Zebrafish Xenografts Unveil Sensitivity to Olaparib beyond BRCA Status




Abstract
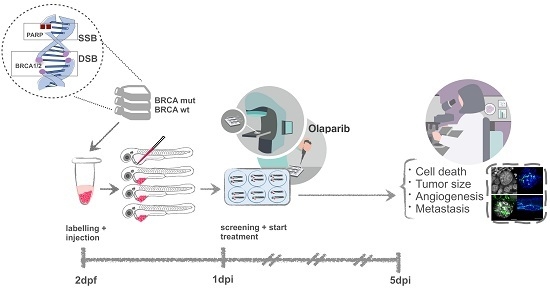
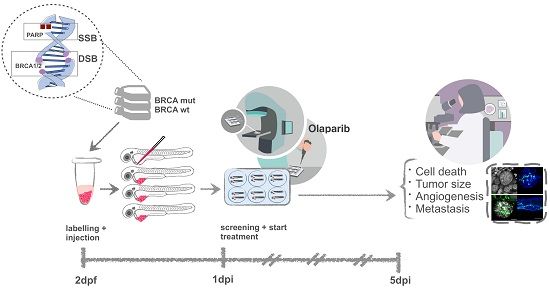{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
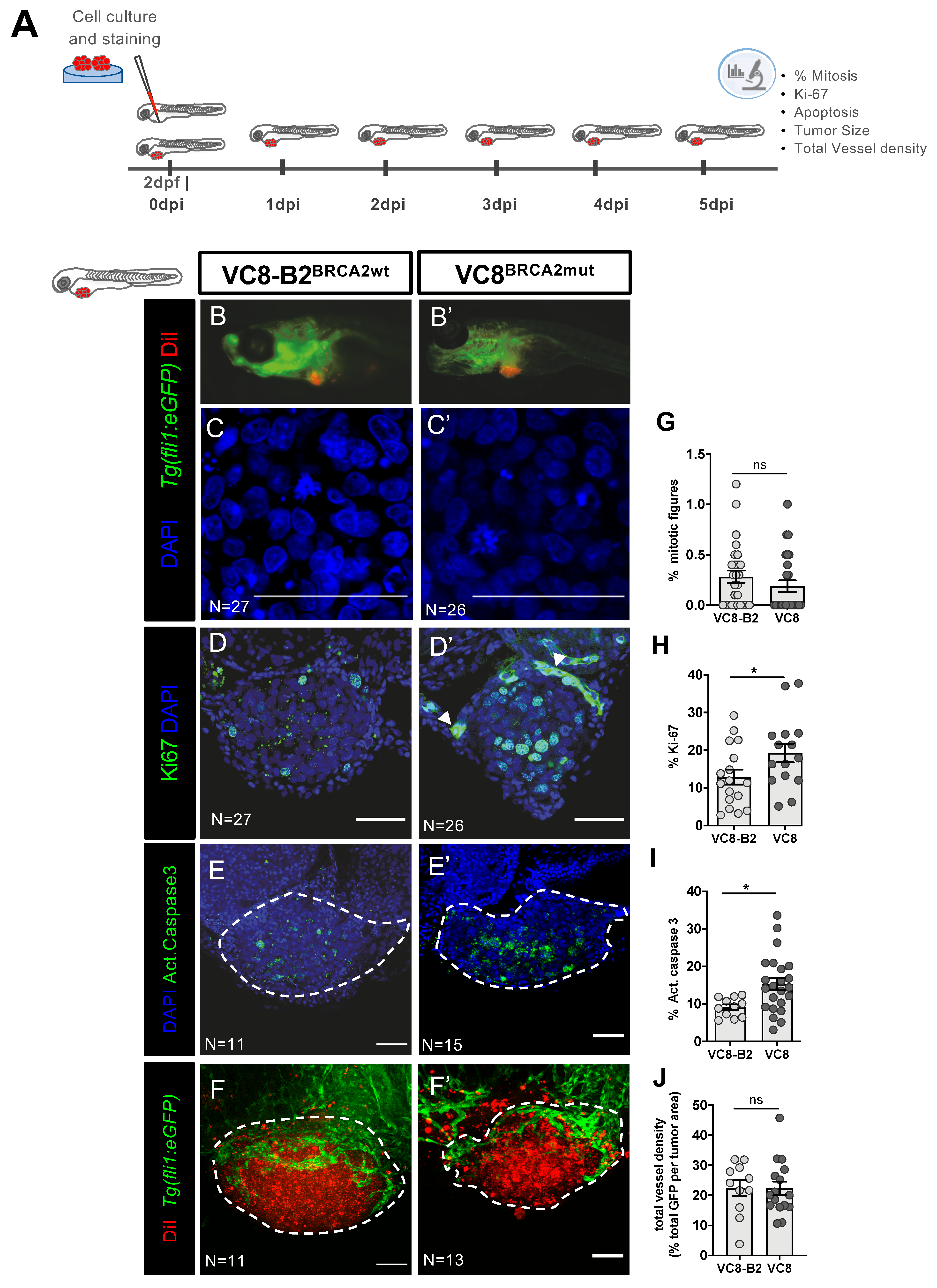
2.1. Phenotypes Associated with Different BRCA2 Status Are Observed in Zebrafish Xenografts
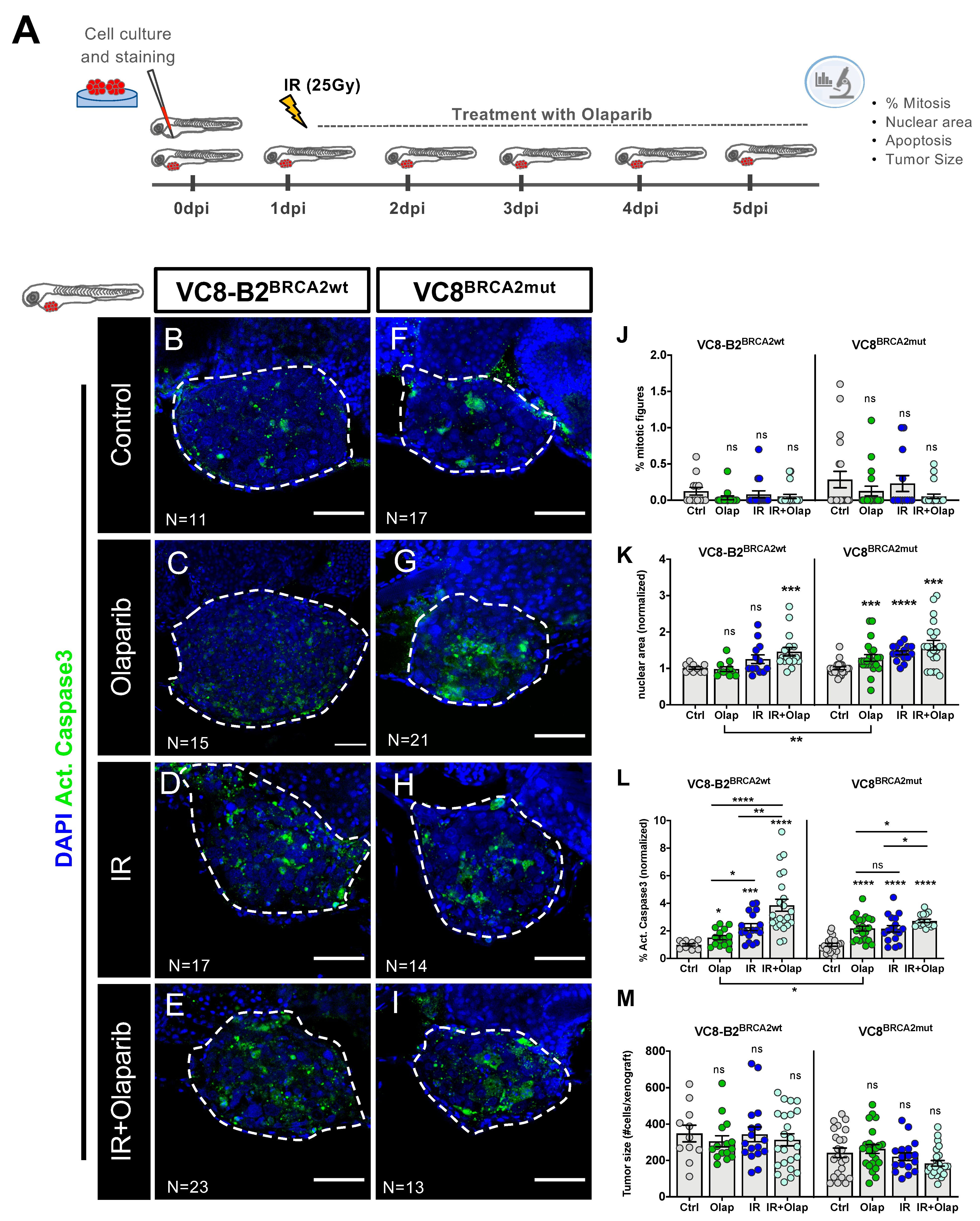
2.2. VC8-B2 and VC8 Zebrafish Xenografts Show Different Sensitivities to Treatment with Olaparib Alone and in Combination with IR
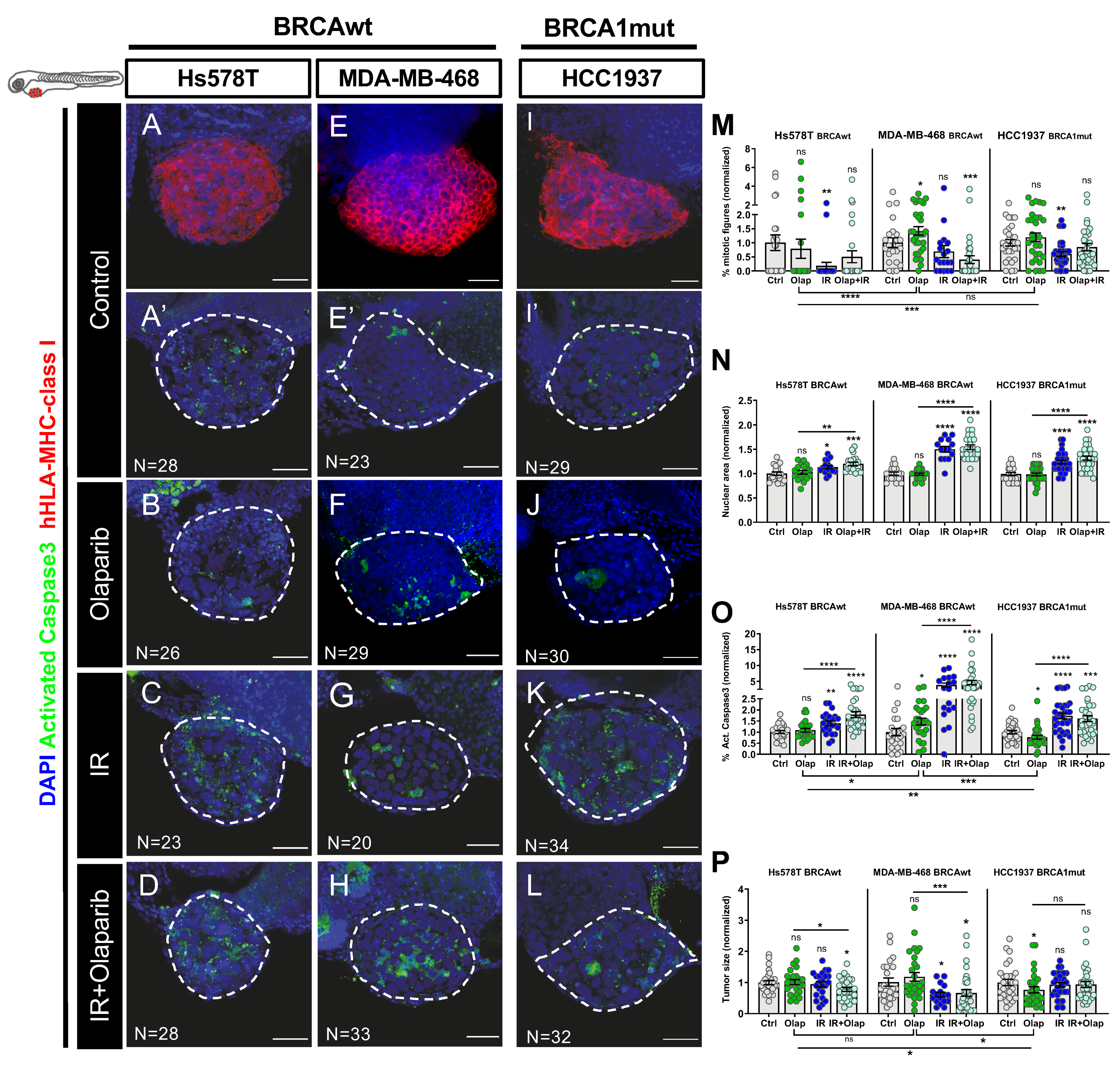
2.3. Human TNBC Zebrafish Xenografts Reveal Different Sensitivities to Olaparib and IR Independently of BRCA Status
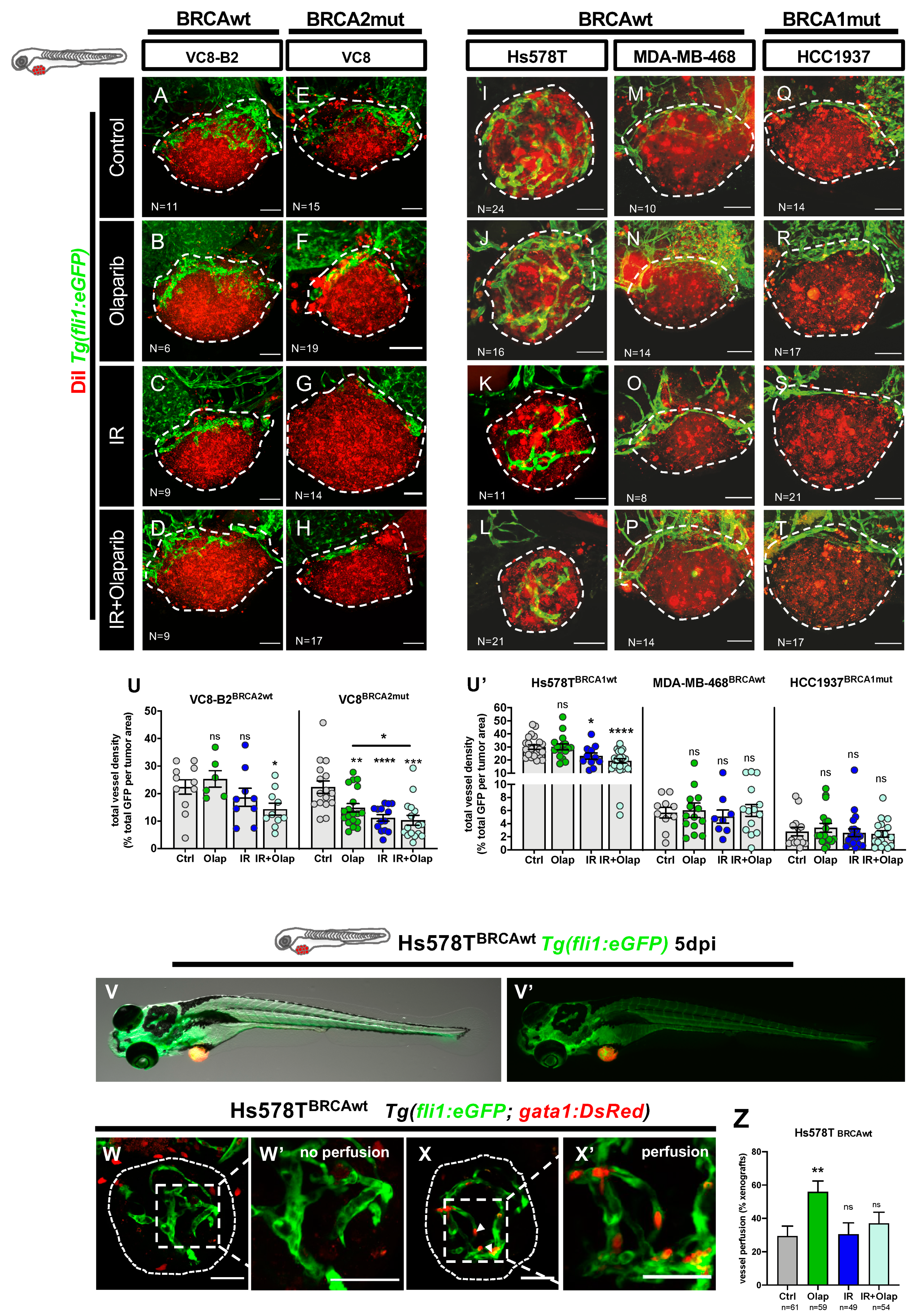
2.4. Olaparib Can Inhibit Angiogenesis or Normalize Tumor Vessels
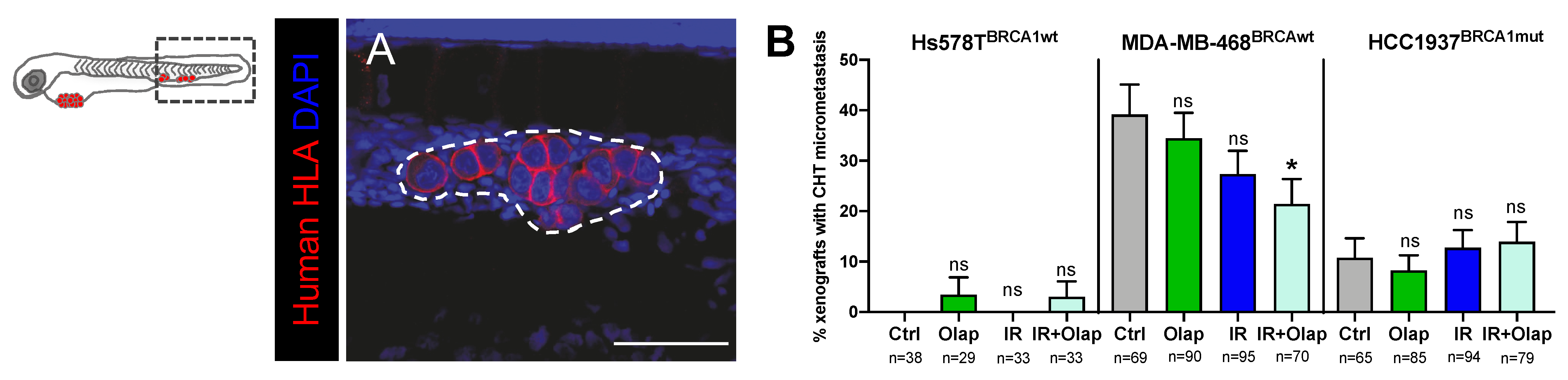
2.5. Combinatorial Treatment of Olaparib and IR Is Able to Reduce Metastatic Spread
3. Discussion
4. Materials and Methods
4.1. Animal Care and Handling
4.2. Zebrafish Lines
4.3. Cell Lines
4.4. Cell Culture
4.5. Cell Labelling
4.6. Zebrafish Xenografts Injection
4.7. Zebrafish Xenografts Irradiation and Drug Administration
4.8. Whole-Mount Immunofluorescence
4.9. Imaging and Quantification
4.10. Metastatic Potential Quantification
4.11. Imaging and Angiogenesis Quantification
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Chen, C.-C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol. 2018, 2, 313–336. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002, 108, 171–182. [Google Scholar] [CrossRef]
- Mersch, J.; Jackson, M.A.; Park, M.; Nebgen, D.; Peterson, S.K.; Singletary, C.; Arun, B.K.; Litton, J.K. Cancers associated with BRCA1 and BRCA 2 mutations other than breast and ovarian. Cancer 2015, 121, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Senkus, E.; Costa, A.; Papadopoulos, E.; Aapro, M.; André, F.; Harbeck, N.; Lopez, B.A.; Barrios, C.H.; Bergh, J.; et al. ESMO Clinical Practice Guidelines Slideset—Advanced Breast Cancer. ESMO 2018. [Google Scholar]
- Morales, J.C.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of Poly (ADP-ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef]
- Min, A.; Im, S.A. PARP inhibitors as therapeutics: Beyond modulation of parylation. Cancers (Basel) 2020, 12, 394. [Google Scholar] [CrossRef] [PubMed]
- Keung, M.Y.; Wu, Y.; Badar, F.; Vadgama, J.V. Response of Breast Cancer Cells to PARP Inhibitors Is Independent of BRCA Status. J. Clin. Med. 2020, 9, 940. [Google Scholar] [CrossRef]
- O’connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef]
- Helleday, T. Clinical Strategies for PARP Inhibitors beyond Homologous Recombination Deficiency. J. Med. Oncol. 2018, 1, 2. [Google Scholar]
- Curtin, N.J.; Drew, Y.; Sharma-Saha, S. Why BRCA mutations are not tumour-agnostic biomarkers for PARP inhibitor therapy. Nat. Rev. Clin. Oncol. 2019, 16, 725–726. [Google Scholar] [CrossRef] [PubMed]
