Essential Thrombocythemia and Acquired von Willebrand Syndrome: The Shadowlands between Thrombosis and Bleeding


Abstract
1. Introduction
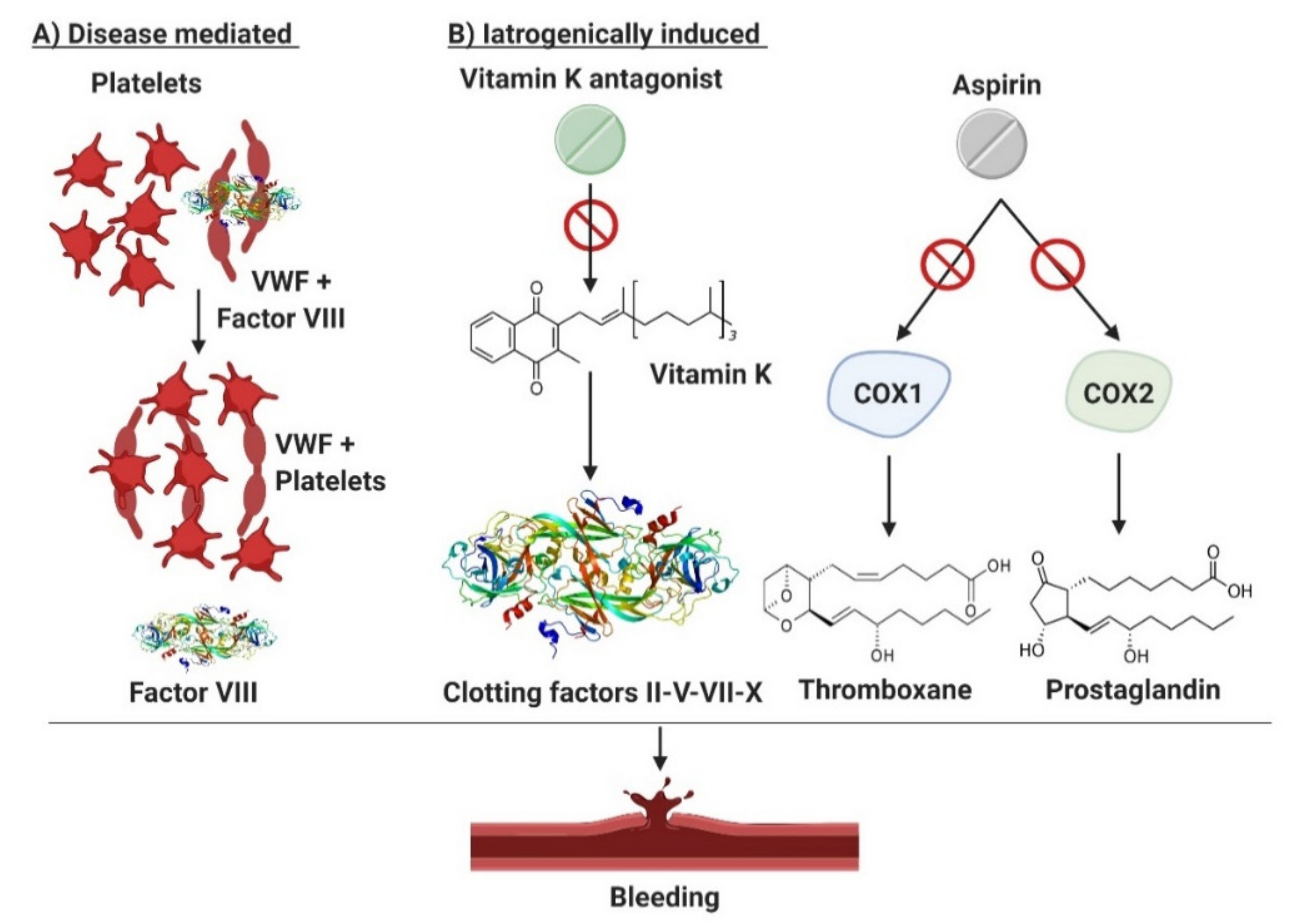
2. Clinical and Therapeutic Pitfalls
3. Risk Factors and Stratification Models
3.1. The Evolution of Prognostication Systems
3.2. Mutational Profile
3.3. Leukocytosis
3.4. Inherited Thrombophilia
4. Cytoreductive Therapies
4.1. Hydroxyurea
4.2. Interferons
4.3. Ruxolitinib
4.4. Therapeutic-Choice Considerations
5. Antiplatelet Therapy
6. Vitamin K Antagonists (VKA)
7. Therapeutic Interventions in Non-High-Risk Patients: A Matter of Controversial Debate
8. Extreme Thrombocytosis and Acquired von Willebrand Syndrome: The Paradox of Hemorrhagic Thrombocythemia
9. Incidence Rates and Risk Factors of Bleeding in Essential Thrombocythemia with Extreme Thrombocytosis
10. Options for Therapeutic Management of Acquired von Willebrand Syndrome in Essential Thrombocythemia
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Campo, E.; Swerdlow, S.H.; Harris, N.L.; Pileri, S.; Stein, H.; Jaffe, E.S. The 2008 WHO classification of lymphoid neoplasms and beyond: Evolving concepts and practical applications. Blood 2011, 117, 5019–5032. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Mehta, J.; Wang, H.; Iqbal, S.U.; Mesa, R. Epidemiology of myeloproliferative neoplasms in the United States. Leuk. Lymphoma 2014, 55, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Boyd, E.M.; Bench, A.J.; Goday-Fernandez, A.; Anand, S.; Vaghela, K.J.; Beer, P.; Scott, M.A.; Bareford, D.; Green, A.R.; Huntly, B.; et al. Clinical utility of routine MPL exon 10 analysis in the diagnosis of essential thrombocythaemia and primary myelofibrosis. Br. J. Haematol. 2010, 149, 250–257. [Google Scholar] [CrossRef]
- Tefferi, A. Myeloproliferative neoplasms: A decade of discoveries and treatment advances. Am. J. Hematol. 2016, 91, 50–58. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Rambaldi, A.; Barosi, G.; Marchioli, R.; Marfisi, R.M.; Finazzi, G.; Guerini, V.; Fabris, F.; et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007, 110, 840–846. [Google Scholar] [CrossRef]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef]
- Nangalia, J.; Green, T.R. The evolving genomic landscape of myeloproliferative neoplasms. Hematol. Am Soc. Hematol. Educ. Program 2014, 2014, 287–296. [Google Scholar] [CrossRef]
- Cervantes, F.; Alvarez-Larran, A.; Talarn, C.; Gomez, M.; Montserrat, E. Myelofibrosis with myeloid metaplasia following essential thrombocythaemia: Actuarial probability, presenting characteristics and evolution in a series of 195 patients. Br. J. Haematol. 2002, 118, 786–790. [Google Scholar] [CrossRef]
- Tefferi, A.; Pardanani, A. Essential Thrombocythemia. N. Engl. J. Med. 2019, 381, 2135–2144. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Tischer, A.; Wassie, E.A.; Finke, C.M.; Belachew, A.A.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A.D. The prognostic advantage of calreticulin mutations in myelofibrosis might be confined to type 1 or type 1-like CALR variants. Blood 2014, 124, 2465–2466. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; Ketterling, R.; Hanson, C.H.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: Clinical, cytogenetic and molecular comparisons. Leukemia 2014, 28, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Shammo, J.M.; Stein, B.L. Mutations in MPNs: Prognostic implications, window to biology, and impact on treatment decisions. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014, 124, 2507–2513. [Google Scholar] [CrossRef]
- Milosevic Feenstra, J.D.; Nivarthi, H.; Gisslinger, H.; Leroy, E.; Rumi, E.; Chachoua, I.; Bagienski, K.; Kubesova, B.; Pietra, D.; Gisslinger, B.; et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 2016, 127, 325–332. [Google Scholar] [CrossRef]
- Cabagnols, X.; Favale, F.; Pasquier, F.; Messaoudi, K.; Defour, J.P.; Ianotto, J.C.; Marzac, C.; Le Couédic, J.P.; Droin, N.; Chachoua, I.; et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood 2016, 127, 333–342. [Google Scholar] [CrossRef]
