Targeting Mononuclear Phagocyte Receptors in Cancer Immunotherapy: New Perspectives of the Triggering Receptor Expressed on Myeloid Cells (TREM-1)



Abstract
1. Introduction
2. MPs in Tumors
2.1. MP Pro- and Anti-Cancer Activities
2.2. Tumor Hypoxia Contributes to MP Pro-Tumoral Phenotype
2.3. Targeting MPs in Cancers
3. MP Activatory/Inhibitory Surface Receptors
3.1. Pattern Recognition Receptor (PRR) and Immunoregulatory Signaling (IRS) Receptors Expressed on MPs
3.2. Role of PRR and IRS Receptors in Tumors
3.3. PRR and IRS Receptors as Therapeutic Targets in Cancer
4. TREM
4.1. TREM-1 Structure and Expression Regulation in MPs
4.2. TREM-1 Putative Ligands and Signaling
5. TREM-1 Role in Inflammatory Responses and Infectious/Non-Infectious Diseases
6. TREM-1 Role in Cancer
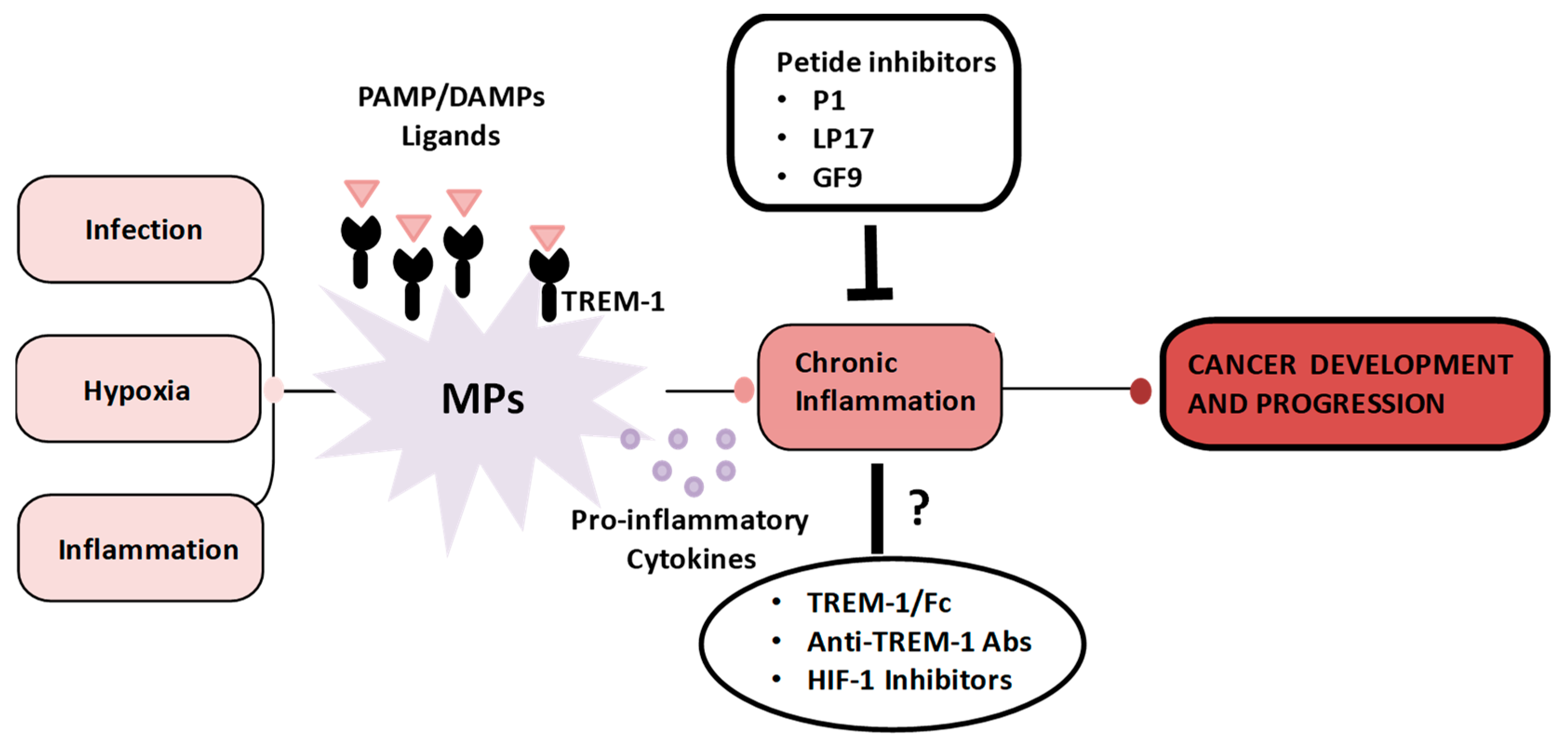
7. TREM-1 Therapeutic Targeting
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TME | tumor microenvironment |
| IBD | inflammatory bowels disease |
| MPs | mononuclear phagocytes |
| PAMPs/DAMPs | pathogen- or damage-associated molecular patterns |
| TREM 1 | triggering receptor expressed on myeloid cells |
| Mn | monocytes |
| TAMs | tumor-associated macrophages |
| TADCs | tumor-associated dendritic cells |
| ROS | reactive oxygen species |
| NO | nitric oxide |
| pO2 | partial oxygen pressure |
| PRRs | pattern recognition receptors |
| IRS | immunoregulatory signaling |
| ADCC | Ab-dependent cell-mediated cytotoxicity |
| TLRs | toll-like receptors |
| MSR | macrophage scavenger receptor 1 |
| MARCO | macrophage receptor with collagenous structure |
| NSCLC | non-small cell lung cancer |
| SIRPα | signal regulatory protein-alpha |
| CD47 | cluster of differentiation 47 |
| PD1 | programmed cell death-1 |
| PDL1 | programmed cell death-1 ligand |
| LILRB1 | leukocyte immunoglobulin-like receptor B1 |
| β2M | β2-microglobulin |
| TIM-3 | T cell Ig and mucin domain 3 |
| HMGB1 | high mobility group box 1 |
| mAbs | monoclonal antibodies |
| FcRs | Fc region of Igs |
| ADP | Ab-dependent phagocytosis |
| CYT | cytoplasmic tail |
| DAP12 | DNA activating protein 12 |
| ITAM | immunoreceptor tyrosine-based activatory motif |
| sTREM-1 | soluble TREM-1 |
| MMP | metalloproteinase |
| LCs | Langerhans cells |
| HRE | hypoxia response element |
| HIF1 | hypoxia-inducible transcription factor |
| HCC | hepatocellular carcinoma |
| siRNA | small interfering RNA |
| HSCs | hepatic stellate cells |
References
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef]
- Bosch, F.X.; Lorincz, A.; Munoz, N.; Meijer, C.J.; Shah, K.V. The causal relation between human papillomavirus and cervical cancer. J. Clin. Pathol. 2002, 55, 244–265. [Google Scholar] [CrossRef]
