Phase I Study of Ficlatuzumab and Cetuximab in Cetuximab-Resistant, Recurrent/Metastatic Head and Neck Cancer

 , ,
, ,
Abstract
1. Introduction
2. Results
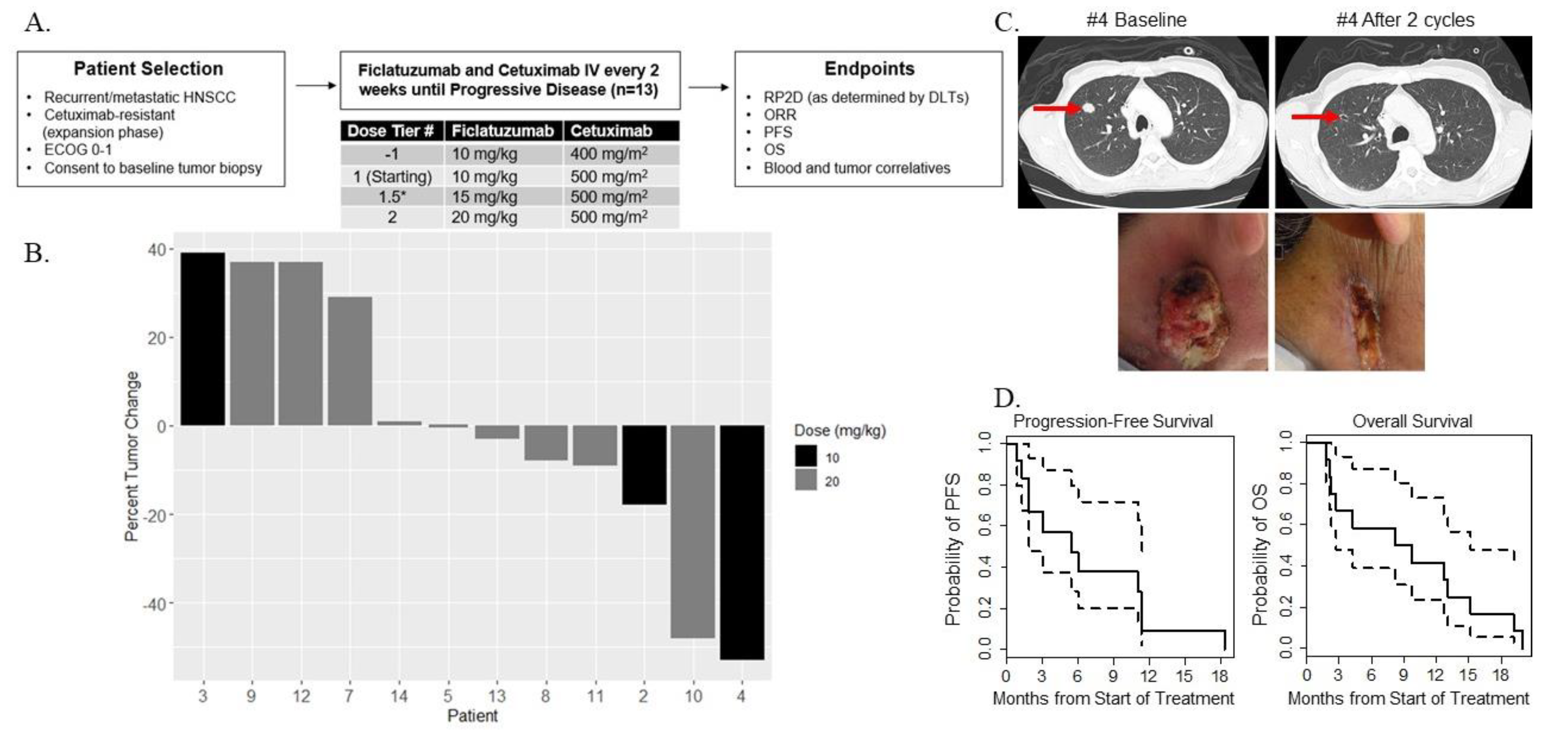
2.1. Patient Characteristics
2.2. Dose-Limiting Toxicities and Recommended Phase II Dose
2.3. Preliminary Oncologic Efficacy
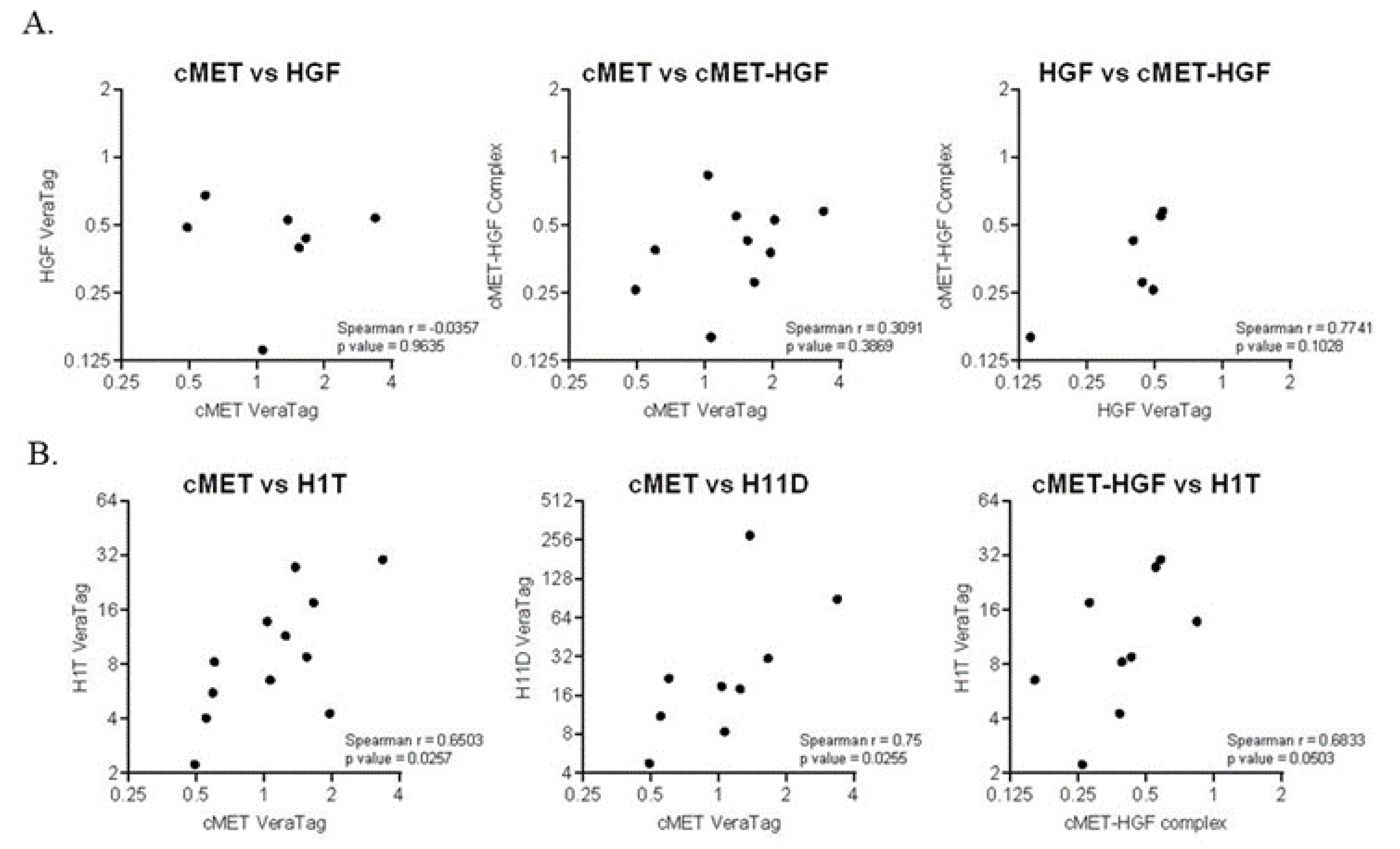
2.4. Prognostic Biomarkers
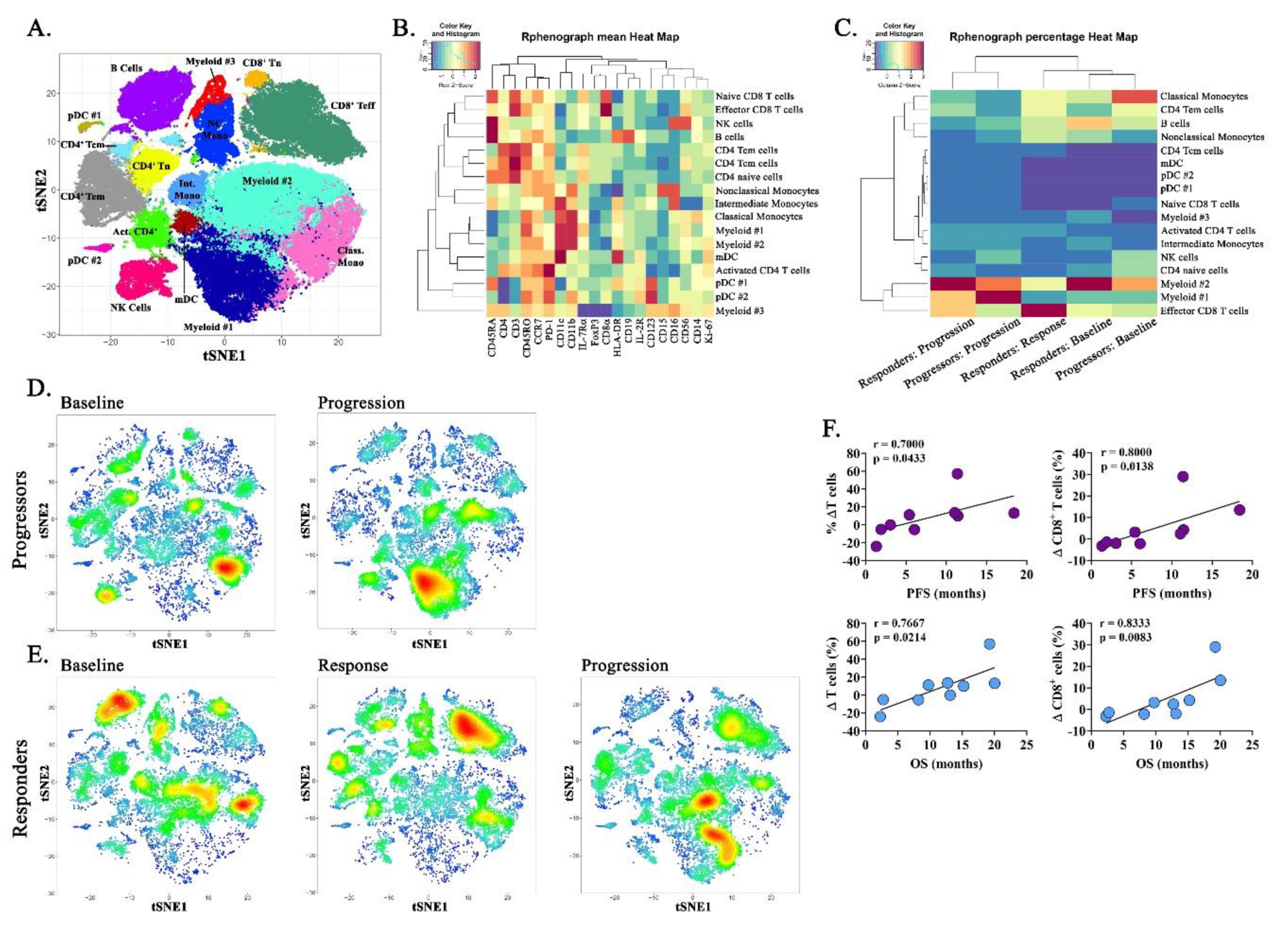
2.5. Immune Correlatives
3. Discussion
4. Materials and Methods
4.1. Human Subjects Considerations
4.2. Study Design and Statistical Considerations
4.3. Treatment Plan
4.4. Biomarker Signaling Correlatives and Statistical Considerations
4.5. Immune Correlatives
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Laversanne, M.; Brewster, D.H.; Mbalawa, C.G.; Kohler, B.; Piñeros, M.; Steliarova-Foucher, E.; Swaminathan, R.; Antoni, S.; et al. Cancer Incidence in Five Continents: Inclusion criteria, highlights from Volume X and the global status of cancer registration. Int. J. Cancer 2015, 137, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenár, É.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.-R.; Cupissol, D.; et al. Platinum-Based Chemotherapy plus Cetuximab in Head and Neck Cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Soulières, D.; le Tourneau, C.; Dinis, J.; Licitra, L.; Ahn, M.-J.; Soria, A.; Machiels, J.-P.; Mach, N.; Mehra, R.; et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): A randomised, open-label, phase 3 study. Lancet 2019, 393, 156–167. [Google Scholar] [CrossRef]
- Grandis, J.R.; Melhem, M.F.; Gooding, W.E.; Holst, V.A.; Wagener, M.M.; Drenning, S.D.; Day, R.; Tweardy, D.J. Levels of TGF-α and EGFR Protein in Head and Neck Squamous Cell Carcinoma and Patient Survival. J. Natl. Cancer Inst. 1998, 90, 824–832. [Google Scholar] [CrossRef]
- Chung, C.H.; Ely, K.; McGavran, L.; Varella-Garcia, M.; Parker, J.; Parker, N.; Jarrett, C.; Carter, J.; Murphy, B.A.; Netterville, J.; et al. Increased Epidermal Growth Factor Receptor Gene Copy Number Is Associated With Poor Prognosis in Head and Neck Squamous Cell Carcinomas. J. Clin. Oncol. 2006, 24, 4170–4176. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Herbst, R.S.; León, X.; Amellal, N.; Baselga, J. Overview of the efficacy of cetuximab in recurrent and/or metastatic squamous cell carcinoma of the head and neck in patients who previously failed platinum-based therapies. Cancer 2008, 112, 2710–2719. [Google Scholar] [CrossRef]