- Abdulkarim, K.; Girodon, F.; Johansson, P.; Maynadie, M.; Kutti, J.; Carli, P.M.; Bovet, E.; Andreasson, B. AML transformation in 56 patients with Ph- MPD in two well defined populations. Eur. J. Haematol. 2009, 82, 106–111. [Google Scholar] [CrossRef]
- Spivak, J.L. Myeloproliferative Neoplasms. N. Engl. J. Med. 2017, 376, 2168–2181. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef]
- Radaelli, F.; Colombi, M.; Calori, R.; Zilioli, V.R.; Bramanti, S.; Iurlo, A.; Zanella, A. Analysis of risk factors predicting thrombotic and/or haemorrhagic complications in 306 patients with essential thrombocythemia. Hematol. Oncol. 2007, 25, 115–120. [Google Scholar] [CrossRef]
- Cervantes, F. Management of essential thrombocythemia. Hematol. Am Soc. Hematol. Educ. Program 2011, 2011, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Cortelazzo, S.; Viero, P.; Finazzi, G.; D’Emilio, A.; Rodeghiero, F.; Barbui, T. Incidence and risk factors for thrombotic complications in a historical cohort of 100 patients with essential thrombocythemia. J. Clin. Oncol. 1990, 8, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Besses, C.; Cervantes, F.; Pereira, A.; Florensa, L.; Sole, F.; Hernandez-Boluda, J.C.; Woessner, S.; Sans-Sabrafen, J.; Rozman, C.; Montserrat, E. Major vascular complications in essential thrombocythemia: A study of the predictive factors in a series of 148 patients. Leukemia 1999, 13, 150–154. [Google Scholar] [CrossRef][Green Version]
- Alvarez-Larran, A.; Cervantes, F.; Bellosillo, B.; Giralt, M.; Julia, A.; Hernandez-Boluda, J.C.; Bosch, A.; Hernandez-Nieto, L.; Clapes, V.; Burgaleta, C.; et al. Essential thrombocythemia in young individuals: Frequency and risk factors for vascular events and evolution to myelofibrosis in 126 patients. Leukemia 2007, 21, 1218–1223. [Google Scholar] [CrossRef]
- Stein, B.L.; Martin, K. From Budd-Chiari syndrome to acquired von Willebrand syndrome: Thrombosis and bleeding complications in the myeloproliferative neoplasms. Hematol. Am. Soc. Hematol. Educ. Program 2019, 2019, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Moulard, O.; Mehta, J.; Fryzek, J.; Olivares, R.; Iqbal, U.; Mesa, R.A. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur. J. Haematol. 2014, 92, 289–297. [Google Scholar] [CrossRef]
- Passamonti, F.; Rumi, E.; Pungolino, E.; Malabarba, L.; Bertazzoni, P.; Valentini, M.; Orlandi, E.; Arcaini, L.; Brusamolino, E.; Pascutto, C.; et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am. J. Med. 2004, 117, 755–761. [Google Scholar] [CrossRef]
- Campbell, P.J.; MacLean, C.; Beer, P.A.; Buck, G.; Wheatley, K.; Kiladjian, J.J.; Forsyth, C.; Harrison, C.N.; Green, A.R. Correlation of blood counts with vascular complications in essential thrombocythemia: Analysis of the prospective PT1 cohort. Blood 2012, 120, 1409–1411. [Google Scholar] [CrossRef]
- Tefferi, A.; Fonseca, R.; Pereira, D.L.; Hoagland, H.C. A long-term retrospective study of young women with essential thrombocythemia. Mayo Clin. Proc. 2001, 76, 22–28. [Google Scholar] [CrossRef]
- Falchi, L.; Bose, P.; Newberry, K.J.; Verstovsek, S. Approach to patients with essential thrombocythaemia and very high platelet counts: What is the evidence for treatment? Br. J. Haematol. 2017, 176, 352–364. [Google Scholar] [CrossRef]
- Michiels, J.J. Acquired von Willebrand disease due to increasing platelet count can readily explain the paradox of thrombosis and bleeding in thrombocythemia. Clin. Appl. Thromb. Hemost. 1999, 5, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Lussana, F.; Carobbio, A.; Salmoiraghi, S.; Guglielmelli, P.; Vannucchi, A.M.; Bottazzi, B.; Leone, R.; Mantovani, A.; Barbui, T.; Rambaldi, A. Driver mutations (JAK2V617F, MPLW515L/K or CALR), pentraxin-3 and C-reactive protein in essential thrombocythemia and polycythemia vera. J. Hematol. Oncol. 2017, 10, 54. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, F.; Pereira, A. Does ruxolitinib prolong the survival of patients with myelofibrosis? Blood 2017, 129, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Barosi, G.; Birgegard, G.; Cervantes, F.; Finazzi, G.; Griesshammer, M.; Harrison, C.; Hasselbalch, H.C.; Hehlmann, R.; Hoffman, R.; et al. Philadelphia-negative classical myeloproliferative neoplasms: Critical concepts and management recommendations from European LeukemiaNet. J. Clin. Oncol. 2011, 29, 761–770. [Google Scholar] [CrossRef]
