, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.S.; Hsu, C.C.; Pai, V.C.; Liao, W.Y.; Huang, S.S.; Tan, K.T.; Yen, C.J.; Hsu, S.C.; Chen, W.Y.; Shan, Y.S.; et al. Metronomic chemotherapy prevents therapy-induced stromal activation and induction of tumor-initiating cells. J. Exp. Med. 2016, 213, 2967–2988. [Google Scholar] [CrossRef]
- Chu, D.; Zhao, Q.; Yu, J.; Zhang, F.; Zhang, H.; Wang, Z. Nanoparticle Targeting of Neutrophils for Improved Cancer Immunotherapy. Adv. Healthc. Mater. 2016, 5, 1088–1093. [Google Scholar] [CrossRef]
- Thalin, C.; Demers, M.; Blomgren, B.; Wong, S.L.; von Arbin, M.; von Heijne, A.; Laska, A.C.; Wallén, H.; Wagner, D.D.; Aspberg, S. NETosis promotes cancer-associated arterial microthrombosis presenting as ischemic stroke with troponin elevation. Thromb. Res. 2016, 139, 56–64. [Google Scholar] [CrossRef]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef]
- Demers, M.; Wong, S.L.; Martinod, K.; Gallant, M.; Cabral, J.E.; Wang, Y.; Wagner, D.D. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology 2016, 5, e1134073. [Google Scholar] [CrossRef]
- Hao, N.B.; Lü, M.H.; Fan, Y.H.; Cao, Y.L.; Zhang, Z.R.; Yang, S.M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef]
- Basel, M.T.; Balivada, S.; Wang, H.; Shrestha, T.B.; Seo, G.M.; Pyle, M.; Abayaweera, G.; Dani, R.; Koper, O.B.; Tamura, M.; et al. Cell-delivered magnetic nanoparticles caused hyperthermia-mediated increate servival in a murine pancreatic cancer model. Int. J. Nanomed. 2012, 7, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.R.; Stanton-Maxey, K.J.; Stanley, J.K.; Levin, C.S.; Bardhan, R.; Akin, D.; Badve, S.; Sturgis, J.; Robinson, J.P.; Bashir, R.; et al. A cellular Trojan Horse for delivery of therapeutic nanoparticles into tumors. Nano Lett. 2007, 7, 3759–3765. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Tsai, T.H.; Huang, Y.C.; Shieh HRLiao, H.F.; Chen, Y.J. Differential Immunomodulating Effects of Pegylated Liposomal Doxorubicin Nanoparticles on Human Macrophages. J. Nanosci. Nanotechnol. 2012, 12, 7739–7746. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Zheng, Y.R.; Gadde, S.; Pfirschk, C.; Zope, H.; Engblom, C.; Kohler, R.H.; Iwamoto, Y.; Yang, K.S.; Askevold, B.; et al. Weissleder R Tumour-associated Macrophages Act as a Slow-Release Reservoir of Nano-Therapeutic Pt(IV) Pro-Drug. Nat. Commun. 2015, 6, 8692. [Google Scholar] [CrossRef] [PubMed]
- Doshi, N.; Swiston, A.J.; Gilbert, J.B.; Alcaraz, M.L.; Cohen, R.E.; Rubner, M.F.; Mitragotri, S. Cell-Based Drug Delivery Devices Using Phagocytosis-Resistant Backpacks. Adv. Mater. 2011, 23, H105–H109. [Google Scholar] [CrossRef]
