Good Cop, Bad Cop: Defining the Roles of Δ40p53 in Cancer and Aging

 ,
,  and
and
Abstract
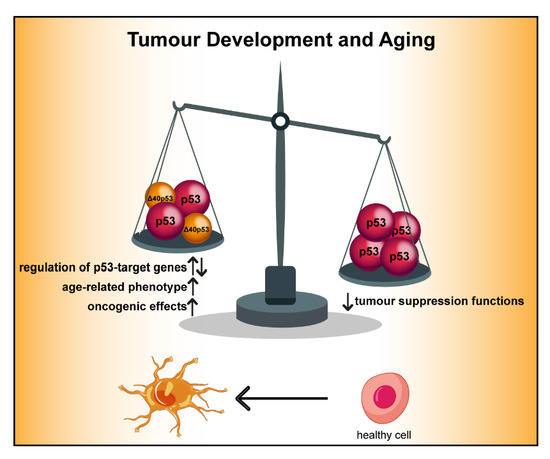
1. Introduction
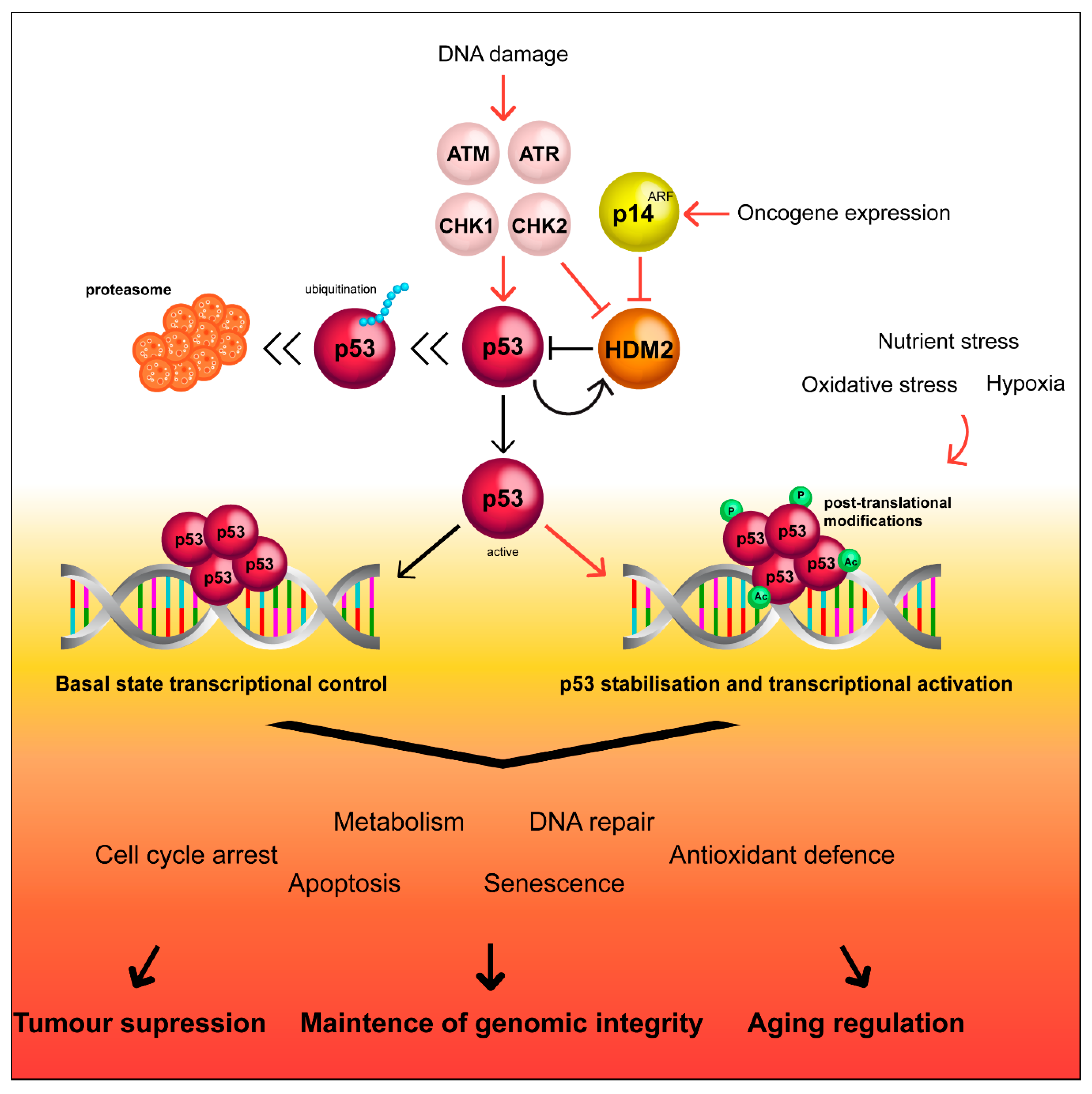
2. The Canonical p53 Pathway
3. p53 Isoforms
4. The ∆40p53 Isoform
4.1. Mechanism of Production
4.2. The In Vitro Function of ∆40p53
4.2.1. Modulation of Transcription of p53-Target Genes
4.2.2. Cell-Cycle Regulation
4.2.3. Apoptosis
4.2.4. Senescence
4.3. Why Is ∆40p53 Functionally Distinct to p53?
4.4. ∆40p53 in Cancer Prognostication and Clinical Outcomes
4.5. The Role of ∆40p53 in Aging
5. Can ∆40p53 Be Targeted to Modulate the p53 Pathway?
5.1. Modulation of ∆40p53 Using Compounds Already Used Clinically (In Vivo)
5.1.1. HDM2 Antagonists
5.1.2. Endoplasmic Reticulum (ER) Stress
5.1.3. 20S Proteasome Targeting
5.2. Modulation of ∆40p53 Using In Vitro Strategies
5.2.1. Transfection and Transduction for Exogenous Expression
5.2.2. Antisense Oligonucleotides
5.2.3. RNA Interference and CRISPR/Cas9
5.2.4. Targeting the G-Quadruplex (G4) Structure
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Norbury, C.J.; Hickson, I.D. Cellular responses to DNA damage. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 367–401. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Hakem, R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008, 27, 589–605. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Avery-Kiejda, K.A.; Zhang, X.D.; Adams, L.J.; Scott, R.J.; Vojtesek, B.; Lane, D.P.; Hersey, P. Small molecular weight variants of p53 are expressed in human melanoma cells and are induced by the DNA-damaging agent cisplatin. Clin. Cancer Res. 2008, 14, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.L.; Schumacher, B. DNA damage responses and p53 in the aging process. Blood 2018, 131, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef]
- Helton, E.S.; Chen, X. p53 modulation of the DNA damage response. J. Cell. Biochem. 2007, 100, 883–896. [Google Scholar] [CrossRef]
- Rozan, L.M.; El-Deiry, W.S. p53 downstream target genes and tumor suppression: A classical view in evolution. Cell Death Differ. 2007, 14, 3–9. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, L. The transcriptional targets of p53 in apoptosis control. Biochem. Biophys. Res. Commun. 2005, 331, 851–858. [Google Scholar] [CrossRef]