- Svrcek, M.; Borralho, N.P.; Villanacci, V.; Beaugerie, L.; Rogler, G.; De Hertogh, G.; Tripathi, M.; Feakins, R. Clinicopathological and Molecular Specificities of Inflammatory Bowel Disease-Related Colorectal Neoplastic Lesions: The Role of Inflammation. J. Crohn’s Colitis 2018, 12, 1486–1498. [Google Scholar] [CrossRef]
- Ponzoni, M.; Pastorino, F.; Di, P.D.; Perri, P.; Brignole, C. Targeting Macrophages as a Potential Therapeutic Intervention: Impact on Inflammatory Diseases and Cancer. Int. J. Mol. Sci. 2018, 19, 1953. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- de Visser, K.E.; Coussens, L.M. The interplay between innate and adaptive immunity regulates cancer development. Cancer Immunol. Immunother. 2005, 54, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- LaGory, E.L.; Giaccia, A.J. The ever-expanding role of HIF in tumour and stromal biology. Nat. Cell Biol. 2016, 18, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Cancer and inflammation: An old intuition with rapidly evolving new concepts. Annu. Rev. Immunol. 2012, 30, 677–706. [Google Scholar] [CrossRef] [PubMed]
- Ben Baruch, A. Inflammation-associated immune suppression in cancer: The roles played by cytokines, chemokines and additional mediators. Semin. Cancer Biol. 2006, 16, 38–52. [Google Scholar] [CrossRef]
- Rahat, M.A.; Marom, B.; Bitterman, H.; Weiss-Cerem, L.; Kinarty, A.; Lahat, N. Hypoxia reduces the output of matrix metalloproteinase-9 (MMP-9) in monocytes by inhibiting its secretion and elevating membranal association. J. Leukoc. Biol. 2006, 79, 706–718. [Google Scholar] [CrossRef]
- Shalapour, S.; Karin, M. Immunity, inflammation, and cancer: An eternal fight between good and evil. J. Clin. Investig. 2015, 125, 3347–3355. [Google Scholar] [CrossRef]
- Murdoch, C.; Giannoudis, A.; Lewis, C.E. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004, 104, 2224–2234. [Google Scholar] [CrossRef]
- Knowles, H.J.; Harris, A.L. Macrophages and the hypoxic tumour microenvironment. Front Biosci. 2007, 12, 4298–4314. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Mantovani, A. Immunology in the clinic review series; focus on cancer: Tumour-associated” macrophages: Undisputed stars of the inflammatory tumour microenvironment. Clin. Exp. Immunol. 2012, 167, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Stout, R.D.; Jiang, C.; Matta, B.; Tietzel, I.; Watkins, S.K.; Suttles, J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 2005, 175, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Bosco, M.C.; Puppo, M.; Blengio, F.; Fraone, T.; Cappello, P.; Giovarelli, M.; Varesio, L. Monocytes and dendritic cells in a hypoxic environment: Spotlights on chemotaxis and migration. Immunobiology 2008, 213, 733–749. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef]
- Davis, M.J.; Tsang, T.M.; Qiu, Y.; Dayrit, J.K.; Freij, J.B.; Huffnagle, G.B.; Olszewski, M.A. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. MBio 2013, 4, e00264-13. [Google Scholar] [CrossRef]
