Hereditary Gastric and Breast Cancer Syndromes Related to CDH1 Germline Mutation: A Multidisciplinary Clinical Review

 ,
,  ,
,  ,
,  , ,
, ,  ,
, 
 ,
,  , , ,
, , ,  add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Hereditary Diffuse Gastric Cancer
2.1. Environmental Factors and GC
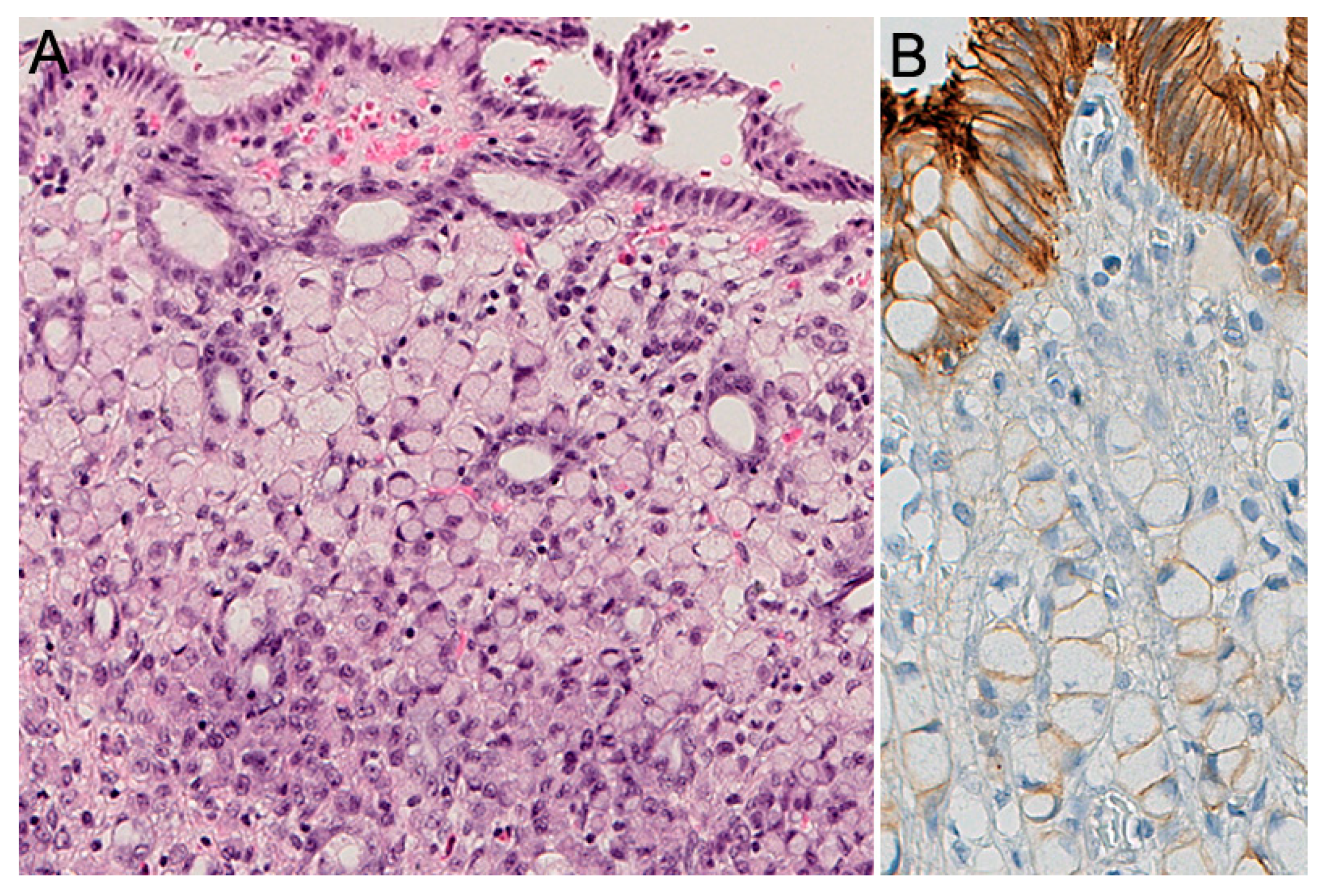
2.2. Pathology of HDGC
2.3. Histopathology of Prophylactic Gastrectomy
2.4. Histopathology: Advanced HDGC
2.5. Histochemical and Immunohistochemical Stains
2.6. Endoscopy
2.7. Prophylactic Gastrectomy
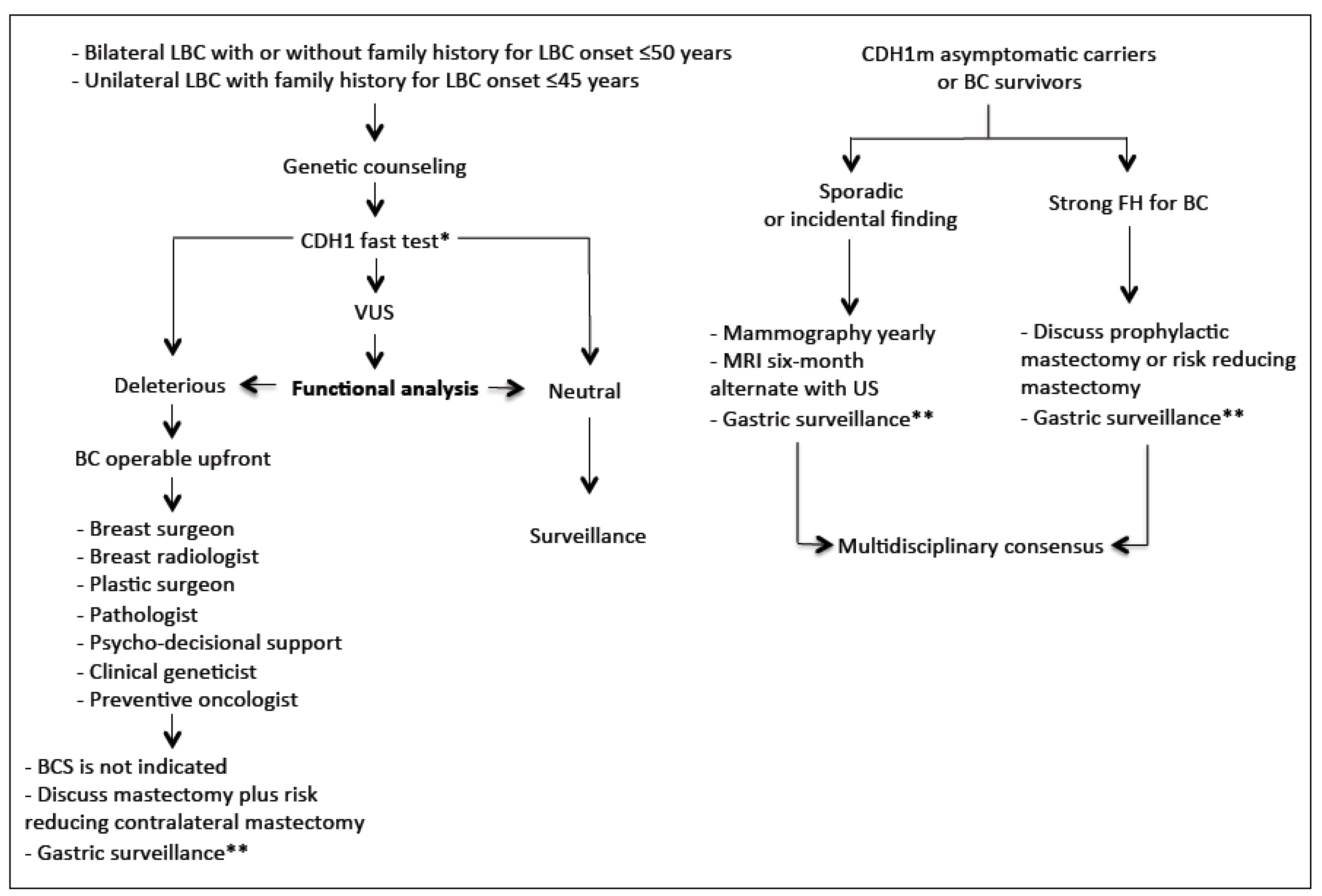
3. Hereditary Lobular Breast Cancer (HLBC)
3.1. Definition
3.2. CDH1 Screening: Preliminary Considerations
3.3. Pathology
3.4. Breast Imaging
3.5. Surgical Management
3.6. Post-Mastectomy Breast Reconstruction
4. Common Managements
4.1. Genetic Counseling
4.2. Psychological Counseling
4.3. CDH1 Missense Variants: Challenging Routine Laboratory Tests
4.4. Bioimaging Strategies to Identify Aberrant E-Cadherin Expression Signatures
5. Others
5.1. Clinical Management of CDH1 Carriers without a Family History of GC and LBC
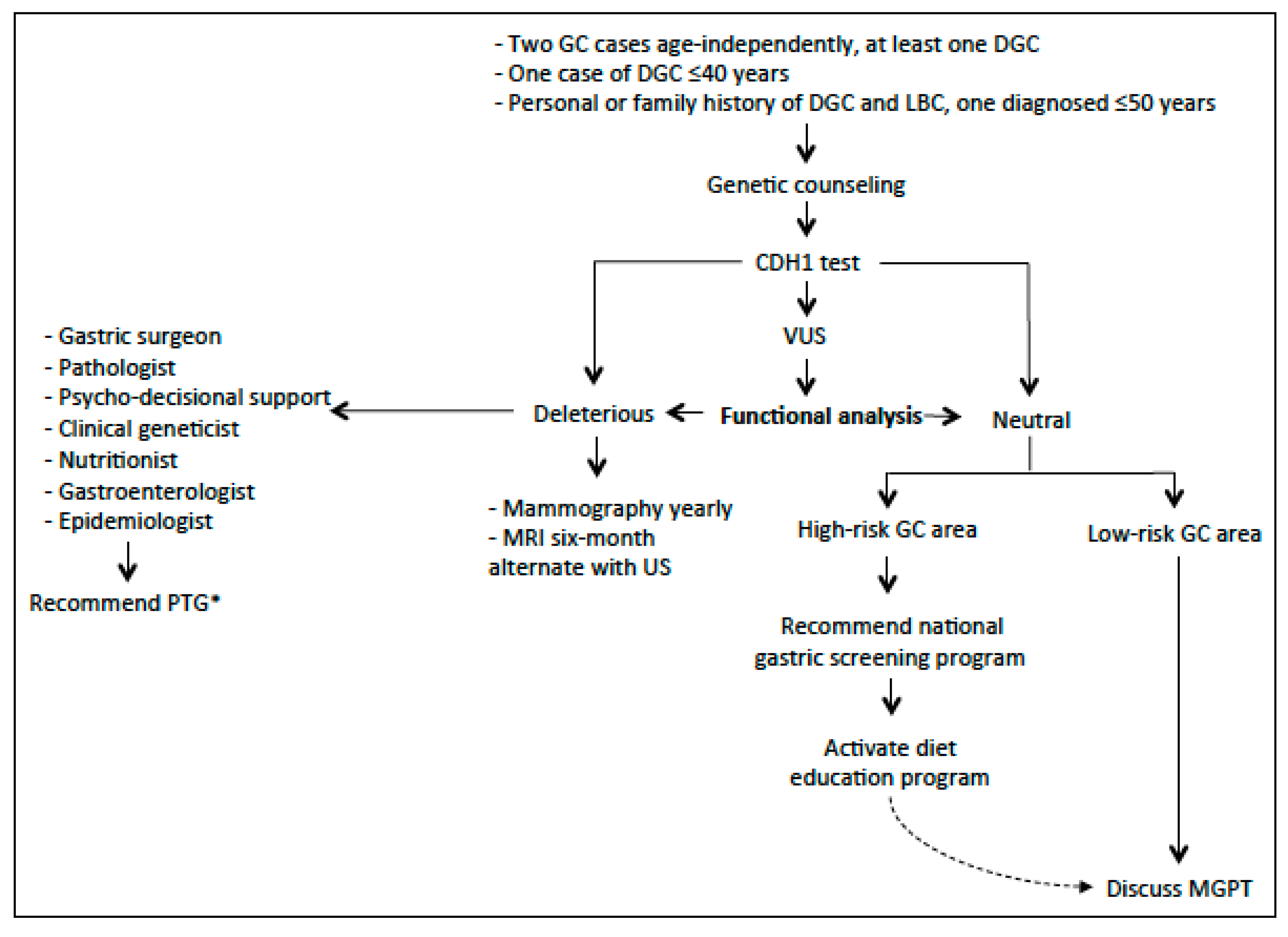
5.2. High and Low-Risk Geographical Regions for GC: Impact on Clinical Management
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guilford, P.; Hopkins, J.; Harraway, J.; McLeod, M.; McLeod, N.; Harawira, P.; Taite, H.; Scoular, R.; Miller, A.; Reeve, A.E. E-cadherin germline mutations in familial gastric cancer. Nature 1998, 392, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.G. Familial gastric cancer. N. Z. Med. J. 1964, 63, 287–296. [Google Scholar]
- Caldas, C.; Carneiro, F.; Lynch, H.T.; Yokota, J.; Wiesner, G.L.; Powell, S.M.; Lewis, F.R.; Huntsman, D.G.; Pharoah, P.D.; Jankowski, J.; et al. Familial gastric cancer: Overview and guidelines for management. J. Med. Genet. 1999, 36, 873–880. [Google Scholar]
- Kaurah, P.; Macmillan, A.; Boyd, N.; Senz, J.; De Luca, A.; Chun, N.; Suriano, G.; Zaor, S.; Van Manen, L.; Gilpin, C.; et al. Founder and Recurrent CDH1 Mutations in Families with Hereditary Diffuse Gastric Cancer. JAMA 2007, 297, 2360–2372. [Google Scholar] [CrossRef] [PubMed]
- Brooks-Wilson, A.R.; Kaurah, P.; Suriano, G.; Leach, S.; Senz, J.; Grehan, N.; Butterfield, Y.S.N.; Jeyes, J.; Schinas, J.; Bacani, J.; et al. Germline E-cadherin mutations in hereditary diffuse gastric cancer: Assessment of 42 new families and review of genetic screening criteria. J. Med. Genet. 2004, 41, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Van Der Post, R.S.; Vogelaar, I.; Carneiro, F.; Guilford, P.; Huntsman, D.; Hoogerbrugge, N.; Caldas, C.; Schreiber, K.E.C.; Hardwick, R.H.; Ausems, M.G.E.M.; et al. Hereditary diffuse gastric cancer: Updated clinical guidelines with an emphasis on germlineCDH1mutation carriers. J. Med. Genet. 2015, 52, 361–374. [Google Scholar] [CrossRef]
- Corso, G.; Figueiredo, J.; La Vecchia, C.; Veronesi, P.; Pravettoni, G.; Macis, D.; Karam, R.; Gullo, R.L.; Provenzano, E.; Toesca, A.; et al. Hereditary lobular breast cancer with an emphasis on E-cadherin genetic defect. J. Med. Genet. 2018, 55, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Hansford, S.; Kaurah, P.; Li-Chang, H.; Woo, M.; Senz, J.; Pinheiro, H.; A Schrader, K.; Schaeffer, D.F.; Shumansky, K.; Zogopoulos, G.; et al. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 mutations and beyond. JAMA Oncol. 2015, 1, 23–32. [Google Scholar] [CrossRef]
- Roberts, M.E.; Ranola, J.M.O.; Marshall, M.L.; Susswein, L.R.; Graceffo, S.; Bohnert, K.; Tsai, G.; Klein, R.T.; Hruska, K.S.; Shirts, B.H. Comparison of CDH1 Penetrance Estimates in Clinically Ascertained Families vs Families Ascertained for Multiple Gastric Cancers. JAMA Oncol. 2019, 5, 1325. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Lyons, K.; Le, L.C.; Pham, Y.T.-H.; Borron, C.; Park, J.Y.; Tran, C.T.; Tran, T.V.; Tran, H.T.-T.; Vu, K.T.; Do, C.D.; et al. Gastric cancer: Epidemiology, biology, and prevention: A mini review. Eur. J. Cancer Prev. 2019, 28, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Pelucchi, C.; Lunet, N.; Adany, S.B.C.R.; Zhang, Z.; Praud, D.; Boffetta, P.; Levi, F.; Matsuo, K.; Ito, H.; Hu, J.; et al. The stomach cancer pooling (StoP) project: Study design and presentation. Eur. J. Cancer Prev. 2015, 24, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Praud, D.; Rota, M.; Pelucchi, C.; Bertuccio, P.; Rosso, T.; Galeone, C.; Zhang, Z.; Matsuo, K.; Ito, H.; Hu, J.; et al. Cigarette smoking and gastric cancer in the Stomach Cancer Pooling (StoP) Project. Eur. J. Cancer Prev. 2018, 27, 124–133. [Google Scholar] [CrossRef]