- Barabutis, N.; Schally, A.V.; Siejka, A. p53, GHRH, inflammation and cancer. EBioMedicine 2018, 37, 557–562. [Google Scholar] [CrossRef]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef] [PubMed]
- Courtois, S.; Verhaegh, G.; North, S.; Luciani, M.G.; Lassus, P.; Hibner, U.; Oren, M.; Hainaut, P. DeltaN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene 2002, 21, 6722–6728. [Google Scholar] [CrossRef]
- Bourougaa, K.; Naski, N.; Boularan, C.; Mlynarczyk, C.; Candeias, M.M.; Marullo, S.; Fahraeus, R. Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA translation of the p53 isoform p53/47. Mol. Cell 2010, 38, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Stephen, C.W.; Luciani, M.G.; Fahraeus, R. p53 Stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat. Cell Biol. 2002, 4, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.J.; Hrstka, R.; Candeias, M.; Bourougaa, K.; Vojtesek, B.; Fahraeus, R. Stress-dependent changes in the properties of p53 complexes by the alternative translation product p53/47. Cell Cycle 2008, 7, 950–959. [Google Scholar] [CrossRef]
- Sharpless, N.E.; DePinho, R. p53: Good cop/bad cop. Cell 2002, 110, 9–12. [Google Scholar] [CrossRef]
- Hafsi, H.; Santos-Silva, D.; Courtois-Cox, S.; Hainaut, P. Effects of Δ40p53, an isoform of p53 lacking the N-terminus, on transactivation capacity of the tumor suppressor protein p53. BMC Cancer 2013, 13, 134. [Google Scholar] [CrossRef]
- Ungewitter, E.; Scrable, H. Delta40p53 controls the switch from pluripotency to differentiation by regulating IGF signaling in ESCs. Genes Dev. 2010, 24, 2408–2419. [Google Scholar] [CrossRef] [PubMed]
- Pehar, M.; Ko, M.H.; Li, M.; Scrable, H.; Puglielli, L. p44, the ‘longevity-assurance’ isoform of p53, regulates tau phosphorylation and is activated in an age-dependent fashion. Aging Cell 2014, 13, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Pehar, M.; O’Riordan, K.J.; Burns-Cusato, M.; Andrzejewski, M.E.; Del Alcazar, C.G.; Burger, C.; Scrable, H.; Puglielli, L. Altered longevity-assurance activity of p53:p44 in the mouse causes memory loss, neurodegeneration and premature death. Aging Cell 2010, 9, 174–190. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J. p53 and its isoforms in cancer. Br. J. Cancer 2007, 97, 277–282. [Google Scholar] [CrossRef]
- IARC Tp53 Database, 2014. Available online: http://p53.iarc.fr/SelectedStatistics.aspx (accessed on 1 April 2020).