- Barbui, T.; Finazzi, G.; Carobbio, A.; Thiele, J.; Passamonti, F.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; Bertozzi, I.; et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis). Blood 2012, 120, 5128–5133. [Google Scholar] [CrossRef]
- Barbui, T.; Vannucchi, A.M.; Buxhofer-Ausch, V.; De Stefano, V.; Betti, S.; Rambaldi, A.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J. 2015, 5, e369. [Google Scholar] [CrossRef]
- Haider, M.; Gangat, N.; Lasho, T.; Abou Hussein, A.K.; Elala, Y.C.; Hanson, C.; Tefferi, A. Validation of the revised International Prognostic Score of Thrombosis for Essential Thrombocythemia (IPSET-thrombosis) in 585 Mayo Clinic patients. Am. J. Hematol. 2016, 91, 390–394. [Google Scholar] [CrossRef]
- Alvarez-Larran, A.; Martinez, D.; Arenillas, L.; Rubio, A.; Arellano-Rodrigo, E.; Hernandez Boluda, J.C.; Papaleo, N.; Caballero, G.; Martinez, C.; Ferrer-Marin, F.; et al. Essential thrombocythaemia with mutation in MPL: Clinicopathological correlation and comparison with JAK2V617F-mutated and CALR-mutated genotypes. J. Clin. Pathol. 2018, 71, 975–980. [Google Scholar] [CrossRef]
- Rumi, E.; Pietra, D.; Ferretti, V.; Klampfl, T.; Harutyunyan, A.S.; Milosevic, J.D.; Them, N.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014, 123, 1544–1551. [Google Scholar] [CrossRef]
- Horvat, I.; Boban, A.; Zadro, R.; Antolic, M.R.; Serventi-Seiwerth, R.; Roncevic, P.; Radman, I.; Sertic, D.; Vodanovic, M.; Pulanic, D.; et al. Influence of Blood Count, Cardiovascular Risks, Inherited Thrombophilia, and JAK2 V617F Burden Allele on Type of Thrombosis in Patients With Philadelphia Chromosome Negative Myeloproliferative Neoplasms. Clin. Lymphoma Myeloma Leuk. 2019, 19, 53–63. [Google Scholar] [CrossRef]
- Passamonti, F.; Thiele, J.; Girodon, F.; Rumi, E.; Carobbio, A.; Gisslinger, H.; Kvasnicka, H.M.; Ruggeri, M.; Randi, M.L.; Gangat, N.; et al. A prognostic model to predict survival in 867 World Health Organization-defined essential thrombocythemia at diagnosis: A study by the International Working Group on Myelofibrosis Research and Treatment. Blood 2012, 120, 1197–1201. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients With Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Accurso, V.; Mancuso, S.; Carlisi, M.; Raso, S.; Tarantino, G.; Di Piazza, F.; Perez, A.; Russo, A.; Siragusa, S. Comparison between thrombotic risk scores in essential thrombocythemia and survival implications. Hematol. Oncol. 2019, 37, 434–437. [Google Scholar] [CrossRef]
- Hauschner, H.; Bokstad Horev, M.; Misgav, M.; Nagar, M.; Seligsohn, U.; Rosenberg, N.; Koren-Michowitz, M. Platelets from Calreticulin mutated essential thrombocythemia patients are less reactive than JAK2 V617F mutated platelets. Am. J. Hematol. 2019. [Google Scholar] [CrossRef]
- Cottin, L.; Riou, J.; Orvain, C.; Ianotto, J.C.; Boyer, F.; Renard, M.; Truchan-Graczyk, M.; Murati, A.; Jouanneau-Courville, R.; Allangba, O.; et al. Sequential mutational evaluation of CALR -mutated myeloproliferative neoplasms with thrombocytosis reveals an association between CALR allele burden evolution and disease progression. Br. J. Haematol. 2020, 188, 935–944. [Google Scholar] [CrossRef]
- Carobbio, A.; Ferrari, A.; Masciulli, A.; Ghirardi, A.; Barosi, G.; Barbui, T. Leukocytosis and thrombosis in essential thrombocythemia and polycythemia vera: A systematic review and meta-analysis. Blood Adv. 2019, 3, 1729–1737. [Google Scholar] [CrossRef]
- Buxhofer-Ausch, V.; Steurer, M.; Sormann, S.; Schloegl, E.; Schimetta, W.; Gisslinger, B.; Schalling, M.; Krauth, M.T.; Thiele, J.; Ruckser, R.; et al. Impact of white blood cells on thrombotic risk in patients with optimized platelet count in essential thrombocythemia. Eur. J. Haematol. 2018. [Google Scholar] [CrossRef]