- Allegra, C.J.; Jessup, J.M.; Somerfield, M.R.; Hamilton, S.R.; Hammond, E.H.; Hayes, D.F.; McAllister, P.K.; Morton, R.F.; Schilsky, R.L. American Society of Clinical Oncology Provisional Clinical Opinion: Testing for KRAS Gene Mutations in Patients With Metastatic Colorectal Carcinoma to Predict Response to Anti–Epidermal Growth Factor Receptor Monoclonal Antibody Therapy. J. Clin. Oncol. 2009, 27, 2091–2096. [Google Scholar] [CrossRef]
- Licitra, L.; Störkel, S.; Kerr, K.M.; van Cutsem, E.; Pirker, R.; Hirsch, F.R.; Vermorken, J.; von Heydebreck, A.; Esser, R.; Celik, I.; et al. Predictive value of epidermal growth factor receptor expression for first-line chemotherapy plus cetuximab in patients with head and neck and colorectal cancer: Analysis of data from the EXTREME and CRYSTAL studies. Eur. J. Cancer 2013, 49, 1161–1168. [Google Scholar] [CrossRef]
- Licitra, L.; Mesia, R.; Rivera, F.; Remenár, É.; Hitt, R.; Erfán, J.; Rottey, S.; Kawecki, A.; Zabolotnyy, D.; Benasso, M.; et al. Evaluation of EGFR gene copy number as a predictive biomarker for the efficacy of cetuximab in combination with chemotherapy in the first-line treatment of recurrent and/or metastatic squamous cell carcinoma of the head and neck: EXTREME study. Ann. Oncol. 2011, 22, 1078–1087. [Google Scholar] [CrossRef]
- Peruzzi, B.; Bottaro, D.P. Targeting the c-Met Signaling Pathway in Cancer. Clin. Cancer Res. 2006, 12, 3657–3660. [Google Scholar] [CrossRef]
- Knowles, L.M.; Stabile, L.P.; Egloff, A.M.; Rothstein, M.E.; Thomas, S.M.; Gubish, C.T.; Lerner, E.C.; Seethala, R.R.; Suzuki, S.; Quesnelle, K.M.; et al. HGF and c-Met participate in paracrine tumorigenic pathways in head and neck squamous cell cancer. Clin. Cancer Res. 2009, 15, 3740–3750. [Google Scholar] [CrossRef]
- Mandal, M.; Myers, J.N.; Lippman, S.M.; Johnson, F.M.; Williams, M.D.; Rayala, S.K.; Ohshiro, K.; Rosenthal, D.I.; Weber, R.S.; Gallick, G.E.; et al. Epithelial to mesenchymal transition in head and neck squamous carcinoma. Cancer 2008, 112, 2088–2100. [Google Scholar] [CrossRef]
- Basu, D.; Nguyen, T.-T.K.; Montone, K.T.; Zhang, G.; Wang, L.-P.; Diehl, J.A.; Rustgi, A.K.; Lee, J.T.; Weinstein, G.S.; Herlyn, M. Evidence for mesenchymal-like sub-populations within squamous cell carcinomas possessing chemoresistance and phenotypic plasticity. Oncogene 2010, 29, 4170–4182. [Google Scholar] [CrossRef]