- Rota, M.; Pelucchi, C.; Bertuccio, P.; Matsuo, K.; Zhang, Z.; Ito, H.; Hu, J.; Johnson, K.C.; Palli, D.; Ferraroni, M.; et al. Alcohol consumption and gastric cancer risk-A pooled analysis within the StoP project consortium. Int. J. Cancer 2017, 141, 1950–1962. [Google Scholar] [CrossRef] [PubMed]
- Rota, M.; Alicandro, G.; Pelucchi, C.; Bonzi, R.; Bertuccio, P.; Hu, J.; Zhang, Z.; Johnson, K.C.; Palli, D.; Ferraroni, M.; et al. Education and gastric cancer risk—An individual participant data meta-analysis in the StoP project consortium. Int. J. Cancer 2019, 146, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Ferro, A.; Rosato, V.; Rota, M.; Costa, A.R.; Morais, S.; Pelucchi, C.; Johnson, K.C.; Hu, J.; Palli, D.; Ferraroni, M.; et al. Meat intake and risk of gastric cancer in the Stomach cancer Pooling (StoP) project. Int. J. Cancer 2019, 147, 45–55. [Google Scholar] [CrossRef]
- Bertuccio, P.; Alicandro, G.; Rota, M.; Pelucchi, C.; Bonzi, R.; Galeone, C.; Bravi, F.; Johnson, K.C.; Hu, J.; Palli, D.; et al. Citrus fruit intake and gastric cancer: The stomach cancer pooling (StoP) project consortium. Int. J. Cancer 2019, 144, 2936–2944. [Google Scholar] [CrossRef]
- Carneiro, F.; Huntsman, D.G.; Smyrk, T.C.; A Owen, D.; Seruca, R.; Pharoah, P.; Caldas, C.; Sobrinho-Simões, M. Model of the early development of diffuse gastric cancer in E-cadherin mutation carriers and its implications for patient screening. J. Pathol. 2004, 203, 681–687. [Google Scholar] [CrossRef]
- Carneiro, F.; Guilford, P.; Oliveira, C.; van der Post, R.S. Hereditary Diffuse Gastric Cancer. In WHO Classification of Tumours Editorial Board. Digestive System Tumours, 5th ed.; WHO classification of tumours series; International Agency for Research on Cancer: Lyon, France, 2019; Volume 1. [Google Scholar]
- Gullo, I.; Devezas, V.; Baptista, M.; Garrido, L.; Castedo, S.; Morais, R.; Wen, X.; Rios, E.; Pinheiro, J.; Pinto-Ribeiro, I.; et al. Phenotypic heterogeneity of hereditary diffuse gastric cancer: Report of a family with early-onset disease. Gastrointest. Endosc. 2018, 87, 1566–1575. [Google Scholar] [CrossRef]
- Thompson, I.W.; Day, D.W.; A Wright, N. Subnuclear vacuolated mucous cells: A novel abnormality of simple mucin-secreting cells of non-specialized gastric mucosa and Brunner’s glands. Histopathology 1987, 11, 1067–1081. [Google Scholar] [CrossRef]
- Wang, K.; Weinrach, D.; Lal, A.; Musunuri, S.; Ramirez, J.; Ozer, O.; Keh, P.; Rao, M.S. Signet-ring cell change versus signet-ring cell carcinoma: A comparative analysis. Am. J. Surg. Pathol. 2003, 27, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.P.; Gullo, I.; Wen, X.; Devezas, V.; Baptista, M.; Oliveira, C.; Carneiro, F. Pathological features of total gastrectomy specimens from asymptomatic hereditary diffuse gastric cancer patients and implications for clinical management. Histopathology 2018, 73, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Wickremeratne, T.; Lee, C.H.; Kirk, J.; Charlton, A.; Thomas, G.; John, G.K. Prophylactic gastrectomy in a 16-year-old. Eur. J. Gastroenterol. Hepatol. 2014, 26, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.; Murrell, A.; Ito, Y.; Maia, A.-T.; Hyland, S.; Oliveira, C.; Save, V.; Carneiro, F.; Paterson, A.; Grehan, N.; et al. Mechanisms and sequelae of E-cadherin silencing in hereditary diffuse gastric cancer. J. Pathol. 2008, 216, 295–306. [Google Scholar] [CrossRef]
- Blair, V.; Martin, I.; Shaw, D.; Winship, I.; Kerr, D.; Arnold, J.; Harawira, P.; McLeod, M.; Parry, S.; Charlton, A.; et al. Hereditary Diffuse Gastric Cancer: Diagnosis and Management. Clin. Gastroenterol. Hepatol. 2006, 4, 262–275. [Google Scholar] [CrossRef]