- Beckerman, R.; Prives, C. Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2010, 2, a000935. [Google Scholar] [CrossRef] [PubMed]
- Laptenko, O.; Prives, C. Transcriptional regulation by p53: One protein, many possibilities. Cell Death Differ. 2006, 13, 951–961. [Google Scholar] [CrossRef]
- Alarcon-Vargas, D.; Ronai, Z. p53-Mdm2—The affair that never ends. Carcinogenesis 2002, 23, 541–547. [Google Scholar] [CrossRef]
- Chao, C. Mechanisms of p53 degradation. Clin. Chim. Acta 2015, 438, 139–147. [Google Scholar] [CrossRef]
- Sparks, A.; Dayal, S.; Das, J.; Robertson, P.; Menendez, S.; Saville, M. The degradation of p53 and its major E3 ligase Mdm2 is differentially dependent on the proteasomal ubiquitin receptor S5a. Oncogene 2014, 33, 4685–4696. [Google Scholar] [CrossRef]
- Junttila, M.R.; Evan, G. p53—A Jack of all trades but master of none. Nat. Rev. Cancer 2009, 9, 821–829. [Google Scholar] [CrossRef]
- Jacobs, J.J.; Keblusek, P.; Robanus-Maandag, E.; Kristel, P.; Lingbeek, M.; Nederlof, P.M.; van Welsem, T.; van de Vijver, M.J.; Koh, E.Y.; Daley, G.Q.; et al. Senescence bypass screen identifies TBX2, which represses Cdkn2a (p19(ARF)) and is amplified in a subset of human breast cancers. Nat. Genet. 2000, 26, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Ozenne, P.; Eymin, B.; Brambilla, E.; Gazzeri, S. The ARF tumor suppressor: Structure, functions and status in cancer. Int. J. Cancer 2010, 127, 2239–2247. [Google Scholar] [CrossRef] [PubMed]
- Pflaum, J.; Schlosser, S.; Müller, M. p53 Family and Cellular Stress Responses in Cancer. Front. Oncol. 2014, 4, 285. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, A.W.; Royds, J.A.; Jackson, P. The p53 story: Layers of complexity. Carcinogenesis 2005, 26, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Meek, D. Regulation of the p53 response and its relationship to cancer. Biochem. J. 2015, 469, 325–346. [Google Scholar] [CrossRef]
- Wang, B.; Xiao, Z.; Ko, H.L.; Ren, E.-C. The p53 response element and transcriptional repression. Cell Cycle 2010, 9, 870–879. [Google Scholar] [CrossRef]
- Wang, B.; Xiao, Z.; Ren, E. Redefining the p53 response element. Proc. Natl. Acad. Sci. USA 2009, 106, 14373–14378. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef]
- Harris, S.L.; Levine, A. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef]
- Miyashita, T.; Reed, J. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [PubMed]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, L.; Hwang, P.M.; Kinzler, K.W.; Vogelstein, B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 2001, 7, 673–682. [Google Scholar] [CrossRef]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef]
- Carvajal, L.A.; Manfredi, J. Another fork in the road—Life or death decisions by the tumour suppressor p53. EMBO Rep. 2013, 14, 414–421. [Google Scholar] [CrossRef]
- Leslie, P.L.; Franklin, D.A.; Liu, Y.; Zhang, Y. p53 Regulates the Expression of LRP1 and Apoptosis through a Stress Intensity-Dependent MicroRNA Feedback Loop. Cell Rep. 2018, 24, 1484–1495. [Google Scholar] [CrossRef]
- Paek, A.L.; Liu, J.C.; Loewer, A.; Forrester, W.C.; Lahav, G. Cell-to-Cell Variation in p53 Dynamics Leads to Fractional Killing. Cell 2016, 165, 631–642. [Google Scholar] [CrossRef]
- Feng, Z. p53 regulation of the IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring Harb. Perspect. Biol. 2010, 2, a001057. [Google Scholar] [CrossRef]
- Ghosh, A.; Stewart, D.; Matlashewski, G. Regulation of human p53 activity and cell localization by alternative splicing. Mol. Cell Biol. 2004, 24, 7987–7997. [Google Scholar] [CrossRef]
- Marcel, V.; Tran, P.L.; Sagne, C.; Martel-Planche, G.; Vaslin, L.; Teulade-Fichou, M.P.; Hall, J.; Mergny, J.L.; Hainaut, P.; Van Dyck, E. G-quadruplex structures in Tp53 intron 3: Role in alternative splicing and in production of p53 mRNA isoforms. Carcinogenesis 2011, 32, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Perriaud, L.; Marcel, V.; Sagne, C.; Favaudon, V.; Guedin, A.; De Rache, A.; Guetta, C.; Hamon, F.; Teulade-Fichou, M.P.; Hainaut, P.; et al. Impact of G-quadruplex structures and intronic polymorphisms rs17878362 and rs1642785 on basal and ionizing radiation-induced expression of alternative p53 transcripts. Carcinogenesis 2014, 35, 2706–2715. [Google Scholar] [CrossRef] [PubMed]