- De Stefano, V.; Za, T.; Rossi, E.; Fiorini, A.; Ciminello, A.; Luzzi, C.; Chiusolo, P.; Sica, S.; Leone, G. Influence of the JAK2 V617F mutation and inherited thrombophilia on the thrombotic risk among patients with essential thrombocythemia. Haematologica 2009, 94, 733–737. [Google Scholar] [CrossRef]
- Trifa, A.P.; Cucuianu, A.; Popp, R.A.; Coada, C.A.; Costache, R.M.; Militaru, M.S.; Vesa, S.C.; Pop, I.V. The relationship between factor V Leiden, prothrombin G20210A, and MTHFR mutations and the first major thrombotic episode in polycythemia vera and essential thrombocythemia. Ann. Hematol. 2014, 93, 203–209. [Google Scholar] [CrossRef]
- Harrison, C.N.; Campbell, P.J.; Buck, G.; Wheatley, K.; East, C.L.; Bareford, D.; Wilkins, B.S.; van der Walt, J.D.; Reilly, J.T.; Grigg, A.P.; et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N. Engl. J. Med. 2005, 353, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Cortelazzo, S.; Finazzi, G.; Ruggeri, M.; Vestri, O.; Galli, M.; Rodeghiero, F.; Barbui, T. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N. Engl. J. Med. 1995, 332, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Gisslinger, H.; Gotic, M.; Holowiecki, J.; Penka, M.; Thiele, J.; Kvasnicka, H.M.; Kralovics, R.; Petrides, P.E. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: The ANAHYDRET Study, a randomized controlled trial. Blood 2013, 121, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Kiladjian, J.J.; Chomienne, C.; Fenaux, P. Interferon-alpha therapy in bcr-abl-negative myeloproliferative neoplasms. Leukemia 2008, 22, 1990–1998. [Google Scholar] [CrossRef]
- Quintas-Cardama, A.; Kantarjian, H.; Manshouri, T.; Luthra, R.; Estrov, Z.; Pierce, S.; Richie, M.A.; Borthakur, G.; Konopleva, M.; Cortes, J.; et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J. Clin. Oncol. 2009, 27, 5418–5424. [Google Scholar] [CrossRef]
- Czech, J.; Cordua, S.; Weinbergerova, B.; Baumeister, J.; Crepcia, A.; Han, L.; Maie, T.; Costa, I.G.; Denecke, B.; Maurer, A.; et al. JAK2V617F but not CALR mutations confer increased molecular responses to interferon-alpha via JAK1/STAT1 activation. Leukemia 2019, 33, 995–1010. [Google Scholar] [CrossRef]
- Masarova, L.; Patel, K.P.; Newberry, K.J.; Cortes, J.; Borthakur, G.; Konopleva, M.; Estrov, Z.; Kantarjian, H.; Verstovsek, S. Pegylated interferon alfa-2a in patients with essential thrombocythaemia or polycythaemia vera: A post-hoc, median 83 month follow-up of an open-label, phase 2 trial. Lancet Haematol. 2017, 4, e165–e175. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Kosiorek, H.E.; Prchal, J.T.; Rambaldi, A.; Berenzon, D.; Yacoub, A.; Harrison, C.N.; McMullin, M.F.; Vannucchi, A.M.; Ewing, J.; et al. Results of the Myeloproliferative Neoplasms - Research Consortium (MPN-RC) 112 Randomized Trial of Pegylated Interferon Alfa-2a (PEG) Versus Hydroxyurea (HU) Therapy for the Treatment of High Risk Polycythemia Vera (PV) and High Risk Essential Thrombocythemia (ET). Blood 2018, 132, 577. [Google Scholar] [CrossRef]
- Alvarez-Larran, A.; Pereira, A.; Cervantes, F.; Arellano-Rodrigo, E.; Hernandez-Boluda, J.C.; Ferrer-Marin, F.; Angona, A.; Gomez, M.; Muina, B.; Guillen, H.; et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood 2012, 119, 1363–1369. [Google Scholar] [CrossRef]
- Yacoub, A.; Mascarenhas, J.; Kosiorek, H.; Prchal, J.T.; Berenzon, D.; Baer, M.R.; Ritchie, E.; Silver, R.T.; Kessler, C.; Winton, E.; et al. Pegylated interferon alfa-2a for polycythemia vera or essential thrombocythemia resistant or intolerant to hydroxyurea. Blood 2019, 134, 1498–1509. [Google Scholar] [CrossRef]
- Gisslinger, H.; Zagrijtschuk, O.; Buxhofer-Ausch, V.; Thaler, J.; Schloegl, E.; Gastl, G.A.; Wolf, D.; Kralovics, R.; Gisslinger, B.; Strecker, K.; et al. Ropeginterferon alfa-2b, a novel IFNalpha-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood 2015, 126, 1762–1769. [Google Scholar] [CrossRef] [PubMed]
- Gisslinger, H.; Klade, C.; Georgiev, P.; Krochmalczyk, D.; Gercheva-Kyuchukova, L.; Egyed, M.; Rossiev, V.; Dulicek, P.; Illes, A.; Pylypenko, H.; et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): A randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol. 2020, 7, e196–e208. [Google Scholar] [CrossRef]