- Xu, H.; Stabile, L.P.; Gubish, C.T.; Gooding, W.E.; Grandis, J.R.; Siegfried, J.M. Dual blockade of EGFR and c-Met abrogates redundant signaling and proliferation in head and neck carcinoma cells. Clin. Cancer Res. 2011, 17, 4425–4438. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Jagadeeswaran, R.; Faoro, L.; Janamanchi, V.; Nallasura, V.; El Dinali, M.; Yala, S.; Kanteti, R.; Cohen, E.E.; Lingen, M.W.; et al. The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res. 2009, 69, 3021–3031. [Google Scholar] [CrossRef]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Yano, S.; Yamada, T.; Takeuchi, S.; Tachibana, K.; Minami, Y.; Yatabe, Y.; Mitsudomi, T.; Tanaka, H.; Kimura, T.; Kudoh, S.; et al. Hepatocyte Growth Factor Expression in EGFR Mutant Lung Cancer with Intrinsic and Acquired Resistance to Tyrosine Kinase Inhibitors in a Japanese Cohort. J. Thorac. Oncol. 2011, 6, 2011–2017. [Google Scholar] [CrossRef]
- Takahashi, N.; Yamada, Y.; Furuta, K.; Honma, Y.; Iwasa, S.; Takashima, A.; Kato, K.; Hamaguchi, T.; Shimada, Y. Serum levels of hepatocyte growth factor and epiregulin are associated with the prognosis on anti-EGFR antibody treatment in KRAS wild-type metastatic colorectal cancer. Br. J. Cancer 2014, 110, 2716–2727. [Google Scholar] [CrossRef]
- Yamada, T.; Takeuchi, S.; Kita, K.; Bando, H.; Nakamura, T.; Matsumoto, K.; Yano, S. Hepatocyte Growth Factor Induces Resistance to Anti-Epidermal Growth Factor Receptor Antibody in Lung Cancer. J. Thorac. Oncol. 2012, 7, 272–280. [Google Scholar] [CrossRef]
- Tanaka, H.; Kimura, T.; Kudoh, S.; Mitsuoka, S.; Watanabe, T.; Suzumura, T.; Tachibana, K.; Noguchi, M.; Yano, S.; Hirata, K. Reaction of plasma hepatocyte growth factor levels in non-small cell lung cancer patients treated with EGFR-TKIs. Int. J. Cancer 2011, 129, 1410–1416. [Google Scholar] [CrossRef]
- Trotta, A.M.; Ottaiano, A.; Romano, C.; Nasti, G.; Nappi, A.; de Divitiis, C.; Napolitano, M.; Zanotta, S.; Casaretti, R.; D’Alterio, C.; et al. Prospective Evaluation of Cetuximab-Mediated Antibody-Dependent Cell Cytotoxicity in Metastatic Colorectal Cancer Patients Predicts Treatment Efficacy. Cancer Immunol. Res. 2016, 4, 366–374. [Google Scholar] [CrossRef]
- López-Albaitero, A.; Lee, S.C.; Morgan, S.; Grandis, J.R.; Gooding, W.E.; Ferrone, S.; Ferris, R.L. Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol. Immunother. 2009, 58, 1853–1864. [Google Scholar] [CrossRef]
- Faden, D.L.; Concha-Benavente, F.; Chakka, A.B.; McMichael, E.L.; Chandran, U.; Ferris, R.L. Immunogenomic correlates of response to cetuximab monotherapy in head and neck squamous cell carcinoma. Head Neck 2019, 41, 2591–2601. [Google Scholar] [CrossRef]