- Lazar, V.; Hazard, F.; Bertin, F.; Janin, N.; Bellet, D.; Bressac, B. Simple sequence repeat polymorphism within the p53 gene. Oncogene 1993, 8, 1703–1705. [Google Scholar] [PubMed]
- Sagne, C.; Marcel, V.; Amadou, A.; Hainaut, P.; Olivier, M.; Hall, J. A meta-analysis of cancer risk associated with the Tp53 intron 3 duplication polymorphism (rs17878362): Geographic and tumor-specific effects. Cell Death Dis. 2013, 4, e492. [Google Scholar] [CrossRef]
- Sagne, C.; Marcel, V.; Bota, M.; Martel-Planche, G.; Nobrega, A.; Palmero, E.I.; Perriaud, L.; Boniol, M.; Vagner, S.; Cox, D.G.; et al. Age at cancer onset in germline Tp53 mutation carriers: Association with polymorphisms in predicted G-quadruplex structures. Carcinogenesis 2014, 35, 807–815. [Google Scholar] [CrossRef]
- Morten, B.C.; Wong-Brown, M.W.; Scott, R.J.; Avery-Kiejda, K. The presence of the intron 3 16 bp duplication polymorphism of p53 (rs17878362) in breast cancer is associated with a low Delta40p53:p53 ratio and better outcome. Carcinogenesis 2016, 37, 81–86. [Google Scholar] [CrossRef]
- Johannes, G.; Carter, M.S.; Eisen, M.B.; Brown, P.O.; Sarnow, P. Identification of eukaryotic mRNAs that are translated at reduced cap binding complex eIF4F concentrations using a cDNA microarray. Proc. Natl. Acad. Sci. USA 1999, 96, 13118–13123. [Google Scholar] [CrossRef]
- Ray, P.S.; Grover, R.; Das, S. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep. 2006, 7, 404–410. [Google Scholar] [CrossRef]
- Yang, D.Q.; Halaby, M.J.; Zhang, Y. The identification of an internal ribosomal entry site in the 5′-untranslated region of p53 mRNA provides a novel mechanism for the regulation of its translation following DNA damage. Oncogene 2006, 25, 4613–4619. [Google Scholar] [CrossRef]
- Candeias, M.M.; Powell, D.J.; Roubalova, E.; Apcher, S.; Bourougaa, K.; Vojtesek, B.; Bruzzoni-Giovanelli, H.; Fahraeus, R. Expression of p53 and p53//47 are controlled by alternative mechanisms of messenger RNA translation initiation. Oncogene 2006, 25, 6936–6947. [Google Scholar] [CrossRef]
- Solomon, H.; Bräuning, B.; Fainer, I.; Ben-Nissan, G.; Rabani, S.; Goldfinger, N.; Moscovitz, O.; Shakked, Z.; Rotter, V.; Sharon, M. Post-translational regulation of p53 function through 20S proteasome-mediated cleavage. Cell Death Differ. 2017, 24, 2187–2198. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.; Vousden, K. Proteolytic cleavage of human p53 by calpain: A potential regulator of protein stability. Mol. Cell. Biol. 1997, 17, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Ota, A.; Nakao, H.; Sawada, Y.; Karnan, S.; Wahiduzzaman, M.; Inoue, T.; Kobayashi, Y.; Yamamoto, T.; Ishii, N.; Ohashi, T.; et al. Delta40p53alpha suppresses tumor cell proliferation and induces cellular senescence in hepatocellular carcinoma cells. J. Cell Sci. 2017, 130, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Hrstka, R.; Powell, D.J.; Kvardova, V.; Roubalova, E.; Bourougaa, K.; Candeias, M.M.; Sova, P.; Zak, F.; Fahraeus, R.; Vojtesek, B. The novel platinum (IV) complex LA-12 induces p53 and p53/47 responses that differ from the related drug, cisplatin. Anti-Cancer Drug 2008, 19, 369–379. [Google Scholar] [CrossRef]
- Takahashi, R.; Markovic, S.N.; Scrable, H. Dominant Effects of Δ40p53 on p53 Function and Melanoma Cell Fate. J. Investig. Dermatol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.S.; Zhao, H.W.; Qiao, L.X.; Shan, J.Q.; Hou, Q.S.; Chen, D.X.; Guo, H. Effect of Delta40p53 isoform on enhancing the pro-apoptotic function of p53 in tumor cells. Zhonghua Zhong Liu Za Zhi 2017, 39, 332–338. [Google Scholar]
- Melo Dos Santos, N.; de Oliveira, G.A.P.; Ramos Rocha, M.; Pedrote, M.M.; Diniz da Silva Ferretti, G.; Pereira Rangel, L.; Morgado-Diaz, J.A.; Silva, J.L.; Gimba, E.R.P. Loss of the p53 transactivation domain results in high amyloid aggregation of the Delta40p53 isoform in endometrial carcinoma cells. J. Biol. Chem. 2019, 294, 9430–9439. [Google Scholar] [CrossRef]