- Kralovics, R.; Klade, C.; Georgiev, P.; Krochmalczyk, D.; Gercheva-Kyuchukova, L.; Egyed, M.; Rossiev, V.; Dulicek, P.; Illes, A.; Pylypenko, H.; et al. Ropeginterferon Alpha-2b is efficacious and reduces variant Tet2 allele burden in patients with polycythaemia vera and Tet2 mutation: genetic analysis of Phase III PROUD-PV/CONTINUATION-PV studies. Hemasphere 2020, 4, 1–1168. [Google Scholar]
- Verstovsek, S.; Passamonti, F.; Rambaldi, A.; Barosi, G.; Rumi, E.; Gattoni, E.; Pieri, L.; Zhen, H.; Granier, M.; Assad, A.; et al. Ruxolitinib for essential thrombocythemia refractory to or intolerant of hydroxyurea: Long-term phase 2 study results. Blood 2017, 130, 1768–1771. [Google Scholar] [CrossRef]
- Harrison, C.N.; Mead, A.J.; Panchal, A.; Fox, S.; Yap, C.; Gbandi, E.; Houlton, A.; Alimam, S.; Ewing, J.; Wood, M.; et al. Ruxolitinib vs best available therapy for ET intolerant or resistant to hydroxycarbamide. Blood 2017, 130, 1889–1897. [Google Scholar] [CrossRef]
- Harrison, C. Pregnancy and its management in the Philadelphia negative myeloproliferative diseases. Br. J. Haematol. 2005, 129, 293–306. [Google Scholar] [CrossRef]
- Harrison, C.N.; Bareford, D.; Butt, N.; Campbell, P.; Conneally, E.; Drummond, M.; Erber, W.; Everington, T.; Green, A.R.; Hall, G.W.; et al. Guideline for investigation and management of adults and children presenting with a thrombocytosis. Br. J. Haematol. 2010, 149, 352–375. [Google Scholar] [CrossRef]
- Beauverd, Y.; Radia, D.; Cargo, C.; Knapper, S.; Drummond, M.; Pillai, A.; Harrison, C.; Robinson, S. Pegylated interferon alpha-2a for essential thrombocythemia during pregnancy: Outcome and safety. A case series. Haematologica 2016, 101, e182–e184. [Google Scholar] [CrossRef]
- Singh, N.; Kumar, S.; Roy, K.K.; Sharma, V.; Jalak, A. Successful maternal and fetal outcome in a rare case of essential Thrombocythemia with pregnancy using Interferon alpha. Platelets 2012, 23, 319–321. [Google Scholar] [CrossRef][Green Version]
- Martinelli, P.; Martinelli, V.; Agangi, A.; Maruotti, G.M.; Paladini, D.; Ciancia, R.; Rotoli, B. Interferon alfa treatment for pregnant women affected by essential thrombocythemia: Case reports and a review. Am. J. Obs. Gynecol. 2004, 191, 2016–2020. [Google Scholar] [CrossRef]
- Antonioli, E.; Guglielmelli, P.; Pieri, L.; Finazzi, M.; Rumi, E.; Martinelli, V.; Vianelli, N.; Luigia Randi, M.; Bertozzi, I.; De Stefano, V.; et al. Hydroxyurea-related toxicity in 3,411 patients with Ph’-negative MPN. Am. J. Hematol. 2012, 87, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Anagrelide, a therapy for thrombocythemic states: Experience in 577 patients Anagrelide Study Group. Am. J. Med. 1992, 92, 69–76. [CrossRef]
- Jurgens, D.J.; Moreno-Aspitia, A.; Tefferi, A. Anagrelide-associated cardiomyopathy in polycythemia vera and essential thrombocythemia. Haematologica 2004, 89, 1394–1395. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.N. Essential thrombocythaemia: Challenges and evidence-based management. Br. J. Haematol. 2005, 130, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Michiels, J.J.; Berneman, Z.; Schroyens, W.; Koudstaal, P.J.; Lindemans, J.; Neumann, H.A.; van Vliet, H.H. Platelet-mediated erythromelalgic, cerebral, ocular and coronary microvascular ischemic and thrombotic manifestations in patients with essential thrombocythemia and polycythemia vera: A distinct aspirin-responsive and coumadin-resistant arterial thrombophilia. Platelets 2006, 17, 528–544. [Google Scholar] [CrossRef]
- Alvarez-Larran, A.; Pereira, A.; Guglielmelli, P.; Hernandez-Boluda, J.C.; Arellano-Rodrigo, E.; Ferrer-Marin, F.; Samah, A.; Griesshammer, M.; Kerguelen, A.; Andreasson, B.; et al. Antiplatelet therapy versus observation in low-risk essential thrombocythemia with a CALR mutation. Haematologica 2016, 101, 926–931. [Google Scholar] [CrossRef]
- Chu, D.K.; Hillis, C.M.; Leong, D.P.; Anand, S.S.; Siegal, D.M. Benefits and Risks of Antithrombotic Therapy in Essential Thrombocythemia: A Systematic Review. Ann. Intern. Med. 2017, 167, 170–180. [Google Scholar] [CrossRef]