- Charlton, A.; Blair, V.; Shaw, D.; Parry, S.; Guilford, P.; Martin, I.G. Hereditary diffuse gastric cancer: Predominance of multiple foci of signet ring cell carcinoma in distal stomach and transitional zone. Gut 2004, 53, 814–820. [Google Scholar] [CrossRef]
- Chun, Y.S.; Lindor, N.M.; Smyrk, T.C.; Petersen, B.T.; Burgart, L.J.; Guilford, P.J.; Donohue, J.H. Germline E-cadherin gene mutations: Is prophylactic total gastrectomy indicated? Cancer 2001, 92, 181–187. [Google Scholar] [CrossRef]
- Huntsman, D.G.; Carneiro, F.; Lewis, F.R.; MacLeod, P.M.; Hayashi, A.; Monaghan, K.G.; Maung, R.; Seruca, R.; Jackson, C.E.; Caldas, C. Early Gastric Cancer in Young, Asymptomatic Carriers of Germ-Line E-Cadherin Mutations. N. Engl. J. Med. 2001, 344, 1904–1909. [Google Scholar] [CrossRef]
- Rogers, W.M.; Dobo, E.; Norton, J.A.; Van Dam, J.; Jeffrey, R.B.; Huntsman, D.G.; Kingham, K.; Chun, N.; Ford, J.M.; Longacre, T.A. Risk-reducing Total Gastrectomy for Germline Mutations in E-cadherin (CDH1): Pathologic Findings with Clinical Implications. Am. J. Surg. Pathol. 2008, 32, 799–809. [Google Scholar] [CrossRef]
- Fujita, H.; Lennerz, J.K.; Chung, D.C.; Patel, D.; Deshpande, V.; Yoon, S.S.; Lauwers, G.Y. Endoscopic Surveillance of Patients with Hereditary Diffuse Gastric Cancer: Biopsy recommendations after topographic distribution of cancer foci in a series of 10 CDH1-mutated gastrectomies. Am. J. Surg. Pathol. 2012, 36, 1709–1717. [Google Scholar] [CrossRef]
- Pandalai, P.K.; Lauwers, G.Y.; Chung, D.C.; Patel, D.; Yoon, S.S. Prophylactic total gastrectomy for individuals with germline CDH1 mutation. Surgery 2011, 149, 347–355. [Google Scholar] [CrossRef]
- Bardram, L.; Hansen, T.V.O.; Gerdes, A.-M.; Timshel, S.; Friis-Hansen, L.; Federspiel, B. Prophylactic total gastrectomy in hereditary diffuse gastric cancer: Identification of two novel CDH1 gene mutations—A clinical observational study. Fam. Cancer 2014, 13, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, F.; Oliveira, C.; Suriano, G.; Seruca, R. Molecular pathology of familial gastric cancer, with an emphasis on hereditary diffuse gastric cancer. J. Clin. Pathol. 2007, 61, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.C.; Di Pietro, M.; O’Donovan, M.; Richardson, S.; Debiram, I.; Dwerryhouse, S.; Hardwick, R.H.; Tischkowitz, M.; Caldas, C.; Ragunath, K.; et al. Prospective cohort study assessing outcomes of patients from families fulfilling criteria for hereditary diffuse gastric cancer undergoing endoscopic surveillance. Gastrointest. Endosc. 2014, 80, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Frebourg, T.; Oliveira, C.; Hochain, P.; Karam, R.; Manouvrier, S.; Graziadio, C.; Vekemans, M.; Hartmann, A.; Baert-Desurmont, S.; Aandrelex, C.; et al. Cleft lip/palate and CDH1/E-cadherin mutations in families with hereditary diffuse gastric cancer. J. Med. Genet. 2006, 43, 138–142. [Google Scholar] [CrossRef]
- Kluijt, I.; Siemerink, E.J.; Ausems, M.G.; Van Os, T.A.; De Jong, D.; Correia, J.S.; Van Krieken, J.H.J.; Ligtenberg, M.J.; Figueiredo, J.; Van Riel, E.; et al. CDH1-related hereditary diffuse gastric cancer syndrome: Clinical variations and implications for counseling. Int. J. Cancer 2011, 131, 367–376. [Google Scholar] [CrossRef]
- Jacobs, M.F.; Dust, H.; Koeppe, E.S.; Wong, S.; Mulholland, M.; Choi, E.-Y.; Appelman, H.; Stoffel, E.M. Outcomes of Endoscopic Surveillance in Individuals with Genetic Predisposition to Hereditary Diffuse Gastric Cancer. Gastroenterology 2019, 157, 87–96. [Google Scholar] [CrossRef]
- Mills, S.E. Histology for Pathologists, 3rd ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Van Der Kaaij, R.T.; Van Kessel, J.P.; Van Dieren, J.M.; Snaebjörnsson, P.; Balagué, O.; Van Coevorden, F.; Van Der Kolk, L.E.; Sikorska, K.; Cats, A.; Van Sandick, J.W. Outcomes after prophylactic gastrectomy for hereditary diffuse gastric cancer. Br. J. Surg. 2018, 105, e176–e182. [Google Scholar] [CrossRef]
- Lee, A.F.; Rees, H.; Owen, D.A.; Huntsman, D.G. Periodic Acid-Schiff Is Superior to Hematoxylin and Eosin for Screening Prophylactic Gastrectomies from CDH1 Mutation Carriers. Am. J. Surg. Pathol. 2010, 34, 1007–1013. [Google Scholar] [CrossRef]