- Avery-Kiejda, K.A.; Morten, B.; Wong-Brown, M.W.; Mathe, A.; Scott, R. The relative mRNA expression of p53 isoforms in breast cancer is associated with clinical features and outcome. Carcinogenesis 2014, 35, 586–596. [Google Scholar] [CrossRef]
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Kenzelmann Broz, D.; Basak, S.; Park, E.J.; McLaughlin, M.E.; et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011, 145, 571–583. [Google Scholar] [CrossRef]
- Khan, D.; Katoch, A.; Das, A.; Sharathchandra, A.; Lal, R.; Roy, P.; Das, S.; Chattopadhyay, S.; Das, S. Reversible induction of translational isoforms of p53 in glucose deprivation. Cell Death Differ. 2015, 22, 1203–1218. [Google Scholar] [CrossRef]
- McLure, K.G.; Lee, P. How p53 binds DNA as a tetramer. EMBO J. 1998, 17, 3342–3350. [Google Scholar] [CrossRef] [PubMed]
- Graupner, V.; Schulze-Osthoff, K.; Essmann, F.; Janicke, R. Functional characterization of p53beta and p53gamma, two isoforms of the tumor suppressor p53. Cell Cycle 2009, 8, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Ohki, R.; Kawase, T.; Ohta, T.; Ichikawa, H.; Taya, Y. Dissecting functional roles of p53 N-terminal transactivation domains by microarray expression analysis. Cancer Sci. 2007, 98, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Hayman, L.; Chaudhry, W.R.; Revin, V.V.; Zhelev, N.; Bourdon, J. What is the potential of p53 isoforms as a predictive biomarker in the treatment of cancer? Expert Rev. Mol. Diagn. 2019, 19, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, G.; Berger, A.; Berger, R.; Zoric, A.; Braicu, E.I.; Reimer, D.; Fiegl, H.; Marth, C.; Zeimet, A.G.; Ulmer, H.; et al. The N-terminally truncated p53 isoform Delta40p53 influences prognosis in mucinous ovarian cancer. Int. J. Gynecol. Cancer 2012, 22, 372–379. [Google Scholar] [CrossRef]
- Oh, L.; Hainaut, P.; Blanchet, S.; Ariffin, H. Expression of p53 N-terminal isoforms in B-cell precursor acute lymphoblastic leukemia and its correlation with clinicopathological profiles. BMC Cancer 2020, 20, 110. [Google Scholar] [CrossRef]
- Takahashi, R.; Giannini, C.; Sarkaria, J.N.; Schroeder, M.; Rogers, J.; Mastroeni, D.; Scrable, H. p53 isoform profiling in glioblastoma and injured brain. Oncogene 2013, 32, 3165–3174. [Google Scholar] [CrossRef]
- Bischof, K.; Knappskog, S.; Stefansson, I.; McCormack, E.M.; Trovik, J.; Werner, H.M.J.; Woie, K.; Gjertsen, B.T.; Bjorge, L. High expression of the p53 isoform gamma is associated with reduced progression-free survival in uterine serous carcinoma. BMC Cancer 2018, 18, 684. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Miller, L.D.; Smeds, J.; George, J.; Vega, V.B.; Vergara, L.; Ploner, A.; Pawitan, Y.; Hall, P.; Klaar, S.; Liu, E.T.; et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc. Natl. Acad. Sci. USA 2005, 102, 13550–13555. [Google Scholar] [CrossRef]
- Coutant, C.; Rouzier, R.; Qi, Y.; Lehmann-Che, J.; Bianchini, G.; Iwamoto, T.; Hortobagyi, G.N.; Symmans, W.F.; Uzan, S.; Andre, F.; et al. Distinct p53 gene signatures are needed to predict prognosis and response to chemotherapy in ER-positive and ER-negative breast cancers. Clin. Cancer Res. 2011, 17, 2591–2601. [Google Scholar] [CrossRef] [PubMed]
- Knezovic Florijan, M.; Ozretic, P.; Bujak, M.; Pezze, L.; Ciribilli, Y.; Kastelan, Z.; Slade, N.; Hudolin, T. The role of p53 isoforms’ expression and p53 mutation status in renal cell cancer prognosis. In Urologic Oncology: Seminars and Original Investigations; Elsevier: Amsterdam, The Netherlands, 2019; Volume 37, p. 578-e1. [Google Scholar]
- Lasham, A.; Tsai, P.; Fitzgerald, S.J.; Mehta, S.Y.; Knowlton, N.S.; Braithwaite, A.W.; Print, C. Accessing a New Dimension in Tp53 Biology: Multiplex Long Amplicon Digital PCR to Specifically Detect and Quantitate Individual Tp53 Transcripts. Cancers 2020, 12, 769. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Dichtel-Danjoy, M.L.; Sagne, C.; Hafsi, H.; Ma, D.; Ortiz-Cuaran, S.; Olivier, M.; Hall, J.; Mollereau, B.; Hainaut, P.; et al. Biological functions of p53 isoforms through evolution: Lessons from animal and cellular models. Cell Death Differ. 2011, 18, 1815–1824. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, W.; Teresky, A.K.; Hernando, E.; Cordon-Cardo, C.; Levine, A. Declining p53 function in the aging process: A possible mechanism for the increased tumor incidence in older populations. Proc. Natl. Acad. Sci. USA 2007, 104, 16633–16638. [Google Scholar] [CrossRef]