- Rocca, B.; Tosetto, A.; Betti, S.; Soldati, D.; Petrucci, G.; Rossi, E.; Timillero, A.; Cavalca, V.; Porro, B.; Iurlo, A.; et al. A randomized, double-blind trial of three aspirin regimens to optimize antiplatelet therapy in essential thrombocythemia. Blood 2020. [Google Scholar] [CrossRef]
- Barbui, T.; De Stefano, V.; Falanga, A.; Finazzi, G.; Martinelli, I.; Rodeghiero, F.; Vannucchi, A.M.; Barosi, G. Addressing and proposing solutions for unmet clinical needs in the management of myeloproliferative neoplasm-associated thrombosis: A consensus-based position paper. Blood Cancer J. 2019, 9, 61. [Google Scholar] [CrossRef]
- De Stefano, V.; Za, T.; Rossi, E.; Vannucchi, A.M.; Ruggeri, M.; Elli, E.; Micò, C.; Tieghi, A.; Cacciola, R.R.; Santoro, C.; et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: Incidence, risk factors, and effect of treatments. Haematologica 2008, 93, 372–380. [Google Scholar] [CrossRef]
- De Stefano, V.; Ruggeri, M.; Cervantes, F.; Alvarez-Larrán, A.; Iurlo, A.; Randi, M.L.; Elli, E.; Finazzi, M.C.; Finazzi, G.; Zetterberg, E.; et al. High rate of recurrent venous thromboembolism in patients with myeloproliferative neoplasms and effect of prophylaxis with vitamin K antagonists. Leukemia 2016, 30, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; De Stefano, V. Management of venous thromboembolism in myeloproliferative neoplasms. Curr. Opin. Hematol. 2017, 24, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, I.; De Stefano, V.; Carobbio, A.; Randi, M.L.; Santarossa, C.; Rambaldi, A.; Finazzi, M.C.; Cervantes, F.; Arellano-Rodrigo, E.; Rupoli, S.; et al. Cerebral vein thrombosis in patients with Philadelphia-negative myeloproliferative neoplasms. An European Leukemia Net study. Am. J. Hematol. 2014, 89, E200–E205. [Google Scholar] [CrossRef] [PubMed]
- Sant’Antonio, E.; Guglielmelli, P.; Pieri, L.; Primignani, M.; Randi, M.L.; Santarossa, C.; Rumi, E.; Cervantes, F.; Delaini, F.; Carobbio, A.; et al. Splanchnic vein thromboses associated with myeloproliferative neoplasms: An international, retrospective study on 518 cases. Am. J. Hematol. 2020, 95, 156–166. [Google Scholar] [CrossRef] [PubMed]
- The Hokusai-VTE Investigators. Edoxaban versus Warfarin for the Treatment of Symptomatic Venous Thromboembolism. N. Engl. J. Med. 2013, 369, 1406–1415. [Google Scholar] [CrossRef]
- Ellis, M.H.; Lavi, N.; Vannucchi, A.; Harrison, C. Treatment of thromboembolic events coincident with the diagnosis of myeloproliferative neoplasms: A physician survey. Thromb. Res. 2014, 134, 251–254. [Google Scholar] [CrossRef]
- Godfrey, A.L.; Campbell, P.J.; MacLean, C.; Buck, G.; Cook, J.; Temple, J.; Wilkins, B.S.; Wheatley, K.; Nangalia, J.; Grinfeld, J.; et al. Hydroxycarbamide Plus Aspirin Versus Aspirin Alone in Patients With Essential Thrombocythemia Age 40 to 59 Years Without High-Risk Features. J. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Boddu, P.; Falchi, L.; Hosing, C.; Newberry, K.; Bose, P.; Verstovsek, S. The role of thrombocytapheresis in the contemporary management of hyperthrombocytosis in myeloproliferative neoplasms: A case-based review. Leuk. Res. 2017, 58, 14–22. [Google Scholar] [CrossRef]
- Epstein, E.; Goedel, A. Hämorrhagische Thrombocythämie bei vasculärer Schrumpfmilz. Virchows Archiv Pathologische Anatomie Physiologie Klinische Medizin 1934, 292, 233–248. [Google Scholar] [CrossRef]
- Federici, A.B. Acquired von Willebrand syndrome: An underdiagnosed and misdiagnosed bleeding complication in patients with lymphoproliferative and myeloproliferative disorders. Semin. Hematol. 2006, 43, S48–S58. [Google Scholar] [CrossRef]
- Michiels, J.J.; Berneman, Z.; Schroyens, W.; Finazzi, G.; Budde, U.; van Vliet, H.H. The paradox of platelet activation and impaired function: Platelet-von Willebrand factor interactions, and the etiology of thrombotic and hemorrhagic manifestations in Essential thrombocythemia and polycythemia vera. Semin. Thromb. Hemost. 2006, 32, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Smock, K.J.; Divgi, A.B. Polycythemia vera-associated acquired von Willebrand syndrome despite near-normal platelet count. Am. J. Hematol. 2010, 85, 545. [Google Scholar] [CrossRef] [PubMed]