- Papaccio, F.; Della-Corte, C.M.; Viscardi, G.; di Liello, R.; Esposito, G.; Sparano, F.; Ciardiello, F.; Morgillo, F. HGF/MET and the Immune System: Relevance for Cancer Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3595. [Google Scholar] [CrossRef]
- Okunishi, K.; Dohi, M.; Nakagome, K.; Tanaka, R.; Mizuno, S.; Matsumoto, K.; Miyazaki, J.-I.; Nakamura, T.; Yamamoto, K. A novel role of hepatocyte growth factor as an immune regulator through suppressing dendritic cell function. J. Immunol. 2005, 175, 4745–4753. [Google Scholar] [CrossRef]
- Benkhoucha, M.; Santiago-Raber, M.-L.; Schneiter, G.; Chofflon, M.; Funakoshi, H.; Nakamura, T.; Lalive, P.H. Hepatocyte growth factor inhibits CNS autoimmunity by inducing tolerogenic dendritic cells and CD25+Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2010, 107, 6424–6429. [Google Scholar] [CrossRef]
- Bauman, J.E.; Arias-Pulido, H.; Lee, S.-J.; Fekrazad, M.H.; Ozawa, H.; Fertig, E.J.; Howard, J.; Bishop, J.; Wang, H.; Olson, G.T.; et al. A phase II study of temsirolimus and erlotinib in patients with recurrent and/or metastatic, platinum-refractory head and neck squamous cell carcinoma. Oral Oncol. 2013, 49, 461–467. [Google Scholar] [CrossRef]
- To, Y.; Dohi, M.; Matsumoto, K.; Tanaka, R.; Sato, A.; Nakagome, K.; Nakamura, T.; Yamamoto, K. A Two-way Interaction between Hepatocyte Growth Factor and Interleukin-6 in Tissue Invasion of Lung Cancer Cell Line. Am. J. Respir. Cell Mol. Boil. 2002, 27, 220–226. [Google Scholar] [CrossRef]
- Tarhini, A.A.; Rafique, I.; Floros, T.; Tran, P.; Gooding, W.E.; Villaruz, L.C.; Burns, T.F.; Friedland, D.M.; Petro, D.P.; Farooqui, M.; et al. Phase 1/2 study of rilotumumab (AMG 102), a hepatocyte growth factor inhibitor, and erlotinib in patients with advanced non-small cell lung cancer. Cancers 2017, 123, 2936–2944. [Google Scholar] [CrossRef]
- Zhang, Y.; Doshi, S.; Zhu, M. Pharmacokinetics and pharmacodynamics of rilotumumab: A decade of experience in preclinical and clinical cancer research. Br. J. Clin. Pharmacol. 2015, 80, 957–964. [Google Scholar] [CrossRef]
- Tan, E.H.; Lim, W.-T.; Ahn, M.-J.; Ng, Q.-S.; Ahn, J.S.; Tan, D.S.-W.; Sun, J.-M.; Han, M.; Payumo, F.C.; McKee, K.; et al. Phase 1b Trial of Ficlatuzumab, a Humanized Hepatocyte Growth Factor Inhibitory Monoclonal Antibody, in Combination With Gefitinib in Asian Patients With NSCLC. Clin. Pharmacol. Drug Dev. 2018, 7, 532–542. [Google Scholar] [CrossRef]