- Gelmon, K.A.; Hirte, H.W.; Robidoux, A.; Tonkin, K.S.; Tischkowitz, M.; Swenerton, K.; Huntsman, D.; Carmichael, J.; Macpherson, E.; Oza, A.M. Can we define tumors that will respond to PARP inhibitors? A phase II correlative study of olaparib in advanced serous ovarian cancer and triple-negative breast cancer. J. Clin. Oncol. 2010, 28, 3002. [Google Scholar] [CrossRef]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for homologous recombination deficiency in cancer. J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef]
- Martin-Broto, J. The tip of the iceberg: Predicting PARP inhibitor efficacy in prostate cancer. Lancet Oncol. 2020, 21, 17–19. [Google Scholar]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef]
- Peng, Y.; Liao, Q.; Tan, W.; Peng, C.; Hu, Z.; Chen, Y.; Li, Z.; Li, J.; Zhen, B.; Zhu, W.; et al. The deubiquitylating enzyme USP15 regulates homologous recombination repair and cancer cell response to PARP inhibitors. Nat. Commun. 2019, 10, 1224. [Google Scholar] [CrossRef]
- Ali, R.; Al-Kawaz, A.; Toss, M.S.; Green, A.R.; Miligy, I.M.; Mesquita, K.A.; Seedhouse, C.; Mirza, S.; Band, V.; Rakha, E.A.; et al. Targeting PARP1 in XRCC1-Deficient Sporadic Invasive Breast Cancer or Preinvasive Ductal Carcinoma In Situ Induces Synthetic Lethality and Chemoprevention. Cancer Res. 2018, 78, 6818–6827. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Spentzos, D.; Karlan, B.Y.; Taniguchi, T.; Fountzilas, E.; Francoeur, N.; Levine, D.A.; Cannistra, S.A. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J. Clin. Oncol. 2010, 28, 3555–3561. [Google Scholar] [CrossRef]
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; Van Der Gulden, H.; Van Der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-Δ11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res. 2016, 76, 2778–2790. [Google Scholar] [CrossRef] [PubMed]
- Drost, R.; Dhillon, K.K.; Van Der Gulden, H.; Van Der Heijden, I.; Brandsma, I.; Cruz, C.; Chondronasiou, D.; Castroviejo-Bermejo, M.; Boon, U.; Schut, E.; et al. BRCA1185delAG tumors may acquire therapy resistance through expression of RING-less BRCA1. J. Clin. Investig. 2016, 126, 2903–2918. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; Van De Ven, M.; Van Der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; Van Der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; Van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 causes PARP inhibitor resistance in BRCA1-mutated mouse mammary tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef]
- Xu, G.; Ross Chapman, J.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef]
- Castroviejo-Bermejo, M.; Cruz, C.; Llop-Guevara, A.; Gutiérrez-Enríquez, S.; Ducy, M.; Ibrahim, Y.H.; Gris-Oliver, A.; Pellegrino, B.; Bruna, A.; Guzmán, M.; et al. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol. Med. 2018, 10, e9172. [Google Scholar] [CrossRef]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 15, 1203–1210. [Google Scholar] [CrossRef]