- Zhao, Y.; Haney, M.; Mahajan, V.; Reiner, C.B. Active Targeted Macrophage-mediated Delivery of Catalase to Affected Brain Regions in Models of Parkinson’s Disease. J. Nanomed. Nanotechnol. 2011. [Google Scholar] [CrossRef]
- Batrakova, E.V.; Li, S.; Reynolds, A.D.; Mosley, R.L.; Bronich, T.K.; Kabanov, A.V.; Gendelman, H.E. A Macrophage-Nanozyme Delivery System for Parkinson’s Disease. Bioconjug. Chem. 2007, 18, 1498–1506. [Google Scholar] [CrossRef]
- Seo, G.M.; Rachakatla, R.S.; Balivada, S.; Pyle, M.; Shrestha, T.B.; Basel, M.T.; Myers, C.; Wang, H.; Tamura, M.; Bossmann, S.H.; et al. A self-contained enzyme activating prodrug cytotherapy for preclinical melanoma. Mol. Biol. Rep. 2012, 39, 157–165. [Google Scholar] [CrossRef][Green Version]
- Mitchell, M.J.; King, M.R. Leukocytes as carriers for targeted cancer drug delivery. Expert Opin. Drug Deliv. 2015, 42, 375–392. [Google Scholar] [CrossRef]
- Jones, R.B.; Mueller, S.; Kumari, S.; Vrbanac, V.; Genel, S.; Tager, A.M. Antigen recognition-triggered drug delivery mediated by nanocapsule-functionalized cytotoxic T-cells. Biomaterials 2017, 117, 44–53. [Google Scholar] [CrossRef]
- Siriwon, N.; Kim, Y.J.; Siegler, E.; Chen, X.; Rohrs, J.A.; Liu, Y.; Wang, P. CAR-T Cells Surface-Engineered with Drug-Encapsulated Nanoparticles Can Ameliorate Intratumoral T-cell Hypofunction. Cancer Immunol. Res. 2018, 6, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.; Mitragotri, S. Prolonged circulation of large polymeric nanoparticles by non-covalent adsorption on erythrocytes. J. Control. Release 2004, 100, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.; Mitragotri, S. Long circulating nanoparticles via adhesion on red blood cells: Mechanism and extended circulation. Exp. Biol. Med. (Maywood) 2007, 232, 958–966. [Google Scholar] [PubMed]
- Anselmo, A.C.; Gupta, V.; Zern, B.J.; Pan, D.; Zakrewsky, M.; Muzykantov, V.; Mitragotri, S. Delivering nanoparticles to lungs while avoiding liver and spleen through adsorption on red blood cells. ACS Nano 2013, 7, 11129–11137. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, M.; Nikolaev, B.; Marchenko, Y.; Yakovleva, L.; Skvortsov, N.; Mazur, A.; Tolstoy, P.; Ryzhov, V.; Multhoff, G. Targeting experimental orthotopic glioblastoma with chitosan-based superparamagnetic iron oxide nanoparticles (CS-DX-SPIONs). Int. J. Nanomed. 2018, 13, 1471–1482. [Google Scholar] [CrossRef]
- Sun, D.; Chen, J.; Wang, Y.; Ji, H.; Peng, R.; Jin, L.; Wu, W. Advances in refunctionalization of erythrocyte-based nanomedicine for enhancing cancer-targeted drug delivery. Theranostics 2019, 9, 6885–6900. [Google Scholar] [CrossRef]