- Gao, H.-F.; Li, A.-N.; Yang, J.-J.; Chen, Z.-H.; Xie, Z.; Zhang, X.-C.; Su, J.; Lou, N.-N.; Yan, H.-H.; Han, J.-F.; et al. Soluble c-Met Levels Correlated With Tissue c-Met Protein Expression in Patients With Advanced Non–Small-Cell Lung Cancer. Clin. Lung Cancer 2017, 18, 85–91. [Google Scholar] [CrossRef]
- Mok, T.S.; Geater, S.L.; Su, W.-C.; Tan, E.-H.; Yang, J.C.-H.; Chang, G.-C.; Han, M.; Komarnitsky, P.; Payumo, F.; Garrus, J.E.; et al. A Randomized Phase 2 Study Comparing the Combination of Ficlatuzumab and Gefitinib with Gefitinib Alone in Asian Patients with Advanced Stage Pulmonary Adenocarcinoma. J. Thorac. Oncol. 2016, 11, 1736–1744. [Google Scholar] [CrossRef]
- Patnaik, A.; Weiss, G.J.; Papadopoulos, K.P.; Hofmeister, C.C.; Tibes, R.; Tolcher, A.; Isaacs, R.; Jac, J.; Han, M.; Payumo, F.C.; et al. Phase I ficlatuzumab monotherapy or with erlotinib for refractory advanced solid tumours and multiple myeloma. Br. J. Cancer 2014, 111, 272–280. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, S.; Wang, K.; Sun, S.-Y. MET inhibitors for targeted therapy of EGFR TKI-resistant lung cancer. J. Hematol. Oncol. 2019, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.E.W.; Licitra, L.F.; Burtness, B.; Fayette, J.; Gauler, T.; Clement, P.M.; Grau, J.J.; Del Campo, J.M.; Mailliez, A.; Haddad, R.I.; et al. Biomarkers predict enhanced clinical outcomes with afatinib versus methotrexate in patients with second-line recurrent and/or metastatic head and neck cancer. Ann. Oncol. 2017, 28, 2526–2532. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Elez, M.E.; Herranz, M.; Rico, I.; Prudkin, L.; Andreu, J.; Mateos, J.; Carreras, M.J.; Han, M.; Gifford, J.; et al. A Pharmacodynamic/Pharmacokinetic Study of Ficlatuzumab in Patients with Advanced Solid Tumors and Liver Metastases. Clin. Cancer Res. 2014, 20, 2793–2804. [Google Scholar] [CrossRef] [PubMed]
- Benkhoucha, M.; Molnarfi, N.; Kaya, G.; Belnoue, E.; Bjarnadóttir, K.; Dietrich, P.; Walker, P.R.; Martinvalet, D.; Derouazi, M.; Lalive, P.H. Identification of a novel population of highly cytotoxic c-Met-expressing CD8 + T lymphocytes. EMBO Rep. 2017, 18, 1545–1558. [Google Scholar] [CrossRef]
- Sugimoto, M.A.; Vago, J.; Perretti, M.; Teixeira, M.M. Mediators of the Resolution of the Inflammatory Response. Trends Immunol. 2019, 40, 212–227. [Google Scholar] [CrossRef]