- Van Der Post, R.S.; Gullo, I.; Oliveira, C.; Tang, L.H.; Grabsch, H.I.; O’Donovan, M.; Fitzgerald, R.C.; Van Krieken, J.H.J.; Carneiro, F. Histopathological, Molecular, and Genetic Profile of Hereditary Diffuse Gastric Cancer: Current Knowledge and Challenges for the Future. Adv. Exp. Med. Biol. 2016, 908, 371–391. [Google Scholar] [CrossRef]
- Lee, H.E.; Smyrk, T.C.; Zhang, L. Histologic and immunohistochemical differences between hereditary and sporadic diffuse gastric carcinoma. Hum. Pathol. 2018, 74, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Godwin, T.D.; Kelly, S.T.; Brew, T.P.; Bougen-Zhukov, N.M.; Single, A.B.; Chen, A.; Stylianou, C.E.; Harris, L.D.; Currie, S.K.; Telford, B.J.; et al. E-cadherin-deficient cells have synthetic lethal vulnerabilities in plasma membrane organisation, dynamics and function. Gastric Cancer 2018, 22, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Van Der Post, R.S.; Oliveira, C.; Guilford, P.; Carneiro, F. Hereditary gastric cancer: What’s new? Update 2013–2018. Fam. Cancer 2019, 18, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Van Dieren, J.M.; Kodach, L.L.; Cats, A. Targeted vs Random Biopsies in Surveillance Endoscopy in Hereditary Diffuse Gastric Cancer Syndrome. Clin. Gastroenterol. Hepatol. 2020, 18, 1647–1648. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M.; Adar, T.; Patel, D.; Lauwers, G.Y.; Yoon, S.S.; Mullen, J.T.; Chung, D.C. Surveillance Endoscopy in the Management of Hereditary Diffuse Gastric Cancer Syndrome. Clin. Gastroenterol. Hepatol. 2019. [Google Scholar] [CrossRef]
- Mi, E.Z.; di Pietro, M.; O’Donovan, M.; Mi, E.M.; Hardwick, R.; Ziauddeen, H.; Fletcher, P.; Caldas, C.; Ragunath, K. A comparative study of endoscopic surveillance in hereditary diffuse gastric cancer according to CDH1 mutation status. Gastrointest. Endosc. 2018, 8, 408–418. [Google Scholar] [CrossRef]
- Artifon, E.L.D.A.; Marinho, F.R.T. Endoscopic screening for hereditary diffuse gastric cancer: One size does not fit all. Gastrointest. Endosc. 2018, 87, 405–407. [Google Scholar] [CrossRef]
- Van Dieren, J.M.; Kodach, L.L.; Hartog, P.D.; Van Der Kolk, L.E.; Sikorska, K.; Van Velthuysen, M.-L.F.; Van Sandick, J.W.; Koemans, W.J.; Snaebjornsson, P.; Cats, A. Gastroscopic surveillance with targeted biopsies compared with random biopsies in CDH1 mutation carriers. Endoscopy 2020. [Google Scholar] [CrossRef]
- Goetz, M. Characterization of lesions in the stomach: Will confocal laser endomicroscopy replace the pathologist? Best Pract. Res. Clin. Gastroenterol. 2015, 29, 589–599. [Google Scholar] [CrossRef]
- Laszkowska, M.; Silver, E.R.; Schrope, B.; Kastrinos, F.; Wang, T.C.; Hur, C. Optimal Timing of Total Gastrectomy to Prevent Diffuse Gastric Cancer in Individuals with Pathogenic Variants in CDH1. Clin. Gastroenterol. Hepatol. 2020, 18, 822–829. [Google Scholar] [CrossRef]
- Xicola, R.M.; Li, S.; Rodriguez, N.; Reinecke, P.; Karam, R.; Speare, V.; Black, M.H.; LaDuca, H.; Llor, X. Clinical features and cancer risk in families with pathogenic CDH1 variants irrespective of clinical criteria. J. Med. Genet. 2019, 56, 838–843. [Google Scholar] [CrossRef]
- Kumar, S.; Long, J.M.; Ginsberg, G.G.; Katona, B.W. The role of endoscopy in the management of hereditary diffuse gastric cancer syndrome. World J. Gastroenterol. 2019, 25, 2878–2886. [Google Scholar] [CrossRef]
- DiBrito, S.R.; Blair, A.B.; Prasath, V.; Habibi, M.; Harmon, J.W.; Duncan, M.D. Total Gastrectomy for CDH-1 Mutation Carriers: An Institutional Experience. J. Surg. Res. 2020, 247, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Lehnert, T.; Buhl, K. Techniques of reconstruction after total gastrectomy for cancer. Br. J. Surg. 2004, 91, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Corso, G.; Figueiredo, J.; Biffi, R.; Trentin, C.; Bonanni, B.; Feroce, I.; Serrano, D.; Cassano, E.; Annibale, B.; Melo, S.; et al. E-cadherin germline mutation carriers: Clinical management and genetic implications. Cancer Metastasis Rev. 2014, 33, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://clinicaltrials.gov/ct2/show/NCT04206891 (accessed on 20 December 2019).