- Costa, B.; Ferreira, S.; Póvoa, V.; Cardoso, M.J.; Vieira, S.; Stroom, J.; Fidalgo, P.; Rio-Tinto, R.; Figueiredo, N.; Parés, O.; et al. Developments in zebrafish avatars as radiotherapy sensitivity reporters—Towards personalized medicine. EBioMedicine 2020, 51, 102578. [Google Scholar] [CrossRef]
- Fior, R.; Póvoa, V.; Mendes, R.V.; Carvalho, T.; Gomes, A.; Figueiredo, N.; Ferreira, F.R. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proc. Natl. Acad. Sci. USA 2017, 114, E8234–E8243. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Estrada, M.F.; Mendes, R.V.; Fior, R. Zebrafish Avatars towards Personalized Medicine—A Comparative Review between Avatar Models. Cells 2020, 9, 293. [Google Scholar] [CrossRef] [PubMed]
- Fazio, M.; Ablain, J.; Chuan, Y.; Langenau, D.M.; Zon, L.I. Zebrafish patient avatars in cancer biology and precision cancer therapy. Nat. Rev. Cancer 2020, 20, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Martí, J.M.; Fernández-Cortés, M.; Serrano-Sáenz, S.; Zamudio-Martinez, E.; Delgado-Bellido, D.; Garcia-Diaz, A.; Oliver, F.J. The multifactorial role of PARP-1 in tumor microenvironment. Cancers (Basel) 2020, 12, 739. [Google Scholar] [CrossRef] [PubMed]
- Weaver, A.N.; Yang, E.S.; Annunziata, C.; Zhu, X.-D.; Yang, W.-M. Beyond DNA repair: Additional functions of PARP-1 in cancer. Front. Oncol. 2013, 3, 290. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Soria, J.C.; Lord, C.J.; Postel-Vinay, S. Beyond DNA repair: The novel immunological potential of PARP inhibitors. Mol. Cell. Oncol. 2019, 6, 1–4. [Google Scholar] [CrossRef]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M.G. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef]
- Kraakman-van der Zwet, M.; Wiegant, W.W.; Zdzienicka, M.Z. Brca2 (XRCC11) deficiency results in enhanced mutagenesis. Mutagenesis 2003, 18, 521–525. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, H.; Lord, C.J.; Tutt, A.H.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Sun, X.; Kaufman, P.D. Ki-67: More than a proliferation marker. Chromosoma 2018, 127, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.A.; Federici, M.G.; Reuter, V.E.; Boyd, J. Cell proliferation and apoptosis in BRCA-associated hereditary ovarian cancer. Gynecol. Oncol. 2002, 85, 431–434. [Google Scholar] [CrossRef] [PubMed]
- van der Groep, P.; van Diest, P.J.; Smolders, Y.H.C.M.; Ausems, M.G.E.M.; van der Luijt, R.B.; Menko, F.H.; Bart, J.; de Vries, E.G.E.; van der Wall, E. HIF-1α Overexpression in Ductal Carcinoma In Situ of the Breast in BRCA1 and BRCA2 Mutation Carriers. PLoS ONE 2013, 8, e56055. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, C.; Malfettone, A.; Ranieri, G.; Danza, K.; Simone, G.; Paradiso, A.; Mangia, A. VEGF, HIF-1α Expression and MVD as an Angiogenic Network in Familial Breast Cancer. PLoS ONE 2013, 8, e53070. [Google Scholar] [CrossRef]