- Wang, D.; Dong, H.; Li, M.; Cao, Y.; Yang, F.; Zhang, K.; Dai, W.; Wang, C.; Zhang, X. Erythrocyte-Cancer Hybrid Membrane Camouflaged Hollow Copper Sulfide Nanoparticles for Prolonged Circulation Life and Homotypic-Targeting Photothermal/Chemotherapy of Melanoma. ACS Nano 2018, 12, 5241–5252. [Google Scholar] [CrossRef]
- Guo, Y.Y.; Wang, D.; Song, Q.; Wu, T.; Zhuang, X.; Bao, Y.; Kong, M.; Qi, Y.; Tan, S.; Zhang, Z. Erythrocyte Membrane-Enveloped Polymeric Nanoparticles as Nanovaccine for Induction of Antitumor Immunity against Melanoma. ACS Nano 2015, 9, 6918–6933. [Google Scholar] [CrossRef]
- Gage, F.H.; Temple, S. Neural stem cells: Generating and regenerating the brain. Neuron 2013, 80, 588–601. [Google Scholar] [CrossRef]
- Bagó, J.R.; Alfonso-Pecchio, A.; Okolie, O.; Dumitru, R.; Rinkenbaugh, A.; Baldwin, A.S.; Miller, C.R.; Magness, S.T.; Hingtgen, S.D. Therapeutically engineered induced neural stem cells are tumour-homing and inhibit progression of glioblastoma. Nat. Commun. 2016, 7, 10593. [Google Scholar] [CrossRef]
- Rachakatla, R.S.; Balivada, S.; Seo, G.M.; Myers, C.B.; Wang, H.; Samarakoon, T.N.; Dani, R.; Pyle, M.; Kroh, F.O.; Walker, B.; et al. Attenuation of mouse melanoma by A/C magnetic field after delivery of bi-magnetic nanoparticles by neural progenitor cells. ACS Nano 2010, 4, 7093–7104. [Google Scholar] [CrossRef] [PubMed]
- Kevin, S.; Carbajal, K.S.; Schaumburg, C.; Strieter, R.; Kane, J.; Lane, T.E. Migration of engrafted neural stem cells is mediated by CXCL12 signaling through CXCR4 in a viral model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2010, 107, 11068–11073. [Google Scholar]
- Jiang, Z.; Li, Y.; Ji, X.; Tang, Y.; Yu, H.; Ding, L.; Yu, M.; Cui, Q.; Zhang, M.; Ma, Y.; et al. Protein profiling identified key chemokines that regulate the maintenance of human pluripotent stem cells. Sci. Rep. 2017, 7, 14510. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Bagó, J.R.; Sheets, K.T.; Hingtgen, S.D. Neural stem cell therapy for cancer. Methods 2016, 99, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Gnecchi, M.; Melo, L.G. Bone marrow-derived mesenchymal stem cells: Isolation, expansion, characterization, viral transduction, and production of conditioned medium. Methods Mol. Biol. 2009, 482, 281–294. [Google Scholar] [PubMed]
- Hassan, G.; Kasem, I.; Soukkarieh, C.; Aljamali, M. A Simple Method to Isolate and Expand Human Umbilical Cord Derived Mesenchymal Stem Cells: Using Explant Method and Umbilical Cord Blood Serum. Int. J. Stem Cells 2017, 10, 184–192. [Google Scholar] [CrossRef]