- Corso, G.; De Scalzi, A.; Feroce, I.; Veronesi, P.; Bonanni, B.; Galimberti, V. Clinical criteria revision for hereditary lobular breast cancer associated with E-cadherin germline mutations. Pers. Med. 2018, 15, 153–155. [Google Scholar] [CrossRef]
- Breast, T. WHO Classification of Tumours, 5th ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2019. [Google Scholar]
- Guerini-Rocco, E.; Fusco, N. Premalignant and Pre-invasive Lesions of the Breast. In Methods in Molecular Biology; Springer Science and Business Media LLC: Berlin, Germany, 2017; pp. 103–120. [Google Scholar]
- Marotti, J.D.; Schnitt, S.J. Genotype-Phenotype Correlations in Breast Cancer. Surg. Pathol. Clin. 2018, 11, 199–211. [Google Scholar] [CrossRef]
- Mirandola, S.; Pellini, F.; Granuzzo, E.; Lorenzi, M.; Accordini, B.; Ulgelmo, M.; Invento, A.; Lombardi, D.; Caldana, M.; Pollini, G.P. Multidisciplinary management of CDH1 germinal mutation and prophylactic management hereditary lobular breast cancer: A case report. Int. J. Surg. Case Rep. 2019, 58, 92–95. [Google Scholar] [CrossRef]
- Sickles, E.A. The subtle and atypical mammographic features of invasive lobular carcinoma. Radiology 1991, 178, 25–26. [Google Scholar] [CrossRef]
- Kerlikowske, K.; Grady, D.; Barclay, J.; Sickles, E.A.; Ernster, V. Effect of Age, Breast Density, and Family History on the Sensitivity of First Screening Mammography. JAMA 1996, 276, 33. [Google Scholar] [CrossRef]
- Robertson, C.L. A private breast imaging practice: Medical audit of 25,788 screening and 1,077 diagnostic examinations. Radiology 1993, 187, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Hilleren, D.J.; Andersson, I.T.; Lindholm, K.; Linnell, F.S. Invasive lobular carcinoma: Mammographic findings in a 10-year experience. Radiology 1991, 178, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Krecke, K.N.; Gisvold, J.J. Invasive lobular carcinoma of the breast: Mammographic findings and extent of disease at diagnosis in 184 patients. AJR Am. J. Roentgenol. 1993, 161, 957–960. [Google Scholar] [CrossRef] [PubMed]
- Le Gal, M.; Ollivier, L.; Asselain, B.; Meunier, M.; Laurent, M.; Vielh, P.; Neuenschwander, S. Mammographic features of 455 invasive lobular carcinomas. Radiology 1992, 185, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Paramagul, C.P.; A Helvie, M.; Adler, D.D. Invasive lobular carcinoma: Sonographic appearance and role of sonography in improving diagnostic sensitivity. Radiology 1995, 195, 231–234. [Google Scholar] [CrossRef]
- Selinko, V.L.; Middleton, L.P.; Dempsey, P.J. Role of sonography in diagnosing and staging invasive lobular carcinoma. J. Clin. Ultrasound 2004, 32, 323–332. [Google Scholar] [CrossRef]
- Mann, R.M.; Hoogeveen, Y.L.; Blickman, J.G.; Boetes, C. MRI compared to conventional diagnostic work-up in the detection and evaluation of invasive lobular carcinoma of the breast: A review of existing literature. Breast Cancer Res. Treat. 2007, 107, 1–14. [Google Scholar] [CrossRef]
- Mann, R.M.; Kuhl, C.K.; Kinkel, K.; Boetes, C. Breast MRI: Guidelines from the European Society of Breast Imaging. Eur. Radiol. 2008, 18, 1307–1318. [Google Scholar] [CrossRef]
- McGuire, K.P.; Mamounas, E.P. Management of Hereditary Breast Cancer: ASCO, ASTRO, and SSO Guideline. Ann. Surg. Oncol. 2020, 27, 1721–1723. [Google Scholar] [CrossRef]
- Jakub, J.; Peled, A.W.; Gray, R.J.; Greenup, R.A.; Kiluk, J.V.; Sacchini, V.; McLaughlin, S.A.; Tchou, J.C.; Vierkant, R.A.; Degnim, A.C.; et al. Oncologic Safety of Prophylactic Nipple-Sparing Mastectomy in a Population with BRCA Mutations: A Multi-institutional Study. JAMA Surg. 2018, 153, 123–129. [Google Scholar] [CrossRef]
- Valachis, A.; Nearchou, A.D.; Lind, P. Surgical management of breast cancer in BRCA-mutation carriers: A systematic review and meta-analysis. Breast Cancer Res. Treat. 2014, 144, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Tsangaris, T.N.; Kouprina, N.; Wu, L.S.-F.; Balch, C.M.; Vang, R.; Argani, P. The superficial margin of the skin-sparing mastectomy for breast carcinoma: Factors predicting involvement and efficacy of additional margin sampling. Ann. Surg. Oncol. 2008, 15, 1330–1340. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Muller, T.; Baratte, A.; Bruant-Rodier, C.; Bodin, F.; Mathelin, C. Oncological safety of nipple-sparing prophylactic mastectomy: A review of the literature on 3716 cases. Ann. Chir. Plast. Esthet. 2017, 63, e6–e13. [Google Scholar] [CrossRef]
- Weber, W.P.; Haug, M.; Kurzeder, C.; Bjelic-Radisic, V.; Koller, R.; Reitsamer, R.; Fitzal, F.; Biazus, J.; Brenelli, F.; Urban, C.; et al. Oncoplastic Breast Consortium consensus conference on nipple-sparing mastectomy. Breast Cancer Res. Treat. 2018, 172, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Headon, H.L.; Kasem, A.; Mokbel, K. The Oncological Safety of Nipple-Sparing Mastectomy: A Systematic Review of the Literature with a Pooled Analysis of 12,358 Procedures. Arch. Plast. Surg. 2016, 43, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, V.; Morigi, C.; Bagnardi, V.; Corso, G.; Vicini, E.; Fontana, S.K.R.; Naninato, P.; Ratini, S.; Magnoni, F.; Toesca, A.; et al. Oncological Outcomes of Nipple-Sparing Mastectomy: A Single-Center Experience of 1989 Patients. Ann. Surg. Oncol. 2018, 25, 3849–3857. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, V.; Vicini, E.; Corso, G.; Morigi, C.; Fontana, S.; Sacchini, V.; Veronesi, P. Nipple-sparing and skin-sparing mastectomy: Review of aims, oncological safety and contraindications. Breast 2017, 34 (Suppl. S1), S82–S84. [Google Scholar] [CrossRef]
- Van Verschuer, V.M.; Mureau, M.A.; Gopie, J.P.; Vos, E.L.; Verhoef, C.; Menke-Pluijmers, M.B.; Koppert, L.B. Patient satisfaction and nipple-areola sensitivity after bilateral prophylactic mastectomy and immediate implant breast reconstruction in a high breast cancer risk population: Nipple-sparing mastectomy versus skin-sparing mastectomy. Ann. Plast. Surg. 2016, 77, 145–152. [Google Scholar] [CrossRef]
- Razdan, S.N.; Patel, V.; Jewell, S.; McCarthy, C.M. Quality of life among patients after bilateral prophylactic mastectomy: A systematic review of patient-reported outcomes. Qual. Life Res. 2015, 25, 1409–1421. [Google Scholar] [CrossRef]
- Franceschini, G.; Masetti, R. What The Surgeons Should Know About The Bilateral Prophylactic Mastectomy in BRCA Mutation Carriers. Eur. J. Breast Health 2019, 15, 135–136. [Google Scholar] [CrossRef]
- Corso, G.; De Lorenzi, F.; Vicini, E.; Pagani, G.; Veronesi, P.; Sargenti, M.; Magnoni, F.; Naninato, P.; Maisonneuve, P.; Sangalli, C.; et al. Nipple-sparing mastectomy with different approaches: Surgical incisions, complications, and cosmetic results. Preliminary results of 100 consecutive patients at a single center. J. Plast. Reconstr. Aesthetic Surg. 2018, 71, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Toesca, A.; Invento, A.; Massari, G.; Girardi, A.; Peradze, N.; Lissidini, G.; Sangalli, C.; Maisonneuve, P.; Manconi, A.; Gottardi, A.; et al. Update on the Feasibility and Progress on Robotic Breast Surgery. Ann. Surg. Oncol. 2019, 26, 3046–3051. [Google Scholar] [CrossRef] [PubMed]
- European Institute of Oncology. Robotic Nipple-Sparing Mastectomy vs. Conventional Open Technique; ClinicalTrials.gov identifier: NCT 03440398. Available online: www.clinicaltrials.gov/ct2/show/NCT03440398 (accessed on 22 February 2018).