- Lawson, N.D.; Weinstein, B.M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 2002, 248, 307–318. [Google Scholar] [CrossRef]
- Basourakos, S.P.; Li, L.; Aparicio, A.M.; Corn, P.G.; Kim, J.; Thompson, T.C. Combination Platinum-based and DNA Damage Response-targeting Cancer Therapy: Evolution and Future Directions. Curr. Med. Chem. 2016, 24, 1586–1606. [Google Scholar] [CrossRef] [PubMed]
- Kedar, P.S.; Stefanick, D.F.; Horton, J.K.; Wilson, S.H. Increased PARP-1 association with DNA in alkylation damaged, PARP-inhibited mouse fibroblasts. Mol. Cancer Res. 2012, 10, 360–368. [Google Scholar] [CrossRef]
- Michels, J.; Vitale, I.; Senovilla, L.; Enot, D.P.; Garcia, P.; Lissa, D.; Olaussen, K.A.; Brenner, C.; Soria, J.C.; Castedo, M.; et al. Synergistic interaction between cisplatin and PARP inhibitors in non-small cell lung cancer. Cell Cycle 2013, 12, 877–883. [Google Scholar] [CrossRef]
- Mao, Y.; Huang, X.; Shuang, Z.; Lin, G.; Wang, J.; Duan, F.; Chen, J.; Li, S. PARP inhibitor olaparib sensitizes cholangiocarcinoma cells to radiation. Cancer Med. 2018, 7, 1285–1296. [Google Scholar] [CrossRef]
- Shelton, J.W.; Waxweiler, T.V.; Landry, J.; Gao, H.; Xu, Y.; Wang, L.; El-Rayes, B.; Shu, H.K.G. In vitro and in vivo enhancement of chemoradiation using the oral parp inhibitor ABT-888 in colorectal cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 469–476. [Google Scholar] [CrossRef]
- Donawho, C.K.; Luo, Y.; Luo, Y.; Penning, T.D.; Bauch, J.L.; Bouska, J.J.; Bontcheva-Diaz, V.D.; Cox, B.F.; DeWeese, T.L.; Dillehay, L.E.; et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin. Cancer Res. 2007, 13, 2728–2737. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Mason, K.A.; Ang, K.K.; Buchholz, T.; Valdecanas, D.; Mathur, A.; Buser-Doepner, C.; Toniatti, C.; Milas, L. MK-4827, a PARP-1/-2 inhibitor, strongly enhances response of human lung and breast cancer xenografts to radiation. Investig. New Drugs 2012, 30, 2113–2120. [Google Scholar] [CrossRef] [PubMed]
- Senra, J.M.; Telfer, B.A.; Cherry, K.E.; Mccrudden, C.M.; Hirst, D.G.; Connor, M.J.O.; Wedge, S.R.; Stratford, I.J. Inhibition of poly (ADP-ribose) polymerase-1 by olaparib (AZD2281) increases the radiosensitivity of a lung tumor xenograft. Mol. Cancer Ther. 2012, 10, 1949–1958. [Google Scholar] [CrossRef]
- European Medicines Agency. Lynparza: EPAR—Product Information. ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS. 2014. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/lynparza#product-information-section (accessed on 3 May 2020).
- Chan, T.A.; Hermeking, H.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B. 14-3-3σ is required to prevent mitotic catastrophe after DNA damage. Nature 1999, 401, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T.; Soutoglou, E. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat. Rev. Mol. Cell Biol. 2009, 10, 243–254. [Google Scholar] [CrossRef]