- Gao, H.; Priebe, W.; Glod, J.; Banerjee, D. Activation of signal transducers and activators of transcription 3 and focal adhesion kinase by stromalcell-derived factor 1 is required for migration of human mesenchymal stem cellsin response to tumor cell-conditioned medium. Stem Cells 2009, 27, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, S.; Teixeira, V.H.; Kalber, T.; Jose, R.J.; Floto, R.A.; Janes, S.M. Macrophage migration inhibitory factor-CXCR4 is the dominant chemotactic axis in human mesenchymal stem cell recruitment to tumors. J. Immunol. 2015, 194, 3463–3474. [Google Scholar] [CrossRef]
- Phillips, R.J.; Burdick, M.D.; Lutz, M.; Belperio, J.A.; Keane, M.P.; Strieter, R.M. The stromal derived factor-1/CXCL12-CXC chemokinereceptor 4 biological axis in non-small cell lung cancer metastases. Am. J. Respir. Crit. Care Med. 2003, 167, 1676–1686. [Google Scholar] [CrossRef]
- Kamimura, A.; Kamachi, M.; Nishihira, J.; Ogura, S.; Isobe, H.; Dosaka-Akita, H.; Ogata, A.; Shindoh, M.; Ohbuchi, T.; Kawakami, Y. Intracellular dis-tribution of macrophage migration inhibitory factor predicts the prognosis ofpatients with adenocarcinoma of the lung. Cancer 2000, 89, 334–341. [Google Scholar] [CrossRef]
- Han, I.; Lee, M.R.; Nam, K.W.; Oh, J.H.; Moon, K.C.; Kim, H.S. Expression of macrophage migration inhibitory factor relates to survival in high-grade osteosarcoma. Clin. Orthop. Relat. Res. 2008, 466, 2107–2113. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Siegler, K.L.; Bellino, M.A.; Tannenbaum, M. Macrophagemigration inhibitory factor evaluation compared with prostate specific antigen asa biomarker in patients with prostate carcinoma. Cancer 2002, 94, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Tomiyasu, M.; Yoshino, I.; Suemitsu, R.; Okamoto, T.; Sugimachi, K. Quantification of macrophage migration inhibitory factor mRNA expression innon-small cell lung cancer tissues and its clinical significance. Clin. Cancer Res. 2002, 8, 3755–3760. [Google Scholar] [PubMed]
- Otsu, K.; Das, S.; Houser, S.D.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Concentration-dependent inhibition of angiogenesis by mesenchymal stem cells. Blood 2009, 113, 4197–4205. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Xu, Z.; Zhao, T.; Zhao, Z.; Shi, M.; Zhao, R.C.; Ye, L.; Zhang, X. Suppression of tumorigenesis by human mesenchymal stem cells in a hepatoma model. Cell Res. 2008, 18, 500–507. [Google Scholar] [CrossRef]
- Hamada, H.; Kobune, M.; Nakamura, K.; Kawano, Y.; Kato, K.; Honmou, O.; Houkin, K.; Matsunaga, T.; Niitsu, Y. Mesenchymal stem cells (MSC) as therapeutic cytoreagents for gene therapy. Cancer Sci. 2005, 96, 149–156. [Google Scholar] [CrossRef]