- Ilonzo, N.; Tsang, A.; Tsantes, S.; Estabrook, A.; Ma, A.M.T. Breast reconstruction after mastectomy: A ten-year analysis of trends and immediate postoperative outcomes. Breast 2017, 32, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Veronesi, P.; Ballardini, B.; De Lorenzi, F.; Magnoni, F.; Lissidini, G.; Caldarella, P.; Galimberti, V. Immediate breast reconstruction after mastectomy. Breast 2011, 20 (Suppl. S3), S104–S107. [Google Scholar] [CrossRef]
- Kamali, P.; Zettervall, S.L.; Wu, W.; Ibrahim, A.M.S.; Medin, C.; Rakhorst, H.; Schermerhorn, M.; Lee, B.T.; Lin, S.J. Differences in the Reporting of Racial and Socioeconomic Disparities among Three Large National Databases for Breast Reconstruction. Plast. Reconstr. Surg. 2017, 139, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Frey, J.D.; Salibian, A.A.; Karp, N.S.; Choi, M. Implant-Based Breast Reconstruction: Hot Topics, Controversies, and New Directions. Plast. Reconstr. Surg. 2019, 143, 404e–416e. [Google Scholar] [CrossRef]
- Sbitany, H.; Piper, M.; Lentz, R. Prepectoral Breast Reconstruction: A Safe Alternative to Submuscular Prosthetic Reconstruction following Nipple-Sparing Mastectomy. Plast. Reconstr. Surg. 2017, 140, 432–443. [Google Scholar] [CrossRef]
- Li, Y.; Xu, G.; Yu, N.; Huang, J.; Long, X. Prepectoral Versus Subpectoral Implant-Based Breast Reconstruction: A Meta-analysis. Ann. Plast. Surg. 2020. [Google Scholar] [CrossRef]
- Sigalove, S.; Maxwell, G.P.; Sigalove, N.M.; Storm-Dickerson, T.L.; Pope, N.; Rice, J.; Gabriel, A. Prepectoral Implant-Based Breast Reconstruction: Rationale, Indications, and Preliminary Results. Plast. Reconstr. Surg. 2017, 139, 287–294. [Google Scholar] [CrossRef]
- Fitzgerald, R.C.; Hardwick, R.; Huntsman, D.; Carneiro, F.; Guilford, P.; Blair, V.; Chung, D.C.; Norton, J.; Ragunath, K.; Van Krieken, J.H.; et al. Hereditary diffuse gastric cancer: Updated consensus guidelines for clinical management and directions for future research. J. Med. Genet. 2010, 47, 436–444. [Google Scholar] [CrossRef]
- Pharoah, P.D.; Guilford, P.; Caldas, C. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001, 121, 1348–1353. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Seruca, R.; Carneiro, F. Genetics, Pathology, and Clinics of Familial Gastric Cancer. Int. J. Surg. Pathol. 2006, 14, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Senz, J.; Kaurah, P.; Pinheiro, H.; Sanges, R.; Haegert, A.; Corso, G.; Schouten, J.; Fitzgerald, R.; Vogelsang, H.; et al. Germline CDH1 deletions in hereditary diffuse gastric cancer families. Hum. Mol. Genet. 2009, 18, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Plon, S.E.; Eccles, D.M.; Easton, U.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V.; et al. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Lee, K.; Krempely, K.; Roberts, M.E.; Anderson, M.J.; Carneiro, F.; Chao, E.; Dixon, K.; Figueiredo, J.; Ghosh, R.; Huntsman, D.; et al. Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Hum. Mutat. 2018, 39, 1553–1568. [Google Scholar] [CrossRef]
- Kurian, A.W.; Hare, E.E.; Mills, M.A.; Kingham, K.E.; McPherson, L.; Whittemore, A.S.; McGuire, V.; Ladabaum, U.; Kobayashi, Y.; Lincoln, S.E.; et al. Clinical Evaluation of a Multiple-Gene Sequencing Panel for Hereditary Cancer Risk Assessment. J. Clin. Oncol. 2014, 32, 2001–2009. [Google Scholar] [CrossRef]
- Kurian, A.W.; Ward, K.C.; Hamilton, A.S.; Deapen, D.M.; Abrahamse, P.; Bondarenko, I.; Li, Y.; Hawley, S.T.; Morrow, M.; Jagsi, R.; et al. Uptake, Results, and Outcomes of Germline Multiple-Gene Sequencing After Diagnosis of Breast Cancer. JAMA Oncol. 2018, 4, 1066. [Google Scholar] [CrossRef]
- Hall, M.J.; Forman, A.D.; Pilarski, R.; Wiesner, G.; Giri, V.N. Gene panel testing for inherited cancer risk. Natl. Compr. Canc. Netw. 2014, 12, 1339–1346. [Google Scholar] [CrossRef]
- Lowstuter, K.; Espenschied, C.R.; Sturgeon, D.; Ricker, C.; Karam, R.; LaDuca, H.; Culver, J.O.; Dolinsky, J.S.; Chao, E.; Sturgeon, J.; et al. Unexpected CDH1 Mutations Identified on Multigene Panels Pose Clinical Management Challenges. JCO Precis. Oncol. 2017, 1, 1–12. [Google Scholar] [CrossRef]
- Cicero, G.; De Luca, R.; Dorangricchia, P.; Coco, G.L.; Guarnaccia, C.; Fanale, D.; Calò, V.; Russo, A. Risk Perception and Psychological Distress in Genetic Counselling for Hereditary Breast and/or Ovarian Cancer. J. Genet. Couns. 2017, 26, 999–1007. [Google Scholar] [CrossRef]
- Ringwald, J.; Wochnowski, C.; Bosse, K.; Giel, K.E.; Schaeffeler, N.; Zipfel, S.; Teufel, M. Psychological Distress, Anxiety, and Depression of Cancer-Affected BRCA1/2 Mutation Carriers: A Systematic Review. J. Genet. Couns. 2016, 25, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Esteban, I.; Vilaró, M.; Adrover, E.; Angulo, A.; Carrasco, E.; Gadea, N.; Sanchez, A.; Ocana, T.; Llort, G.; Jover, R.; et al. Psychological impact of multigene cancer panel testing in patients with a clinical suspicion of hereditary cancer across Spain. Psycho-Oncology 2018, 27, 1530–1537. [Google Scholar] [CrossRef] [PubMed]
- Meiser, B.; Quinn, V.F.; Mitchell, G.; Tucker, K.; Watts, K.J.; Rahman, B.; Peate, M.; Saunders, C.; Geelhoed, E.; Gleeson, M.; et al. Psychological outcomes and surgical decisions after genetic testing in women newly diagnosed with breast cancer with and without a family history. Eur. J. Hum. Genet. 2018, 26, 972–983. [Google Scholar] [CrossRef]
- Bechara, A. The role of emotion in decision-making: Evidence from neurological patients with orbitofrontal damage. Brain Cogn. 2004, 55, 30–40. [Google Scholar] [CrossRef]
- Martin, L.N.; Delgado, M.R. The influence of emotion regulation on decision-making under risk. J. Cogn. Neurosci. 2011, 23, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Arnaboldi, P.; Lucchiari, C.; Santoro, L.; Sangalli, C.; Luini, A.; Pravettoni, G. PTSD symptoms as a consequence of breast cancer diagnosis: Clinical implications. SpringerPlus 2014, 3, 392. [Google Scholar] [CrossRef]
- Renzi, C.; Riva, S.; Masiero, M.; Pravettoni, G.; Information, P.E.K.F.C. The choice dilemma in chronic hematological conditions: Why choosing is not only a medical issue? A psycho-cognitive perspective. Crit. Rev. Oncol. 2016, 99, 134–140. [Google Scholar] [CrossRef]
- Fioretti, C.; Mazzocco, K.; Pravettoni, G. Psychological Support in Breast Cancer Patients: A Personalized Approach. In Methods in Molecular Biology; Springer Science and Business Media LLC: Berlin, Germany, 2017; pp. 841–847. [Google Scholar] [CrossRef]
- Kondylakis, H.; Kazantzaki, E.; Koumakis, L.; Genitsaridi, I.; Marias, K.; Gorini, A.; Mazzocco, K.; Pravettoni, G.; Burke, D.; McVie, G.; et al. Development of interactive empowerment services in support of personalised medicine. Ecancermedicalscience 2014, 8, 1–14. [Google Scholar] [CrossRef]