- Somosy, Z. Radiation response of cell organelles. Micron 2000, 31, 165–181. [Google Scholar] [CrossRef]
- Zhang, D.; Jin, Q.; Jiang, C.; Gao, M.; Ni, Y.; Zhang, J. Imaging Cell Death: Focus on Early Evaluation of Tumor Response to Therapy. Bioconjug. Chem. 2020, 31, 1025–1051. [Google Scholar] [CrossRef]
- Montero, J.; Sarosiek, K.A.; Deangelo, J.D.; Maertens, O.; Ryan, J.; Ercan, D.; Piao, H.; Horowitz, N.S.; Berkowitz, R.S.; Matulonis, U.; et al. Drug-Induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell 2015, 160, 977–989. [Google Scholar] [CrossRef]
- Oliveira, B.L.; Stenton, B.J.; VB, U.; de Almeida, C.R.; Conde, J.; Negrão, M.; Schneider, F.S.; Cordeiro, C.; Godinho Ferreira, M.; Caramori, G.F.; et al. Platinum-triggered Bond-cleavage of Pentynoyl amide and N-propargyl handles for Drug-Activation. J. Am. Chem. Soc. 2020, 142, 10869–10880. [Google Scholar] [CrossRef]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of “BRCAness” in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef]
- Slade, D. Mitotic functions of poly(ADP-ribose) polymerases. Biochem. Pharmacol. 2019, 167, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Schoonen, P.M.; Talens, F.; Stok, C.; Gogola, E.; Heijink, A.M.; Bouwman, P.; Foijer, F.; Tarsounas, M.; Blatter, S.; Jonkers, J.; et al. Progression through mitosis promotes PARP inhibitor-induced cytotoxicity in homologous recombination-deficient cancer cells. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kukolj, E.; Kaufmann, T.; Dick, A.E.; Zeillinger, R.; Gerlich, D.W.; Slade, D. PARP inhibition causes premature loss of cohesion in cancer cells. Oncotarget 2017, 8, 103931–103951. [Google Scholar] [CrossRef] [PubMed]
- Lafontaine, J.; Boisvert, J.S.; Glory, A.; Coulombe, S.; Wong, P. Synergy between non-thermal plasma with radiation therapy and olaparib in a panel of breast cancer cell lines. Cancers (Basel) 2020, 12, 348. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Martin-Oliva, D.; Aguilar-Quesada, R.; O’Valle, F.; Muñoz-Gámez, J.A.; Martínez-Romero, R.; García Del Moral, R.; Ruiz De Almodóvar, J.M.; Villuendas, R.; Piris, M.A.; Oliver, F.J. Inhibition of poly(ADP-ribose) polymerase modulates tumor-related gene expression, including hypoxia-inducible factor-1 activation, during skin carcinogenesis. Cancer Res. 2006, 66, 5744–5756. [Google Scholar] [CrossRef]
- Gonzalez-Flores, A.; Aguilar-Quesada, R.; Siles, E.; Pozo, S.; Rodríguez-Lara, M.I.; López-Jiménez, L.; López-Rodríguez, M.; Peralta-Leal, A.; Villar, D.; Martín-Oliva, D.; et al. Interaction between PARP-1 and HIF-2 in the hypoxic response. Oncogene 2014, 33, 891–898. [Google Scholar] [CrossRef]