- Altanerova, U.; Jakubechova, J.; Benejova, K.; Priscakova, P.; Pesta, M.; Pitule, P.; Topolcan, O.; Kausitz, J.; Zduriencikova, M.; Repiska, V.; et al. Prodrug suicide gene therapy for cancer targeted intracellular by mesenchymal stem cell exosomes. Int. J. Cancer 2019, 144, 897–908. [Google Scholar] [CrossRef]
- Poggi, A.; Varesano, S.; Zocchi, M.R. How to Hit Mesenchymal Stromal Cells and Make the Tumor Microenvironment Immunostimulant Rather Than Immunosuppressive. Front. Immunol. 2018, 9, 262. [Google Scholar] [CrossRef]
- Wang, M.; Yuan, Q.; Xie, L. Mesenchymal Stem Cell-Based Immunomodulation: Properties and Clinical Application. Stem Cells Int. 2018, 2018, 3057624. [Google Scholar] [CrossRef]
- Mahasa, K.J.; de Pillis, L.; Ouifki, R.; Eladdadi, A.; Maini, P.; Yoon, A.R.; Yun, C.O. Mesenchymal stem cells used as carrier cells of oncolytic adenovirus results in enhanced oncolytic virotherapy. Sci. Rep. 2020, 10, 425. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Seah, I.; Bougazzoul, O.; Choi, G.; Meeth, K.; Bosenberg, M.W.; Wakimoto, H.; Fisher, D.; Shah, K. Stem cell-released oncolytic herpes simplex virus has therapeutic efficacy in brain metastatic melanomas. Proc. Natl. Acad. Sci. USA 2017, 114, E6157–E6165. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.; Sahebkar, A.; Avan, A.; Jaafari, M.R.; Salehi, R.; Salehi, H.; Baharvand, H.; Rezaei, A.; Hadjati, J.; Pawelek, J.M.; et al. Application of Mesenchymal Stem Cells in Melanoma: A Potential Therapeutic Strategy for Delivery of Targeted Agents. Curr. Med. Chem. 2016, 23, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Mundra, V.; Li, W.; Mahato, R.I. Nanoparticle-mediated drug delivery for treating melanoma. Nanomedicine 2015, 10, 2613–2633. [Google Scholar] [CrossRef]
- Loebinger, M.R.; Kyrtatos, P.G.; Turmaine, M.; Price, A.N.; Pankhurst, Q.; Lythgoe, M.F.; Janes, S.M. Magnetic resonance imaging of mesenchymal stem cells homing to pulmonary metastases using biocompatible magnetic nanoparticles. Cancer Res. 2009, 69, 8862–8867. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, F.; Wang, H.; Niu, G.; Choi, K.Y.; Swierczewska, M.; Zhang, G.; Gao, H.; Wang, Z.; Zhu, L.; et al. Mesenchymal stem cell-based cell engineering with multifunctional mesoporous silica nanoparticles for tumor delivery. Biomaterials 2013, 34, 1772–1780. [Google Scholar] [CrossRef]
- Kang, S.; Bhang, S.H.; Hwang, S.; Yoon, J.K.; Song, J.; Jang, H.K.; Kim, S.; Kim, B.S. Mesenchymal Stem Cells Aggregate and Deliver Gold Nanoparticles to Tumors for Photothermal Therapy. ACS Nano 2015, 9, 9678–9690. [Google Scholar] [CrossRef]