- Chang, J.R.; Ghafouri, M.; Mukerjee, R.; Bagashev, A.; Chabrashvili, T.; Sawaya, B. Role of p53 in neurodegenerative diseases. Neurodegener. Dis. 2012, 9, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Tyner, S.D.; Venkatachalam, S.; Choi, J.; Jones, S.; Ghebranious, N.; Igelmann, H.; Lu, X.; Soron, G.; Cooper, B.; Brayton, C.; et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002, 415, 45–53. [Google Scholar] [CrossRef]
- Dumble, M.; Moore, L.; Chambers, S.M.; Geiger, H.; Van Zant, G.; Goodell, M.A.; Donehower, L. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood 2007, 109, 1736–1742. [Google Scholar] [CrossRef]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A.; Butel, J.S.; Allan, B. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Maier, B.; Gluba, W.; Bernier, B.; Turner, T.; Mohammad, K.; Guise, T.; Sutherland, A.; Thorner, M.; Scrable, H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004, 18, 306–319. [Google Scholar] [CrossRef]
- Costantini, C.; Scrable, H.; Puglielli, L. An aging pathway controls the TrkA to p75NTR receptor switch and amyloid beta-peptide generation. EMBO J. 2006, 25, 1997–2006. [Google Scholar] [CrossRef] [PubMed]
- Hinault, C.; Kawamori, D.; Liew, C.W.; Maier, B.; Hu, J.; Keller, S.R.; Mirmira, R.G.; Scrable, H.; Kulkarni, R. Delta40 Isoform of p53 controls beta-cell proliferation and glucose homeostasis in mice. Diabetes 2011, 60, 1210–1222. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.H.; Helfand, S. New tricks of an old molecule: Lifespan regulation by p53. Aging Cell 2006, 5, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J.; Bhaumik, D. Two faces of p53: Aging and tumor suppression. Nucleic Acids Res. 2007, 35, 7475–7484. [Google Scholar] [CrossRef]
- Ungewitter, E.; Scrable, H. Antagonistic pleiotropy and p53. Mech. Ageing Dev. 2009, 130, 10–17. [Google Scholar] [CrossRef]
- Hebert, L.E.; Scherr, P.A.; Bienias, J.L.; Bennett, D.A.; Evans, D.A. Alzheimer Disease in the US Population. Arch. Neurol. 2003, 60, 1119–1122. [Google Scholar] [CrossRef]
- Lee, V.M.-Y.; Goedert, M.; Trojanowski, J. Neurodegenerative Tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Kurz, A.; Perneczky, R. Neurobiology of cognitive disorders. Curr. Opin. Psychiatry 2009, 22, 546–551. [Google Scholar] [CrossRef]
- Loy, C.T.; Schofield, P.R.; Turner, A.M.; Kwok, J.B.J. Genetics of dementia. Lancet 2014, 383, 828–840. [Google Scholar] [CrossRef]
- Tanzi, R. The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006296. [Google Scholar] [CrossRef]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Duyckaerts, C.; Potier, M.C.; Delatour, B. Alzheimer disease models and human neuropathology: Similarities and differences. Acta Neuropathol. 2008, 115, 5–38. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, K.; Vogt, D.L.; Liang, M.; Shen, Y.; Lamb, B.T.; Pimplikar, S. Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc. Natl. Acad. Sci. USA 2009, 106, 18367–18372. [Google Scholar] [CrossRef] [PubMed]
- Götz, J.; Streffer, J.R.; David, D.; Schild, A.; Hoerndli, F.; Pennanen, L.; Kurosinski, P.; Chen, F. Transgenic animal models of Alzheimer’s disease and related disorders: Histopathology, behavior and therapy. Mol. Psychiatry 2004, 9, 664–683. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef]
- SantaCruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau Suppression in a Neurodegenerative Mouse Model Improves Memory Function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef]
- Li, M.; Pehar, M.; Liu, Y.; Bhattacharyya, A.; Zhang, S.C.; O’Riordan, K.J.; Burger, C.; D’Adamio, L.; Puglielli, L. The amyloid precursor protein (APP) intracellular domain regulates translation of p44, a short isoform of p53, through an IRES-dependent mechanism. Neurobiol. Aging 2015, 36, 2725–2736. [Google Scholar] [CrossRef]
- Aloni-Grinstein, R.; Shetzer, Y.; Kaufman, T.; Rotter, V. p53: The barrier to cancer stem cell formation. FEBS Lett. 2014, 588, 2580–2589. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Lowe, S. Stem cells: The promises and perils of p53. Nature 2009, 460, 1085–1086. [Google Scholar] [CrossRef]