- Zhou, X.; Patel, D.; Sen, S.; Shanmugam, V.; Sidawy, A.; Mishra, L.; Nguyen, B.N. Poly-ADP-ribose polymerase inhibition enhances ischemic and diabetic wound healing by promoting angiogenesis. J. Vasc. Surg. 2017, 65, 1161–1169. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Cao, Y.; Moeller, B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer 2008, 8, 425–437. [Google Scholar] [CrossRef]
- Paris, F.; Fuks, Z.; Kang, A.; Capodieci, P.; Juan, G.; Ehleiter, D.; Haimovitz-Friedman, A.; Cordon-Cardo, C.; Kolesnick, R. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science (80-) 2001, 293, 293–297. [Google Scholar] [CrossRef]
- Grabham, P.; Sharma, P. The effects of radiation on angiogenesis. Vasc. Cell 2013, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar] [CrossRef] [PubMed]
- Traver, D.; Paw, B.H.; Poss, K.D.; Penberthy, W.T.; Lin, S.; Zon, L.I. Transplantation and in vivo imaging of multilineage engraftment in zebrafish bloodless mutants. Nat. Immunol. 2003, 4, 1238–1246. [Google Scholar] [CrossRef]
- Schacke, M.; Kumar, J.; Colwell, N.; Hermanson, K.; Folle, G.A.; Nechaev, S.; Dhasarathy, A.; Lafon-Hughes, L. PARP-1/2 inhibitor olaparib prevents or partially reverts EMT induced by TGF-β in NMuMG cells. Int. J. Mol. Sci. 2019, 20, 518. [Google Scholar] [CrossRef]
- Pu, H.; Horbinski, C.; Hensley, P.J.; Matuszak, E.A.; Atkinson, T.; Kyprianou, N. PARP-1 regulates epithelial-mesenchymal transition (EMT) in prostate tumorigenesis. Carcinogenesis 2014, 35, 2592–2601. [Google Scholar] [CrossRef] [PubMed]
- Sundahl, N.; Duprez, F.; Ost, P.; De Neve, W.; Mareel, M. Effects of radiation on the metastatic process. Mol. Med. 2018, 24, 16. [Google Scholar] [CrossRef]
- Shao, F.; Liu, J.; Duan, Y.; Li, L.; Liu, L.; Zhang, C.; He, S. Efficacy and safety of PARP inhibitors as the maintenance therapy in ovarian cancer: A meta-analysis of nine randomized controlled trials. Biosci. Rep. 2020, 40, BSR20192226. [Google Scholar] [CrossRef]
- Lim, D.; Ngeow, J. Evaluation of the methods to identify patients who may benefit from PARP inhibitor use. Endocr. Relat. Cancer 2016, 23, R267–R285. [Google Scholar] [CrossRef][Green Version]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- McMahon, M.; Frangova, T.G.; Henderson, C.J.; Wolf, C.R. Olaparib, monotherapy or with ionizing radiation, exacerbates DNA damage in normal tissues: Insights from a new p21 reporter mouse. Mol. Cancer Res. 2016, 14, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Verginadis, I.I.; Dey, S.; Lin, L.; Guo, L.; Zheng, Y.; Koumenis, C. Radiosensitization by the PARP inhibitor olaparib in BRCA1-proficient and deficient high-grade serous ovarian carcinomas. Gynecol. Oncol. 2018, 150, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Verbiest, T.; Devery, A.M.; Bokobza, S.M.; Weber, A.M.; Leszczynska, K.B.; Hammond, E.M.; Ryan, A.J. Hypoxia potentiates the radiation-sensitizing effect of olaparib in human non-small cell lung cancer xenografts by contextual synthetic lethality. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Lohse, I.; Kumareswaran, R.; Cao, P.; Pitcher, B.; Gallinger, S.; Bristow, R.G.; Hedley, D.W. Effects of combined treatment with ionizing radiation and the PARP inhibitor olaparib in BRCA mutant and wild type patient-derived pancreatic cancer xenografts. PLoS ONE 2016, 11, e0167272. [Google Scholar] [CrossRef] [PubMed]
- Henneman, L.; Van Miltenburg, M.H.; Michalak, E.M.; Braumuller, T.M.; Jaspers, J.E.; Drenth, A.P.; De Korte-Grimmerink, R.; Gogola, E.; Szuhai, K.; Schlicker, A.; et al. Selective resistance to the PARP inhibitor olaparib in a mouse model for BRCA1-deficient metaplastic breast cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 8409–8414. [Google Scholar] [CrossRef]
- To, C.; Kim, E.H.; Royce, D.B.; Williams, C.R.; Collins, R.M.; Risingsong, R.; Sporn, M.B.; Liby, K.T. The PARP inhibitors, veliparib and olaparib, are effective chemopreventive agents for delaying mammary tumor development in BRCA1-deficient mice. Cancer Prev. Res. 2014, 7, 698–707. [Google Scholar] [CrossRef]
- Cornet, C.; Dyballa, S.; Terriente, J.; Di Giacomo, V. ZeOncoTest: Refining and automating the zebrafish xenograft model for drug discovery in cancer. Pharmaceuticals 2020, 13, 1. [Google Scholar] [CrossRef]
- Xiao, J.; Glasgow, E.; Agarwal, S. Zebrafish Xenografts for Drug Discovery and Personalized Medicine. Trends Cancer 2020. [Google Scholar] [CrossRef]
- Cully, M. Zebrafish earn their drug discovery stripes. Nat. Rev. Drug Discov. 2019, 18, 811–813. [Google Scholar] [CrossRef]
- Yan, C.; Brunson, D.C.; Tang, Q.; Haber, D.A.; Rawls, J.F.; Langenau, D.M. Visualizing Engrafted Human Cancer and Therapy Responses in Immunodeficient Zebrafish. Cell 2019, 1–12. [Google Scholar] [CrossRef]
- Hou, S.Q.; Ouyang, M.; Brandmaier, A.; Hao, H.; Shen, W.H. PTEN in the maintenance of genome integrity: From DNA replication to chromosome segregation. BioEssays 2017, 39, 1700082. [Google Scholar] [CrossRef] [PubMed]
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.S.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.S.; Kohler, R.H.; Landon, M.; Giedt, R.; Weissleder, R. Single cell resolution in vivo imaging of DNA damage following PARP inhibition. Sci. Rep. 2015, 5, 10129. [Google Scholar] [CrossRef] [PubMed]
- Osoegawa, A.; Gills, J.J.; Kawabata, S.; Dennis, P.A. Rapamycin sensitizes cancer cells to growth inhibition by the PARP inhibitor olaparib. Oncotarget 2017, 8, 87044–87053. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; Van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef]
- Barazas, M.; Gasparini, A.; Huang, Y.; Küçükosmanoglu, A.; Annunziato, S.; Bouwman, P.; Sol, W.; Kersbergen, A.; Proost, N.; De Korte-Grimmerink, R.; et al. Radiosensitivity is an acquired vulnerability of PARPI-resistant BRCA1-deficient tumors. Cancer Res. 2019, 79, 452–460. [Google Scholar] [CrossRef]