- Branislava, J.; Arbab, A.S. Cord blood endothelial progenitor cells as therapeutic and imaging probes. Imaging Med. 2012, 4, 477–490. [Google Scholar]
- Keighron, C.; Caomhán, J.; Lyons, C.J.; Creane, M.; O’Brien, T.; Liew, A. Recent Advances in Endothelial Progenitor Cells Toward Their Use in Clinical Translation. Front. Med. 2018, 5, 354. [Google Scholar] [CrossRef]
- Medina, R.J.; Barber, C.L.; Sabatier, F.; Dignat-George, F.; Melero-Martin, J.M.; Khosrotehrani, K.; Ohneda, O.; Randi, A.M.; Chan, J.K.Y.; Yamaguchi, T.; et al. Endothelial progenitors: A consensus statement on nomenclature. Stem Cells Transl. Med. 2017, 6, 1316–1320. [Google Scholar] [CrossRef]
- Medina, R.J.; O’Neill, C.L.; Sweeney, M.; Guduric-Fuchs, J.; Gardiner, T.A.; Simpson, D.A.; Stitt, A.W. Molecular analysis of endothelial progenitor cell (EPC) subtypes reveals two distinct cell populations with different identities. BMC Med. Genom. 2010, 3, 18. [Google Scholar] [CrossRef]
- Prater, D.N.; Case, J.; Ingram, D.A.; Yoder, M.C. Working hypothesis to redefine endothelial progenitor cells. Leukemia 2007, 21, 1141–1149. [Google Scholar] [CrossRef]
- Yoder, M.C.; Mead, L.E.; Prater, D.; Krier, T.R.; Mroueh, K.N.; Li, F.; Krasich, R.; Temm, C.J.; Prchal, J.T.; Ingram, D.A. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood 2007, 109, 1801–1809. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.D. Endothelial progenitor cells—An evolving story. Microvasc. Res. 2010, 79, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Ingram, D.A.; Mead, L.E.; Tanaka, H.; Meade, V.; Fenoglio, A.; Mortell, K.; Pollok, K.; Ferkowicz, M.J.; Gilley, D.; Yoder, M.C. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood 2004, 104, 2752–2760. [Google Scholar] [CrossRef] [PubMed]
- Margheri, F.; Chillà, A.; Laurenzana, A.; Serratì, S.; Mazzanti, B.; Saccardi, R.; Santosuosso, M.; Danza, G.; Sturli, N.; Rosati, F.; et al. Endothelial progenitor cell-dependent angiogenesis requires localization of the full-length form of uPAR in caveolae. Blood 2011, 118, 3743–3755. [Google Scholar] [CrossRef] [PubMed]
- Bieback, K.; Vinci, M.; Elvers-Hornung, S.; Bartol, A.; Gloe, T.; Czabanka, M.; Klüter, H.; Augustin, H.; Vajkoczy, P. Recruitment of human cord blood-derived endothelial colony-forming cells to sites of tumor angiogenesis. Cytotherapy 2013, 15, 726–739. [Google Scholar] [CrossRef]
- Laurenzana, A.; Biagioni, A.; D’Alessio, S.; Bianchini, F.; Chillà, A.; Margheri, F.; Luciani, C.; Mazzanti, B.; Pimpinelli, N.; Torre, E.; et al. Melanoma cell therapy: Endothelial progenitor cells as shuttle of the MMP12 uPAR-degrading enzyme. Oncotarget 2014, 5, 3711–3727. [Google Scholar] [CrossRef]