- Gorini, A.; Pravettoni, G. P5 medicine: A plus for a personalized approach to oncology. Nat. Rev. Clin. Oncol. 2011, 8, 444. [Google Scholar] [CrossRef]
- Glatzer, M.; Panje, C.M.; Sirén, C.; Cihoric, N.; Putora, P.M. Decision Making Criteria in Oncology. Oncology 2018, 18, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tamagawa, R.; Garland, S.N.; Vaska, M.; Carlson, L.E. Who benefits from psychosocial interventions in oncology? A systematic review of psychological moderators of treatment outcome. J. Behav. Med. 2012, 35, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Petrocchi, S.; Iannello, P.; Lecciso, F.; Levante, A.; Antonietti, A.; Schulz, P. Interpersonal trust in the context of doctor-patient relationship: Dyadic analysis with one-with-many design. Soc. Sci. Med. 2019, 235, 112391. [Google Scholar] [CrossRef] [PubMed]
- Herlitz, A.; Munthe, C.; Törner, M.; Forsander, G. The Counseling, Self-Care, Adherence Approach to Person-Centered Care and Shared Decision Making: Moral Psychology, Executive Autonomy, and Ethics in Multi-Dimensional Care Decisions. Health Commun. 2016, 31, 964–973. [Google Scholar] [CrossRef]
- Jones, P.S.; Meleis, A.I. Health is empowerment. ANS Adv. Nurs. Sci. 1993, 15, 1–14. [Google Scholar] [CrossRef]
- Mandelblatt, J.; Kreling, B.; Figeuriedo, M.; Feng, S. What Is the Impact of Shared Decision Making on Treatment and Outcomes for Older Women with Breast Cancer? J. Clin. Oncol. 2006, 24, 4908–4913. [Google Scholar] [CrossRef]
- Shay, L.A.; Lafata, J.E. Where is the evidence? A systematic review of shared decision making and patient outcomes. Med. Decis. Mak. 2014, 35, 114–131. [Google Scholar] [CrossRef]
- Majithia, A.R.; Tsuda, B.; Agostini, M.; Gnanapradeepan, K.; Rice, R.; Peloso, G.; Patel, K.A.; Zhang, X.; Broekema, M.F.; Patterson, N.; et al. Prospective functional classification of all possible missense variants in PPARG. Nat. Genet. 2016, 48, 1570–1575. [Google Scholar] [CrossRef]
- Findlay, G.M.; Daza, R.M.; Martin, B.; Zhang, M.D.; Leith, A.P.; Gasperini, M.; Janizek, J.D.; Huang, X.; Starita, L.M.; Shendure, J. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018, 562, 217–222. [Google Scholar] [CrossRef]
- Raraigh, K.S.; Han, S.T.; Davis-Marcisak, E.F.; Evans, T.A.; Pellicore, M.; McCague, A.F.; Joynt, A.T.; Lu, Z.; Atalar, M.; Sharma, N.; et al. Functional Assays Are Essential for Interpretation of Missense Variants Associated with Variable Expressivity. Am. J. Hum. Genet. 2018, 102, 1062–1077. [Google Scholar] [CrossRef]
- Suriano, G.; Seixas, S.; Rocha, J.; Seruca, R. A model to infer the pathogenic significance of CDH1 germline missense variants. J. Mol. Med. 2006, 84, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, R.C.; Caldas, C. Clinical implications of E-cadherin associated hereditary diffuse gastric cancer. Gut 2004, 53, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, J.; Seruca, R. Germline Missense Mutants in Hereditary Diffuse Gastric Cancer. In Spotlight on Familial and Hereditary Gastric Cancer; Springer Science and Business Media LLC: Berlin, Germany, 2013; pp. 77–86. [Google Scholar] [CrossRef]
- Melo, S.; Figueiredo, J.; Fernandes, M.S.; Gonçalves, M.; De Sá, E.M.; Sanches, J.M.; Seruca, R. Predicting the Functional Impact of CDH1 Missense Mutations in Hereditary Diffuse Gastric Cancer. Int. J. Mol. Sci. 2017, 18, 2687. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Pinheiro, H.; Figueiredo, J.; Seruca, R.; Carneiro, F. Familial gastric cancer: Genetic susceptibility, pathology, and implications for management. Lancet Oncol. 2015, 16, e60–e70. [Google Scholar] [CrossRef]
- Correia, J.S.; Figueiredo, J.; Lopes, R.; Stricher, F.; Oliveira, C.; Serrano, L.; Seruca, R. E-Cadherin Destabilization Accounts for the Pathogenicity of Missense Mutations in Hereditary Diffuse Gastric Cancer. PLoS ONE 2012, 7, e33783. [Google Scholar] [CrossRef]
- Suriano, G.; Oliveira, C.; Ferreira, P.; Machado, J.C.; Bordin, M.C.; De Wever, O.; Bruyneel, E.A.; Moguilevsky, N.; Grehan, N.; Porter, T.R.; et al. Identification of CDH1 germline missense mutations associated with functional inactivation of the E-cadherin protein in young gastric cancer probands. Hum. Mol. Genet. 2003, 12, 575–582. [Google Scholar] [CrossRef]
- Figueiredo, J.; Söderberg, O.; Correia, J.S.; Grannas, K.; Suriano, G.; Seruca, R. The importance of E-cadherin binding partners to evaluate the pathogenicity of E-cadherin missense mutations associated to HDGC. Eur. J. Hum. Genet. 2012, 21, 301–309. [Google Scholar] [CrossRef]
- Sanches, J.M.; Figueiredo, J.; Fonseca, M.; Durães, C.; Melo, S.; Esménio, S.; Seruca, R. Quantification of mutant E-cadherin using bioimaging analysis of in situ fluorescence microscopy. A new approach to CDH1 missense variants. Eur. J. Hum. Genet. 2014, 23, 1072–1079. [Google Scholar] [CrossRef]
- Mestre, T.; Figueiredo, J.; Ribeiro, A.S.; Paredes, J.; Seruca, R.; Sanches, J.M. Quantification of topological features in cell meshes to explore E-cadherin dysfunction. Sci. Rep. 2016, 6, 25101. [Google Scholar] [CrossRef]
- Pinho, S.S.; Seruca, R.; Gärtner, F.; Yamaguchi, Y.; Gu, J.; Taniguchi, N.; Reis, C.A. Modulation of E-cadherin function and dysfunction by N-glycosylation. Cell. Mol. Life Sci. 2010, 68, 1011–1020. [Google Scholar] [CrossRef]
- Pena-Couso, L.; Perea, J.; Melo, S.; Mercadillo, F.; Figueiredo, J.; Sanches, J.M.; Sanchez-Ruiz, A.; Robles, L.; Oliveira, C.; Urioste, M. Clinical and functional characterization of the CDH1 germline variant c.1679C>G in three unrelated families with hereditary diffuse gastric cancer. Eur. J. Hum. Genet. 2018, 26, 1348–1353. [Google Scholar] [CrossRef]
- Correia, J.S.; Figueiredo, J.; Oliveira, C.; Van Hengel, J.; Seruca, R.; Van Roy, F.; Suriano, G. Endoplasmic reticulum quality control: A new mechanism of E-cadherin regulation and its implication in cancer. Hum. Mol. Genet. 2008, 17, 3566–3576. [Google Scholar] [CrossRef]
- Suriano, G.; Oliveira, M.J.; Huntsman, D.; Mateus, A.R.; Ferreira, P.; Casares, F.; Oliveira, C.; Carneiro, F.; Machado, J.C.; Mareel, M.; et al. E-cadherin germline missense mutations and cell phenotype: Evidence for the independence of cell invasion on the motile capabilities of the cells. Hum. Mol. Genet. 2003, 12, 3007–3016. [Google Scholar] [CrossRef]
- Boterberg, T.; E Bracke, M.; A Bruyneel, E.; Mareel, M.; Brooks, S.A.; Schumacher, U. Cell Aggregation Assays. Methods Mol. Med. 2001, 58, 33–45. [Google Scholar] [PubMed]
- Suriano, G.; Mulholland, D.; De Wever, O.; Ferreira, P.; Mateus, A.R.; Bruyneel, E.; Nelson, C.C.; Mareel, M.M.; Yokota, J.; Huntsman, D.; et al. The intracellular E-cadherin germline mutation V832 M lacks the ability to mediate cell–cell adhesion and to suppress invasion. Oncogene 2003, 22, 5716–5719. [Google Scholar] [CrossRef]