- Williams, C.B.; Yeh, E.S.; Soloff, A.C. Tumor-associated macrophages: Unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer 2016, 2, 15025. [Google Scholar] [CrossRef]
- Yen, B.L.-J.; Yen, M.-L.; Hsu, P.-J.; Liu, K.-J.; Wang, C.-J.; Bai, C.-H.; Sytwu, H.-K. Multipotent Human Mesenchymal Stromal Cells Mediate Expansion of Myeloid-Derived Suppressor Cells via Hepatocyte Growth Factor/c-Met and STAT3. Stem Cell Rep. 2013, 1, 139–151. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Stabile, L.; Egloff, A.; Gibson, M.; Gooding, W.; Ohr, J.; Zhou, P.; Rothenberger, N.; Wang, L.; Geiger, J.; Flaherty, J.; et al. IL6 is associated with response to dasatinib and cetuximab: Phase II clinical trial with mechanistic correlatives in cetuximab-resistant head and neck cancer. Oral Oncol. 2017, 69, 38–45. [Google Scholar] [CrossRef]
- Ivanova, A.; Montazer-Haghighi, A.; Mohanty, S.G.; Durham, S.D. Improved up-and-down designs for phase I trials. Stat. Med. 2002, 22, 69–82. [Google Scholar] [CrossRef]
- Tabernero, J.; Ciardiello, F.; Rivera, F.; Rodriguez-Braun, E.; Ramos, F.J.; Martinelli, E.; Vega-Villegas, M.E.; Roselló, S.; Liebscher, S.; Kisker, O.; et al. Cetuximab administered once every second week to patients with metastatic colorectal cancer: A two-part pharmacokinetic/pharmacodynamic phase I dose-escalation study. Ann. Oncol. 2010, 21, 1537–1545. [Google Scholar] [CrossRef]
- Tabernero, J.; Pfeiffer, P.; Cervantes, A. Administration of Cetuximab Every 2 Weeks in the Treatment of Metastatic Colorectal Cancer: An Effective, More Convenient Alternative to Weekly Administration? Oncologist 2008, 13, 113–119. [Google Scholar] [CrossRef]
- Staff, J.; Sherman, E.; Lisa, D.; Agarwal, N.; Algazy, K.; Brockstein, B.; Langer, C.; Lim, D.; Mehra, R.; Rajan, S.K.; et al. A randomized phase II study of cetuximab every 2 weeks at either 500 or 750 mg/m2 for patients with recurrent or metastatic head and neck squamous cell cancer. J. Natl. Compr. Cancer Netw. 2012, 10, 1391–1398. [Google Scholar] [CrossRef]
- Guigay, J.; Fayette, J.; Mesia, R.; Lafond, C.; Saada-Bouzid, E.; Geoffrois, L.; Martin, L.; Cupissol, D.; Capitain, O.; Castanie, H.; et al. TPExtreme randomized trial: TPEx versus Extreme regimen in 1st line recurrent/metastatic head and neck squamous cell carcinoma (R/M HNSCC). J. Clin. Oncol. 2019, 37, 6002. [Google Scholar] [CrossRef]
- Carbone, D.P.; Ding, K.; Roder, H.; Grigorieva, J.; Roder, J.; Tsao, M.-S.; Seymour, L.; Shepherd, F.A. Prognostic and Predictive Role of the VeriStrat® Plasma Test in Patients with Advanced Non-Small Cell Lung Cancer Treated with Erlotinib or Placebo in the NCIC Clinical Trials Group BR.21 Trial. J. Thorac. Oncol. 2012, 7, 1653–1660. [Google Scholar] [CrossRef]