- Margheri, G.; Zoppi, A.; Olmi, R.; Trigari, S.; Traversi, R.; Severi, M.; Bani, D.; Bianchini, F.; Torre, E.; Margheri, F.; et al. Tumor-tropic endothelial colony forming cells (ECFCs) loaded with near-infrared sensitive Au nanoparticles: A “cellular stove” approach to the photoablation of melanoma. Oncotarget 2016, 7, 39846–39860. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, N.; Costeas, P.; Hartwell, L.; Hurley, C.K.; Raffoux, C.; Rosenmayr, A.; Greinix, H. Quality Assurance and Clinical Working Groups of the World Marrow Donor Association.Haematopoietic stem cell donor registries: World Marrow Donor Association recommendations for evaluation of donor health. Bone Marrow Transpl. 2008, 42, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef] [PubMed]



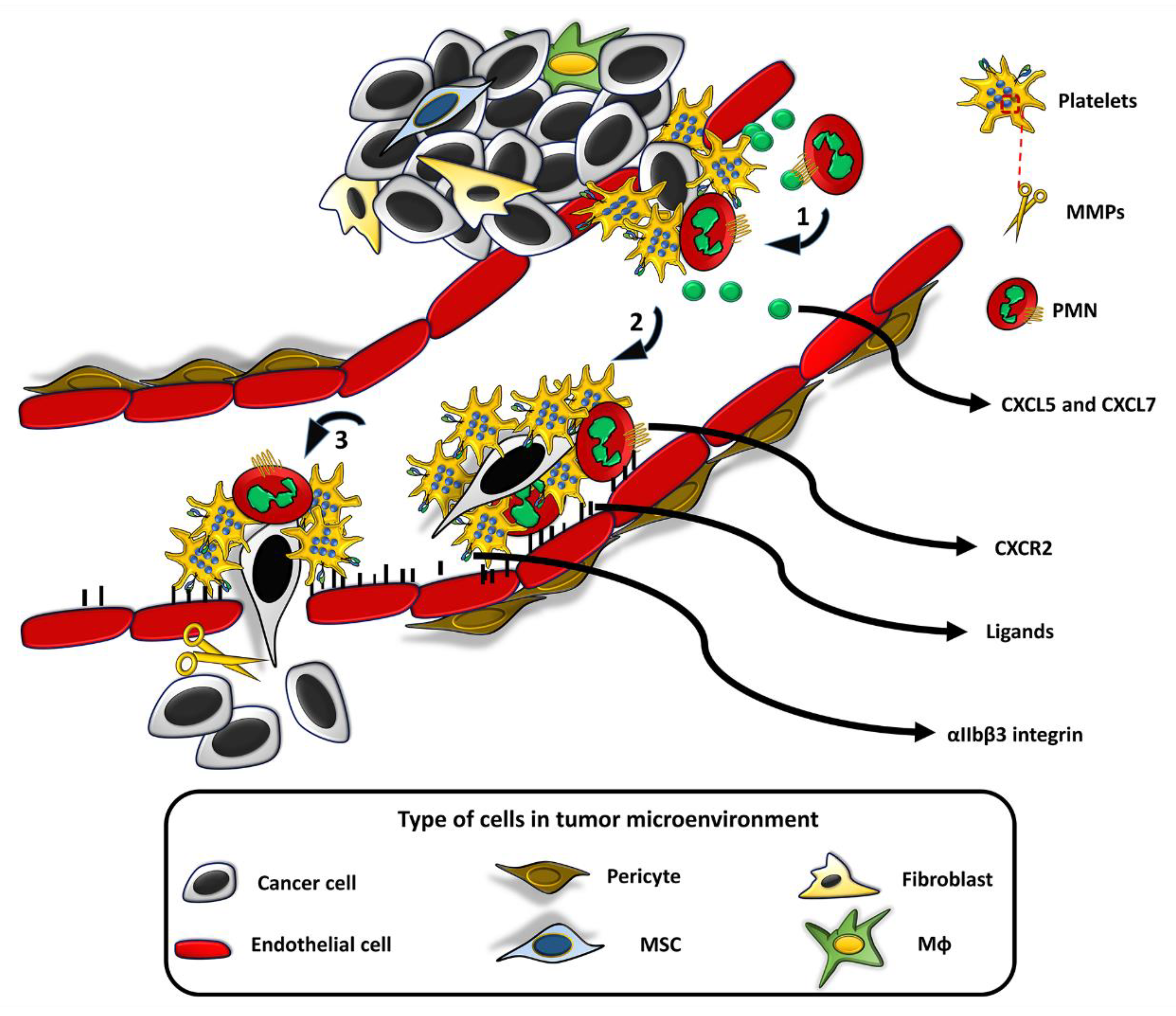{kind=link}
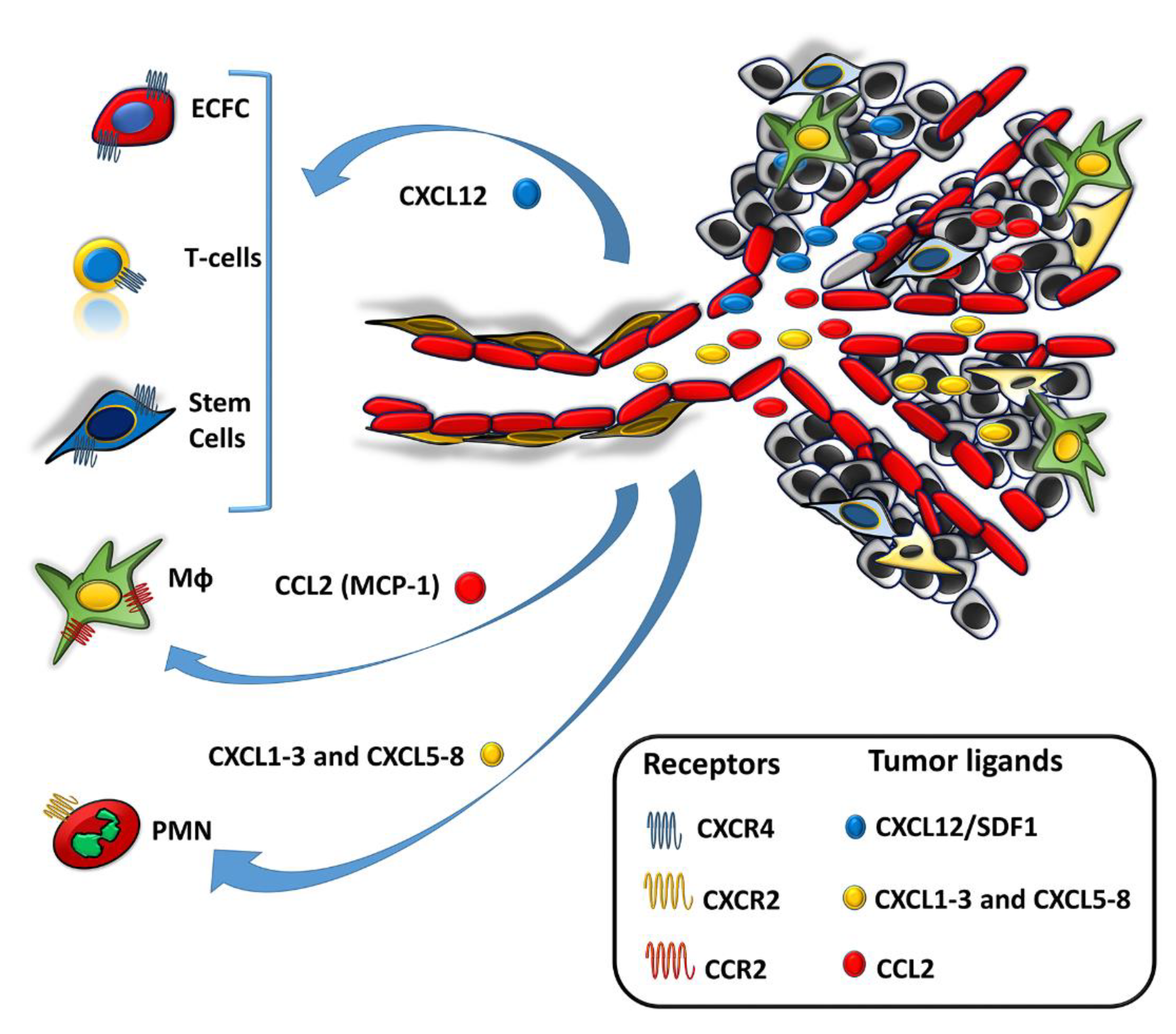{kind=link}
| Cell Type | Delivery System | Therapeutic Mechanism | Target | Tolerability | Life Span | Main Drawabacks | Ref. |
|---|---|---|---|---|---|---|---|
| Platlets | Membrane-coated gold nanostars containing curcumin (ghost-cells) | NIR Controlled release | Melanoma primary tumor; Possible metastasis | YES (source: autologous blood) | 7-–10 days | No proliferation; Issues with purification; | [40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57] |
| Neutrophils | Albumin-NPs loaded with pyropheophorbide-a, and anti-GP75 mAb | Photodynamic therapy | Melanoma primary tumor; Metastasis | Yes (source: autologous blood) | 7 days in vivo Few hours in vitro | Short life | [58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80] |
| Monocytes | SPIONs- or Au nanoshells-loaded; liposome-doxorubicin-loaded | Hyperthermia; Release of encapsulated therapeutic cargos | Pancreatic cancer; breast cancer; experimental lung metastases of melanoma | Yes (source: autologous blood) | 1–3 days for circulating cells; years for tissue-resident Mϕ | Liver, spleen and lungs sequestration; direct toxic effects of NP-chemotherapeutic cargos | [81,82,83,84,85,86,87,88,89] |
| Lymphocytes | Internal or surface-immobilized NP systemic delivery of engineered CAR-T cross-linked to multilamellar liposomal vescicles (cMLV) containing a specific inhibitor (SHC) of immunosuppressor A2aR | Inhibition of the tumor immunosuppressive microenvironment. | Experimental human ovarian cancer; Experimental chronic myelogenous leukemia; Possible metastasis | Yes | 4 days–5 weeks for B cells, months -years for T cells | Acute anaphylaxis; tumor lysis syndrome (TLS); cytokine release syndrome (CRS) | [90,91,92] |