- Kleinman, H.K.; Martin, G.R. Matrigel: Basement membrane matrix with biological activity. Semin. Cancer Biol. 2005, 15, 378–386. [Google Scholar] [CrossRef]
- Kleinman, H.K.; McGarvey, M.L.; Liotta, L.A.; Robey, P.G.; Tryggvason, K.; Martin, G.R. Isolation and characterization of type IV procollagen, laminin, and heparan sulfate proteoglycan from the EHS sarcoma. Biochemistry 1982, 21, 6188–6193. [Google Scholar] [CrossRef]
- Mateus, A.R.; Correia, J.S.; Figueiredo, J.; Heindl, S.; Alves, C.C.; Suriano, G.; Luber, B.; Seruca, R. E-cadherin mutations and cell motility: A genotype–phenotype correlation. Exp. Cell Res. 2009, 315, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Mateus, A.R.; Seruca, R.; Machado, J.C.; Keller, G.; Oliveira, M.J.; Suriano, G.; Luber, B. EGFR regulates RhoA-GTP dependent cell motility in E-cadherin mutant cells. Hum. Mol. Genet. 2007, 16, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, J.W.; Conchello, J.A. Fluorescence microscopy. Nat. Methods 2005, 2, 910–919. [Google Scholar] [CrossRef]
- Muzzey, D.; Van Oudenaarden, A. Quantitative time-lapse fluorescence microscopy in single cells. Annu. Rev. Cell Dev. Biol. 2009, 25, 301–327. [Google Scholar] [CrossRef] [PubMed]
- Sandison, D.R.; Williams, R.M.; Wells, K.S.; Strickler, J.; Webb, W.W. Quantitative Fluorescence Confocal Laser Scanning Microscopy (CLSM). In Handbook of Biological Confocal Microscopy; Springer Science and Business Media LLC: Berlin, Germany, 1995; pp. 39–53. [Google Scholar]
- Nakano, A. Spinning-disk Confocal Microscopy—A Cutting-Edge Tool for Imaging of Membrane Traffic. Cell Struct. Funct. 2002, 27, 349–355. [Google Scholar] [CrossRef]
- Waters, J.C. Accuracy and precision in quantitative fluorescence microscopy. J. Cell Biol. 2009, 185, 1135–1148. [Google Scholar] [CrossRef]
- A Hamilton, N. Quantification and its Applications in Fluorescent Microscopy Imaging. Traffic 2009, 10, 951–961. [Google Scholar] [CrossRef]
- Figueiredo, J.; Rodrigues, I.; Ribeiro, J.; Fernandes, M.S.; Melo, S.; Sousa, B.; Paredes, J.; Oliveira, C.; Sanches, J.M. Geometric compensation applied to image analysis of cell populations with morphological variability: A new role for a classical concept. Sci. Rep. 2018, 8, 10266. [Google Scholar] [CrossRef]
- Fonseca, L.M.G.; Manjunath, B.S. Registration techniques for multisensor remotely sensed imagery. Photogramm. Eng. Remote Sens. 1996, 62, 1049–1056. [Google Scholar]
- Zitová, B.; Flusser, J. Image registration methods: A survey. Image Vis. Comput. 2003, 21, 977–1000. [Google Scholar] [CrossRef]
- Sanches, J.M.; Marques, J. Joint image registration and volume reconstruction for 3D ultrasound. Pattern Recognit. Lett. 2003, 24, 791–800. [Google Scholar] [CrossRef]
- Li, S.; Wakefield, J.; Noble, J.A. Automated Segmentation and Alignment of Mitotic Nuclei for Kymograph Visualisation. In Proceedings of the 2011 IEEE International Symposium on Biomedical Imaging: From Nano to Macro, Chicago, IL, USA, 30 March–2 April 2011; pp. 622–625. [Google Scholar] [CrossRef]
- Figueiredo, J.; Melo, S.; Gamet, K.; Godwin, T.; Seixas, S.; Sanches, J.M.; Guilford, P.; Oliveira, C. E-cadherin signal sequence disruption: A novel mechanism underlying hereditary cancer. Mol. Cancer 2018, 17, 112. [Google Scholar] [CrossRef]
- Ribeiro, A.; Carvalho, F.; Figueiredo, J.; Mestre, T.; Monteiro, J.; Guedes, A.F.; Fonseca, M.; Sanches, J.M.; Seruca, R.; Santos, N.C.; et al. Atomic force microscopy and graph analysis to study the P-cadherin/SFK mechanotransduction signalling in breast cancer cells. Nanoscale 2016, 8, 19390–19401. [Google Scholar] [CrossRef]
- Hamilton, J.G.; Long, J.M.; Brandt, A.C.; Brower, J.; Symecko, H.; Salo-Mullen, E.E.; Christian, S.N.; Harstad, T.; Couch, F.J.; Garber, J.E.; et al. Patients’ Medical and Psychosocial Experiences After Detection of a CDH1 Variant with Multigene Panel Testing. JCO Precis. Oncol. 2019, 1–14. [Google Scholar] [CrossRef]
- Marrelli, D.; Pedrazzani, C.; Corso, G.; Neri, A.; Di Martino, M.; Pinto, E.; Roviello, F. Different Pathological Features and Prognosis in Gastric Cancer Patients Coming From High-Risk and Low-Risk Areas of Italy. Ann. Surg. 2009, 250, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Palli, D.; Russo, A.G.; DeCarli, A. Dietary patterns, nutrient intake and gastric cancer in a high-risk area of Italy. Cancer Causes Control 2001, 12, 163–172. [Google Scholar] [CrossRef] [PubMed]
- A González, C.; Jakszyn, P.; Pera, G.; Agudo, A.; Bingham, S.; Palli, D.; Ferrari, P.; Boeing, H.; Del Giudice, G.; Plebani, M.; et al. Meat Intake and Risk of Stomach and Esophageal Adenocarcinoma within the European Prospective Investigation into Cancer and Nutrition (EPIC). J. Natl. Cancer Inst. 2006, 98, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Corso, G.; Marrelli, D.; Pascale, V.; Vindigni, C.; Roviello, F. Frequency of CDH1 germline mutations in gastric carcinoma coming from high- and low-risk areas: Metanalysis and systematic review of the literature. BMC Cancer 2012, 12, 8. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Markers | DGC | LBC |
|---|---|---|
| Cytokeratin 7 | +/- | + |
| Cytocheratin 20 | -/+ | - |
| GATA3 | - | + |
| CDX2 | +/- | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corso, G.; Montagna, G.; Figueiredo, J.; La Vecchia, C.; Fumagalli Romario, U.; Fernandes, M.S.; Seixas, S.; Roviello, F.; Trovato, C.; Guerini-Rocco, E.; et al. Hereditary Gastric and Breast Cancer Syndromes Related to CDH1 Germline Mutation: A Multidisciplinary Clinical Review. Cancers 2020, 12, 1598. https://doi.org/10.3390/cancers12061598
Corso G, Montagna G, Figueiredo J, La Vecchia C, Fumagalli Romario U, Fernandes MS, Seixas S, Roviello F, Trovato C, Guerini-Rocco E, et al. Hereditary Gastric and Breast Cancer Syndromes Related to CDH1 Germline Mutation: A Multidisciplinary Clinical Review. Cancers. 2020; 12(6):1598. https://doi.org/10.3390/cancers12061598
Chicago/Turabian StyleCorso, Giovanni, Giacomo Montagna, Joana Figueiredo, Carlo La Vecchia, Uberto Fumagalli Romario, Maria Sofia Fernandes, Susana Seixas, Franco Roviello, Cristina Trovato, Elena Guerini-Rocco, and et al. 2020. "Hereditary Gastric and Breast Cancer Syndromes Related to CDH1 Germline Mutation: A Multidisciplinary Clinical Review" Cancers 12, no. 6: 1598. https://doi.org/10.3390/cancers12061598
APA StyleCorso, G., Montagna, G., Figueiredo, J., La Vecchia, C., Fumagalli Romario, U., Fernandes, M. S., Seixas, S., Roviello, F., Trovato, C., Guerini-Rocco, E., Fusco, N., Pravettoni, G., Petrocchi, S., Rotili, A., Massari, G., Magnoni, F., De Lorenzi, F., Bottoni, M., Galimberti, V., ... Bonanni, B. (2020). Hereditary Gastric and Breast Cancer Syndromes Related to CDH1 Germline Mutation: A Multidisciplinary Clinical Review. Cancers, 12(6), 1598. https://doi.org/10.3390/cancers12061598