- Dua, R.; Zhang, J.; Parry, G.; Penuel, E. Detection of Hepatocyte Growth Factor (HGF) Ligand-c-MET Receptor Activation in Formalin-Fixed Paraffin Embedded Specimens by a Novel Proximity Assay. PLoS ONE 2011, 6, e15932. [Google Scholar] [CrossRef]
- Shi, Y.; Huang, W.; Tan, Y.; Jin, X.; Dua, R.; Penuel, E.; Mukherjee, A.; Sperinde, J.; Pannu, H.; Chenna, A.; et al. A Novel Proximity Assay for the Detection of Proteins and Protein Complexes: Quantitation of HER1 and HER2 Total Protein Expression and Homodimerization in Formalin-fixed, Paraffin-Embedded Cell Lines and Breast Cancer Tissue. Diagn. Mol. Pathol. 2009, 18, 11–21. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Chen, H.; Lau, M.C.; Wong, M.T.; Newell, E.W.; Poidinger, M.; Chen, J. Cytofkit: A Bioconductor Package for an Integrated Mass Cytometry Data Analysis Pipeline. PLoS Comput. Boil. 2016, 12, e1005112. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Patient Characteristics | n (%) |
|---|---|
| Age (Median, Range) | 58.4 (46.7–80.1 years) |
| Sex | |
| Male | 10 (77%) |
| Female | 3 (23%) |
| ECOG Performance Status | |
| 0 | 8 (62%) |
| 1 | 5 (38%) |
| Primary Tumor Site | |
| Oral Cavity | 1 (8%) |
| Oropharynx | 4 (31%) |
| Hypopharynx | 2 (15%) |
| Larynx | 5 (38%) |
| External Auditory Canal | 1 (8%) |
| Platinum-Refractory | |
| Yes | 12 (92%) |
| No | 1 (8%) |
| Cetuximab-Resistant at Protocol Entry | |
| Yes | 12 (92%) |
| No | 1 (8%) |
| Tumor HPV Status | |
| p16+ Oropharynx | 1 (8%) |
| p16- Oropharynx and Non-Oropharynx | 12 (92%) |
| Previous Treatment with Anti-PD1/L1 mAb | |
| Yes | 5 (38%) |
| No | 8 (62%) |
| Baseline VeriStrat Status | |
| Good | 4 (33%) |
| Poor | 9 (67%) |
| Adverse Events | NCI CTCAE Grade | |||
|---|---|---|---|---|
| Grade 1–2 n (%) | Grade 3–4 n (%) | |||
| Tier 1 | Tier 2 | Tier 1 | Tier2 | |
| Constitutional | ||||
| Flu-Like Symptoms | 2 (67%) | 3 (30%) | 0 | 0 |
| Dermatologic | ||||
| Acneiform Rash | 3 (100%) | 6 (60%) | 0 | 0 |
| Hepatic | ||||
| Hypoalbuminemia | 0 | 5 (50%) | 1 (33%) | 0 |
| Infection | 1 (33%) | 0 | 0 | 2 (20%) |
| Metabolic | ||||
| Hypomagnesemia | 1 (33%) | 3 (30%) | 0 | 0 |
| Hyponatremia | 1 (33%) | 4 (40%) | 0 | 0 |
| Hypophosphatemia | 2 (67%) | 2 (20%) | 0 | 1 (10%) |
| Vascular | ||||
| Thromboembolism | 0 | 0 | 1 (33%) | 1 (10%) |