- Jain, A.K.; Barton, M. p53: Emerging roles in stem cells, development and beyond. Development 2018, 145, dev158360. [Google Scholar] [CrossRef]
- Davidson, W.; Kari, C.; Ren, Q.; Daroczi, B.; Dicker, A.; Rodeck, U. Differential regulation of p53 function by the N-terminal DeltaNp53 and Delta113p53 isoforms in zebrafish embryos. BMC Dev. Biol. 2010, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Momand, J.; Finlay, C. The p53 tumour suppressor gene. Nature 1991, 351, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Gorska, A.; Swiatkowska, A.; Dutkiewicz, M.; Ciesiolka, J. Modulation of p53 Expression Using Antisense Oligonucleotides Complementary to the 5′-Terminal Region of p53 mRNA In Vitro and in the Living Cells. PLoS ONE 2013, 8, e78863. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef]
- Erba, H.P.; Becker, P.S.; Shami, P.J.; Grunwald, M.R.; Flesher, D.L.; Zhu, M.; Rasmussen, E.; Henary, H.A.; Anderson, A.A.; Wang, E. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 2019, 3, 1939–1949. [Google Scholar] [CrossRef]
- Jones, C.L.; Njomen, E.; Sjögren, B.; Dexheimer, T.S.; Tepe, J. Small Molecule Enhancement of 20S Proteasome Activity Targets Intrinsically Disordered Proteins. ACS Chem. Biol. 2017, 12, 2240–2247. [Google Scholar] [CrossRef]
- Huber, P.E.; Pfisterer, P. In vitro and in vivo transfection of plasmid DNA in the Dunning prostate tumor R3327-AT1 is enhanced by focused ultrasound. Gene Ther. 2000, 7, 1516–1525. [Google Scholar] [CrossRef]
- Jin, L.; Zeng, X.; Liu, M.; Deng, Y.; He, N. Current progress in gene delivery technology based on chemical methods and nano-carriers. Theranostics 2014, 4, 240–255. [Google Scholar] [CrossRef]
- Riley, M.K.; Vermerris, W. Recent Advances in Nanomaterials for Gene Delivery—A Review. Nanomaterials 2017, 7, 94. [Google Scholar] [CrossRef]
- Avery-Kiejda, K.A.; Bowden, N.A.; Croft, A.J.; Scurr, L.L.; Kairupan, C.F.; Ashton, K.A.; Talseth-Palmer, B.A.; Rizos, H.; Zhang, X.D.; Scott, R.J.; et al. p53 in human melanoma fails to regulate target genes associated with apoptosis and the cell cycle and may contribute to proliferation. BMC Cancer 2011, 11, 203. [Google Scholar] [CrossRef]
- Joruiz, S.M.; Bourdon, J.-C. p53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016, 6, a026039. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, L.; Huang, X. Genome modification by CRISPR/Cas9. FEBS J. 2014, 281, 5186–5193. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Cancer | Cell Line | Role/Mechanism of Action | Approach Used to Modulate Δ40p53 Expression | Reference |
|---|---|---|---|---|
| Hepatocellular carcinoma | HuH-1 (TP53WT), HepG2 (TP53WT), HEP3B (TP53−/−, HuH-7 (TP53Y220C), PLC/PRF/5 (TP53R249S) human hepatocellular carcinoma cell lines | Tumour suppressor role Promotion of cellular senescence (increase in percentage of SA-β-gal-positive cells) Suppression of clonogenicity Inhibition of cell proliferation Cell cycle arrest at G1 phase Increase in p21WAF1, HDM2, FAS, IL-8 and GADD45A expression Increase in protein half-life of FLp53 | Vector targeted deletion of TP53 exon 2 Retroviral plasmid transduction with constructs containing ORF of Δ40p53 Knockdown of p53 by using retroviral plasmids and CRISPR/Cas9 Inhibition of HDM2 (MG132 inhibitor) | [64] |
| Melanoma | Mel-FH, Mel-MC, Mel-CV, Mel-RM, MM200, Me1007, Me4405, IgR3, MM186, Mel-KN, Mel-JD, MM283, Mel 4.1, Mel-FC, MM415, MM486, MV3, Sk-Mel-110 and Sk-Mel-28 melanoma cell lines | Expressed at low levels in normal cells but highly expressed in tumour cells Overexpression inhibited transcriptional activation of PUMA and p21WAF1 at the basal level and following treatment with DNA damaging agents | Transduction of construct pCIN4.p47 containing the ORF of Δ40p53 | [5] |
| A375 melanoma cell line | Increase in number of dead cells Reduction of skin cancer cell viability Induction of apoptosis Increase in p53, p21WAF1 and PIDD gene expression Creation of nuclear tetramers with endogenous, serine 15–phosphorylated p53 | Transduction with a construct containing the ORF of Δ40p53 | [66] | |