- Troost, M.M.; van Genderen, P.J. Excessive prolongation of the Ivy bleeding time after aspirin in essential thrombocythemia is also demonstrable in vitro in the high shear stress system PFA-100. Ann. Hematol. 2002, 81, 353–354. [Google Scholar] [CrossRef] [PubMed]
- Fabris, F.; Casonato, A.; Grazia del Ben, M.; De Marco, L.; Girolami, A. Abnormalities of von Willebrand factor in myeloproliferative disease: A relationship with bleeding diathesis. Br. J. Haematol. 1986, 63, 75–83. [Google Scholar] [CrossRef]
- Sanchez-Luceros, A.; Meschengieser, S.S.; Woods, A.I.; Blanco, A.N.; Kempfer, A.C.; Casais, P.; Salviu, M.J.; Lazzari, M.A. Acquired von Willebrand factor abnormalities in myeloproliferative disorders and other hematologic diseases: A retrospective analysis by a single institution. Haematologica 2002, 87, 264–270. [Google Scholar]
- Tefferi, A.; Gangat, N.; Wolanskyj, A.P. Management of extreme thrombocytosis in otherwise low-risk essential thrombocythemia; does number matter? Blood 2006, 108, 2493–2494. [Google Scholar] [CrossRef]
- Palandri, F.; Polverelli, N.; Catani, L.; Sollazzo, D.; Ottaviani, E.; Parisi, S.; Baccarani, M.; Vianelli, N. Bleeding in essential thrombocythaemia: A retrospective analysis on 565 patients. Br. J. Haematol. 2012, 156, 281–284. [Google Scholar] [CrossRef]
- Finazzi, G.; Carobbio, A.; Thiele, J.; Passamonti, F.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; Bertozzi, I.; Vannucchi, A.M.; et al. Incidence and risk factors for bleeding in 1104 patients with essential thrombocythemia or prefibrotic myelofibrosis diagnosed according to the 2008 WHO criteria. Leukemia 2012, 26, 716–719. [Google Scholar] [CrossRef]
- Moxon-Emre, I.; Schlichter, L.C. Neutrophil depletion reduces blood-brain barrier breakdown, axon injury, and inflammation after intracerebral hemorrhage. J. Neuropathol. Exp. Neurol. 2011, 70, 218–235. [Google Scholar] [CrossRef]
- Fang, H.; Sun, C.; Xu, L.; Owen, R.J.; Auth, R.D.; Snoy, P.J.; Frucht, D.M. Neutrophil elastase mediates pathogenic effects of anthrax lethal toxin in the murine intestinal tract. J. Immunol. 2010, 185, 5463–5467. [Google Scholar] [CrossRef]
- Alvarez-Larran, A.; Cervantes, F.; Pereira, A.; Arellano-Rodrigo, E.; Perez-Andreu, V.; Hernandez-Boluda, J.C.; Ayats, R.; Salvador, C.; Muntanola, A.; Bellosillo, B.; et al. Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood 2010, 116, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Budde, U.; Scharf, R.E.; Franke, P.; Hartmann-Budde, K.; Dent, J.; Ruggeri, Z.M. Elevated platelet count as a cause of abnormal von Willebrand factor multimer distribution in plasma. Blood 1993, 82, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Van Genderen, P.J.; Budde, U.; Michiels, J.J.; van Strik, R.; van Vliet, H.H. The reduction of large von Willebrand factor multimers in plasma in essential thrombocythaemia is related to the platelet count. Br. J. Haematol. 1996, 93, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Budde, U.; Schaefer, G.; Mueller, N.; Egli, H.; Dent, J.; Ruggeri, Z.; Zimmerman, T. Acquired von Willebrand’s disease in the myeloproliferative syndrome. Blood 1984, 64, 981–985. [Google Scholar] [CrossRef]
- Federici, A.B.; Rand, J.H.; Mannucci, P.M. Acquired von Willebrand syndrome: An important bleeding complication to be considered in patients with lymphoproliferative and myeloproliferative disorders. Hematol. J. 2001, 2, 358–362. [Google Scholar] [CrossRef]
- El-Moneim, A.A.; Kratz, C.P.; Boll, S.; Rister, M.; Pahl, H.L.; Niemeyer, C.M. Essential versus reactive thrombocythemia in children: Retrospective analyses of 12 cases. Pediatr. Blood Cancer 2007, 49, 52–55. [Google Scholar] [CrossRef]
- Polokhov, D.M.; Ershov, N.M.; Ignatova, A.A.; Ponomarenko, E.A.; Gaskova, M.V.; Zharkov, P.A.; Fedorova, D.V.; Poletaev, A.V.; Seregina, E.A.; Novichkova, G.A.; et al. Platelet function and blood coagulation system status in childhood essential thrombocythemia. Platelets 2019, 1–11. [Google Scholar] [CrossRef]