| Peripheral Edema | 0 | 2 (20%) | 0 | 1 (10%) |
| Head and Neck Lymphedema | 1 (33%) | 2 (20%) | 0 | 0 |
| Progression-Free Survival | |||
| Covariate | HR | 95% CI | p value |
| Circulating Biomarkers | |||
| scMet | 1.92 | 0.95–3.86 | 0.048 |
| HGF | 0.86 | 0.57–1.29 | 0.452 |
| IL6 | 0.59 | 0.27–1.26 | 0.110 (0.337) |
| VeriStrat | 1.54 | 0.41–5.81 | 0.517 (1.0) |
| Tumor Biomarkers | |||
| cMet | 2.09 | 0.70–6.24 | 0.172 |
| HGF | 1.65 | 0.37–7.33 | 0.508 |
| cMet–HGF | 2.11 | 0.54–8.14 | 0.273 |
| H1T | 2.80 | 1.03–7.56 | 0.023 (0.187) |
| H11D | 1.14 | 0.86–1.51 | 0.335 (1.0) |
| Overall Survival | |||
| Covariate | HR | 95% CI | p value |
| Circulating Biomarkers | |||
| scMet | 1.63 | 0.87–3.06 | 0.113 |
| HGF | 0.90 | 0.60–1.34 | 0.599 |
| IL6 | 0.80 | 0.51–1.24 | 0.292 (0.790) |
| VeriStrat | 0.89 | 0.25–3.06 | 0.852 (1.0) |
| Tumor Biomarkers | |||
| cMet | 1.58 | 0.51–4.92 | 0.422 |
| HGF | 1.47 | 0.32–6.61 | 0.618 |
| cMet–HGF | 1.84 | 0.45–7.56 | 0.390 |
| H1T | 1.94 | 0.80–4.71 | 0.13 (0.790) |
| H11D | 1.13 | 0.85–1.50 | 0.39 (0.790) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauman, J.E.; Ohr, J.; Gooding, W.E.; Ferris, R.L.; Duvvuri, U.; Kim, S.; Johnson, J.T.; Soloff, A.C.; Wallweber, G.; Winslow, J.; et al. Phase I Study of Ficlatuzumab and Cetuximab in Cetuximab-Resistant, Recurrent/Metastatic Head and Neck Cancer. Cancers 2020, 12, 1537. https://doi.org/10.3390/cancers12061537
Bauman JE, Ohr J, Gooding WE, Ferris RL, Duvvuri U, Kim S, Johnson JT, Soloff AC, Wallweber G, Winslow J, et al. Phase I Study of Ficlatuzumab and Cetuximab in Cetuximab-Resistant, Recurrent/Metastatic Head and Neck Cancer. Cancers. 2020; 12(6):1537. https://doi.org/10.3390/cancers12061537
Chicago/Turabian StyleBauman, Julie E., James Ohr, William E. Gooding, Robert L. Ferris, Umamaheswar Duvvuri, Seungwon Kim, Jonas T. Johnson, Adam C. Soloff, Gerald Wallweber, John Winslow, and et al. 2020. "Phase I Study of Ficlatuzumab and Cetuximab in Cetuximab-Resistant, Recurrent/Metastatic Head and Neck Cancer" Cancers 12, no. 6: 1537. https://doi.org/10.3390/cancers12061537
APA StyleBauman, J. E., Ohr, J., Gooding, W. E., Ferris, R. L., Duvvuri, U., Kim, S., Johnson, J. T., Soloff, A. C., Wallweber, G., Winslow, J., Gaither-Davis, A., Grandis, J. R., & Stabile, L. P. (2020). Phase I Study of Ficlatuzumab and Cetuximab in Cetuximab-Resistant, Recurrent/Metastatic Head and Neck Cancer. Cancers, 12(6), 1537. https://doi.org/10.3390/cancers12061537