- De Haan, R.; Van Werkhoven, E.; Van Den Heuvel, M.M.; Peulen, H.M.U.; Sonke, G.S.; Elkhuizen, P.; Van Den Brekel, M.W.M.; Tesselaar, M.E.T.; Vens, C.; Schellens, J.H.M.; et al. Study protocols of three parallel phase 1 trials combining radical radiotherapy with the PARP inhibitor olaparib. BMC Cancer 2019, 19, 901. [Google Scholar] [CrossRef]
- Michmerhuizen, A.R.; Pesch, A.M.; Moubadder, L.; Chandler, B.C.; Wilder-Romans, K.; Cameron, M.; Olsen, E.; Thomas, D.G.; Zhang, A.; Hirsh, N.; et al. PARP1 inhibition radiosensitizes models of inflammatory breast cancer to ionizing radiation. Mol. Cancer Ther. 2019, 18, 2063–2073. [Google Scholar] [CrossRef]
- Fulton, B.; Short, S.C.; James, A.; Nowicki, S.; McBain, C.; Jefferies, S.; Kelly, C.; Stobo, J.; Morris, A.; Williamson, A.; et al. PARADIGM-2: Two parallel phase I studies of olaparib and radiotherapy or olaparib and radiotherapy plus temozolomide in patients with newly diagnosed glioblastoma, with treatment stratified by MGMT status. Clin. Transl. Radiat. Oncol. 2018, 8, 12–16. [Google Scholar] [CrossRef]
- Lesueur, P.; Lequesne, J.; Grellard, J.M.; Dugué, A.; Coquan, E.; Brachet, P.E.; Geffrelot, J.; Kao, W.; Emery, E.; Berro, D.H.; et al. Phase I/IIa study of concomitant radiotherapy with olaparib and temozolomide in unresectable or partially resectable glioblastoma: OLA-TMZ-RTE-01 trial protocol. BMC Cancer 2019, 19, 1–11. [Google Scholar] [CrossRef]
- Jannetti, S.A.; Zeglis, B.M.; Zalutsky, M.R.; Reiner, T. Poly(ADP-Ribose)Polymerase (PARP) Inhibitors and Radiation Therapy. Front. Pharmacol. 2020, 11, 170. [Google Scholar] [CrossRef] [PubMed]
- White, R.M.; Sessa, A.; Burke, C.; Bowman, T.; LeBlanc, J.; Ceol, C.; Bourque, C.; Dovey, M.; Goessling, W.; Burns, C.E.; et al. Transparent Adult Zebrafish as a Tool for In Vivo Transplantation Analysis. Cell Stem Cell 2008, 2, 183–189. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varanda, A.B.; Martins-Logrado, A.; Godinho Ferreira, M.; Fior, R. Zebrafish Xenografts Unveil Sensitivity to Olaparib beyond BRCA Status. Cancers 2020, 12, 1769. https://doi.org/10.3390/cancers12071769
Varanda AB, Martins-Logrado A, Godinho Ferreira M, Fior R. Zebrafish Xenografts Unveil Sensitivity to Olaparib beyond BRCA Status. Cancers. 2020; 12(7):1769. https://doi.org/10.3390/cancers12071769
Chicago/Turabian StyleVaranda, Ana Beatriz, Ana Martins-Logrado, Miguel Godinho Ferreira, and Rita Fior. 2020. "Zebrafish Xenografts Unveil Sensitivity to Olaparib beyond BRCA Status" Cancers 12, no. 7: 1769. https://doi.org/10.3390/cancers12071769
APA StyleVaranda, A. B., Martins-Logrado, A., Godinho Ferreira, M., & Fior, R. (2020). Zebrafish Xenografts Unveil Sensitivity to Olaparib beyond BRCA Status. Cancers, 12(7), 1769. https://doi.org/10.3390/cancers12071769