- Kreher, S.; Ochsenreither, S.; Trappe, R.U.; Pabinger, I.; Bergmann, F.; Petrides, P.E.; Koschmieder, S.; Matzdorff, A.; Tiede, A.; Griesshammer, M.; et al. Prophylaxis and management of venous thromboembolism in patients with myeloproliferative neoplasms: Consensus statement of the Haemostasis Working Party of the German Society of Hematology and Oncology (DGHO), the Austrian Society of Hematology and Oncology (OGHO) and Society of Thrombosis and Haemostasis Research (GTH e.V.). Ann. Hematol. 2014, 93, 1953–1963. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| WHO 2008 | WHO 2016 (Revised) |
|---|---|
| Major criteria | Major criteria |
| 1. Platelet count ≥ 450 × 109/L | 1. Platelet count ≥ 450 × 109/L |
| 2. BM biopsy showing proliferation of the megakaryocyte lineage with increased numbers of enlarged, mature megakaryocytes | 2. BM biopsy showing proliferation of the megakaryocyte lineage with increased numbers of enlarged, mature megakaryocytes with hyperlobulated nuclei. No significant left-shift of neutrophil myelopoiesis or erythropoiesis and very rarely minor (grade 1) increase in reticulin fibers |
| 3. Not meeting WHO criteria for CML, PV, PMF, MDS, or other myeloid neoplasms | 3. Not meeting WHO criteria for BCR-ABL1 + CML, PV, PMF, MDS, or other myeloid neoplasms |
| 4. Presence of JAK2 V617F mutation or other clonal marker or lack of evidence of a secondary cause of thrombocytosis | 4. Presence of JAK2, CALR or MPL mutation |
| No minor criteria | Minor criteria 1. Presence of a clonal marker or absence of evidence for reactive thrombocytosis |
| All four major criteria required | All four major criteria or three major and one minor required |
| Traditional ELN Guidelines |
| (a) High-risk: age ≥ 60 years or previous thrombosis |
| (b) Low-risk: none of the above |
| IPSET-thrombosis |
| Risk factors: Age > 60, = 1 point; Cardiovascular risk factors (tobacco use, diabetes, hypercholesterolaemia, hypertension), = 1 point; Previous thrombosis, = 2 point; JAK2 V617F, = 2 points |
| (a) Low risk: 0–1 |
| (b) Intermediate risk: 2 |
| (c) High-risk: ≥3 |
| IPSET- thrombosis (Revised) |
| (a) Very low risk: no thrombosis history, age ≤ 60 years and JAK2/MPL-unmutated |
| (b) Low risk: no thrombosis history, age ≤ 60 years and JAK2/MPL-mutated |
| (c) Intermediate risk: no thrombosis history, age > 60 years and JAK2/MPL-unmutated |
| (d) High risk: thrombosis history or age > 60 years with JAK2/MPL mutation |
| MIPSS-ET |
| Risk factors: Adverse mutations (SRSF2, SF3B1, U2AF1 and TP53) = 2 points; age > 60 years = 4 points, male sex = 1 point and leukocyte count ≥ 11 × 109/L = 1 point |
| (a) Low risk: 0–1 |
| (b) Intermediate risk: 2–3 |
| (c) High-risk: ≥4 |
| Risk Factors |
|---|
| Advanced age (>60 years) |
| Extreme thrombocytosis (platelet count > 1000 × 109/L) |
| Leukocytosis (leukocyte count ≥ 11 × 109/L) |
| Driving mutation: JAK2 V617F is associated with higher risk of bleeding (CALR unclear) |
| History of bleeding/thrombosis |
| Acquired von Willebrand syndrome |
| Splenomegaly and portal hypertension |
| Medication induced (antiplatelet therapy and anticoagulation therapies) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awada, H.; Voso, M.T.; Guglielmelli, P.; Gurnari, C. Essential Thrombocythemia and Acquired von Willebrand Syndrome: The Shadowlands between Thrombosis and Bleeding. Cancers 2020, 12, 1746. https://doi.org/10.3390/cancers12071746
Awada H, Voso MT, Guglielmelli P, Gurnari C. Essential Thrombocythemia and Acquired von Willebrand Syndrome: The Shadowlands between Thrombosis and Bleeding. Cancers. 2020; 12(7):1746. https://doi.org/10.3390/cancers12071746
Chicago/Turabian StyleAwada, Hassan, Maria Teresa Voso, Paola Guglielmelli, and Carmelo Gurnari. 2020. "Essential Thrombocythemia and Acquired von Willebrand Syndrome: The Shadowlands between Thrombosis and Bleeding" Cancers 12, no. 7: 1746. https://doi.org/10.3390/cancers12071746
APA StyleAwada, H., Voso, M. T., Guglielmelli, P., & Gurnari, C. (2020). Essential Thrombocythemia and Acquired von Willebrand Syndrome: The Shadowlands between Thrombosis and Bleeding. Cancers, 12(7), 1746. https://doi.org/10.3390/cancers12071746