| Osteosarcoma | Saos-2 osteosarcoma cell line | Formation of oligomers with p53 Inhibits transcriptional activation of target genes when expressed in excess of FLp53 | Transfection using pcDNA3-TAp53 (mut40—ATG40 > TTG) and pcDNA3-Δ40p53 vectors | [20] |
| Lung cancer | H1299 lung cancer cell line (p53 null) | Formation of oligomers with p53 Suppression of FL-p53 transcriptional activity in an incremental manner Reduction of p21WAF1 protein expression when Δ40p53 is overexpressed Enhancement of the pro-apoptotic activity of p53 | Transfection using pcDNA3-TAp53 (mut40—ATG40 > TTG) and pcDNA3-Δ40p53 vectors | [20] |
| Colon cancer | HCT116-p53(–/–) (endogenous Δ40p53 expression), HCT116-p53(+/+) (wild-type p53) colon carcinoma cell lines | Enhancement of the pro-apoptotic activity of p53 | Transfection with adenoviral delivery of a Δ40p53 plasmid | [67] |
| Endometrial cancer | Ishikawa, RL-95–2, AN3CA and KLE endometrial cancer cell lines | Major component of p53 amyloid aggregates | None | [68] |
| Breast cancer | p53 MT cell lines (T47D, MDA-MB-231, HMT-3522-T42) and wtp53 cell lines (ZR-75-1 and MCF7) | Upregulated in tumour cell lines when compared to normal breast epithelial cell lines | None | [69] |
| Mechanism | Effect on ∆40p53 | Effect on FLp53 * | Reference | |
|---|---|---|---|---|
| Targeting alternative splicing (retention of intron 2) | siRNA targeting transcripts that retain intron 2 | ↓ | ? | Hypothetical |
| Site directed mutagenesis of G in intron 3 | ↑ | ↓ (mRNA) | [52] | |
| stabilisation of G4 through 360A, PhenDC, K+ | ↓ | ↑ (mRNA) | [52] | |
| Damage to non-G4 transcripts through ionizing radiation | ↓ | ? | [52] | |
| Destabilisation of G4 through Na+ | ↑ | ? | [52] | |
| CRISPR interference inhibiting translation of p53 transcripts retaining intron 2 | ↓ | ? | Hypothetical | |
| CRISPR/Cas9 knockout of intron 2 | ↓ | ? | Hypothetical | |
| Targeting translation via IRES (exon 4) | Transfection with constructs containing a CTG substitution at 2nd ATG | ↓ | ↑ (protein) | [17] |
| Transduction with constructs containing ORF of ∆40p53 | ↑ | ? [5]; none (protein) in p53-null cells [50]; ↑ (protein) [62,64]; | [5,51,64,66] | |
| Transfection with constructs containing a hairpin structure near/over first ATG | ↑ | ↓ (protein) | [61] | |
| Transfection with construct containing stop codon upstream of IRES | ↑ | ↓ (protein) | [61] | |
| Antisense oligonucleotides targeting 5’ end of the p53 transcript | ↑/↓ | ↑/↓ (protein) | [115] | |
| Tunicamycin or thapsigargin-induced ER stress | ↑ | ↑ (protein) | [61] | |
| CRISPR/Cas9 homology-directed repair of the 1st or 2nd ATG | ↑/↓ | ? | Hypothetical | |
| Transfection with constructs containing CTG Substitution at 1st ATG | ↑ | ↓ (protein) | [17] | |
| Transduction leading to deletion of the second ATG | ↑ | None (protein) | [21] | |
| CRISPR/Cas9 excision of exon 4 | ↓ | ↓ (protein) | [64] | |
| Other | Nutlin-3 and MG132 inhibition of HDM2 ubiquitination of FLp53 | ↑;↑FLp53:∆40p53 | ↑ (protein) | [17,64,116] |
| Transduction leading to deletion of exon 2 | ↑ | ? | [64] | |
| 20S proteasome enhancement through manipulated chlorpromazine | ↑ | ? | Hypothetical |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steffens Reinhardt, L.; Zhang, X.; Wawruszak, A.; Groen, K.; De Iuliis, G.N.; Avery-Kiejda, K.A. Good Cop, Bad Cop: Defining the Roles of Δ40p53 in Cancer and Aging. Cancers 2020, 12, 1659. https://doi.org/10.3390/cancers12061659
Steffens Reinhardt L, Zhang X, Wawruszak A, Groen K, De Iuliis GN, Avery-Kiejda KA. Good Cop, Bad Cop: Defining the Roles of Δ40p53 in Cancer and Aging. Cancers. 2020; 12(6):1659. https://doi.org/10.3390/cancers12061659
Chicago/Turabian StyleSteffens Reinhardt, Luiza, Xiajie Zhang, Anna Wawruszak, Kira Groen, Geoffry N. De Iuliis, and Kelly A. Avery-Kiejda. 2020. "Good Cop, Bad Cop: Defining the Roles of Δ40p53 in Cancer and Aging" Cancers 12, no. 6: 1659. https://doi.org/10.3390/cancers12061659
APA StyleSteffens Reinhardt, L., Zhang, X., Wawruszak, A., Groen, K., De Iuliis, G. N., & Avery-Kiejda, K. A. (2020). Good Cop, Bad Cop: Defining the Roles of Δ40p53 in Cancer and Aging. Cancers, 12(6), 1659. https://doi.org/10.3390/cancers12061659