| Red blood cells | Loaded with SPIONs in hypotonic solutions; hijacked with SPIONs or polymeric NP; DOX-loaded hollow copper sulfide NPs coated with RBC and melanoma cell membranes; enveloped polymeric nanoplatform | Photothermal therapy (PTT); Chemotherapy; Anti-tumor immunity | Glioblastoma; melanoma; | Yes (source: autologous blood) | 3months | None | [14], [93,94,95,96,97,98,99] |
| NSCs or iPSCs | Loaded with aminosiloxane-porphyrin functionalized magnetic NPs with core/shell Fe/Fe(3)O | Magnetic hyperthermia | Primary melanoma | Yes (NCS: autologous origin); (iPSC:Low immunogenicity) | Self renewing when cultured and expanded | NSC: difficult to prepare; iPSC, potential induction of teratomas after in vivo transplant. | [100,101,102,103,104,105,106] |
| MSCs | Loaded with SPIONs, silica or Au-NP | Magnetic or plasmonic hyperthermia | Breast cancer; glioblastoma; human fibrosarcoma; | Yes: autologous or allogeneic origin , prepared from the wall of umbilical cord vessels | Self renewing when cultured and expanded | Cell-mediated and humoral immune responses to MHC-mismatched MSC | [107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129] |
| ECFCs | Labelled with 111-In loaded with Au-NP | Phothermal therapy | Primary melanoma | Yes: autologous or allogeneic origin, prepared from umbilical cord blood | Self renewing when cultured and expanded | No major cell-mediated and humoral immune responses to MHC-mismatched MSC | [130,131,132,133,134,135,136,137,138,139,140,141] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chillà, A.; Margheri, F.; Biagioni, A.; Del Rosso, T.; Fibbi, G.; Del Rosso, M.; Laurenzana, A. Cell-Mediated Release of Nanoparticles as a Preferential Option for Future Treatment of Melanoma. Cancers 2020, 12, 1771. https://doi.org/10.3390/cancers12071771
Chillà A, Margheri F, Biagioni A, Del Rosso T, Fibbi G, Del Rosso M, Laurenzana A. Cell-Mediated Release of Nanoparticles as a Preferential Option for Future Treatment of Melanoma. Cancers. 2020; 12(7):1771. https://doi.org/10.3390/cancers12071771
Chicago/Turabian StyleChillà, Anastasia, Francesca Margheri, Alessio Biagioni, Tommaso Del Rosso, Gabriella Fibbi, Mario Del Rosso, and Anna Laurenzana. 2020. "Cell-Mediated Release of Nanoparticles as a Preferential Option for Future Treatment of Melanoma" Cancers 12, no. 7: 1771. https://doi.org/10.3390/cancers12071771
APA StyleChillà, A., Margheri, F., Biagioni, A., Del Rosso, T., Fibbi, G., Del Rosso, M., & Laurenzana, A. (2020). Cell-Mediated Release of Nanoparticles as a Preferential Option for Future Treatment of Melanoma. Cancers, 12(7), 1771. https://doi.org/10.3390/cancers12071771