- Bosco, M.C.; Rapisarda, A.; Reffo, G.; Massazza, S.; Pastorino, S.; Varesio, L. Macrophage activating properties of the tryptophan catabolite picolinic acid. Adv. Exp. Med. Biol. 2003, 527, 55–65. [Google Scholar]
- Gratchev, A.; Kzhyshkowska, J.; Kothe, K.; Muller-Molinet, I.; Kannookadan, S.; Utikal, J.; Goerdt, S. Mphi1 and Mphi2 can be re-polarized by Th2 or Th1 cytokines, respectively, and respond to exogenous danger signals. Immunobiology 2006, 211, 473–486. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Raggi, F.; Pelassa, S.; Pierobon, D.; Penco, F.; Gattorno, M.; Novelli, F.; Eva, A.; Varesio, L.; Giovarelli, M.; Bosco, M.C. Regulation of Human Macrophage M1-M2 Polarization Balance by Hypoxia and the Triggering Receptor Expressed on Myeloid Cells-1. Front. Immunol. 2017, 8, 1097. [Google Scholar] [CrossRef] [PubMed]
- Bosco, M.C. Macrophage polarization: Reaching across the aisle? J. Allergy Clin. Immunol. 2019, 143, 1348–1350. [Google Scholar] [CrossRef] [PubMed]
- Granucci, F.; Zanoni, I.; Ricciardi-Castagnoli, P. Central role of dendritic cells in the regulation and deregulation of immune responses. Cell Mol. Life Sci. 2008, 65, 1683–1697. [Google Scholar] [CrossRef] [PubMed]
- Munitz, A. Inhibitory receptors on myeloid cells: New targets for therapy? Pharmacol. Ther. 2010, 125, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Gerber, J.S.; Mosser, D.M. Stimulatory and inhibitory signals originating from the macrophage Fcgamma receptors. Microbes Infect. 2001, 3, 131–139. [Google Scholar] [CrossRef]
- Colonna, M.; Nakajima, H.; Cella, M. A family of inhibitory and activating Ig-like receptors that modulate function of lymphoid and myeloid cells. Semin. Immunol. 2000, 12, 121–127. [Google Scholar] [CrossRef]
- Bosco, M.C.; Raggi, F.; Varesio, L. Therapeutic Potential of Targeting TREM-1 in Inflammatory Diseases and Cancer. Curr. Pharm. Des. 2016, 22, 6209–6233. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Recht, L.; Strober, S. The Promise of Targeting Macrophages in Cancer Therapy. Clin. Cancer Res. 2017, 23, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- Cortese, N.; Donadon, M.; Rigamonti, A.; Marchesi, F. Macrophages at the crossroads of anticancer strategies. Front. Biosci. 2019, 24, 1271–1283. [Google Scholar]
- Klesney-Tait, J.; Turnbull, I.R.; Colonna, M. The TREM receptor family and signal integration. Nat. Immunol. 2006, 7, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Bouchon, A.; Dietrich, J.; Colonna, M. Cutting edge: Inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J. Immunol. 2000, 164, 4991–4995. [Google Scholar] [CrossRef]
- Bennaceur, K.; Chapman, J.; Brikci-Nigassa, L.; Sanhadji, K.; Touraine, J.L.; Portoukalian, J. Dendritic cells dysfunction in tumour environment. Cancer Lett. 2008, 272, 186–196. [Google Scholar] [CrossRef]
- Schmieder, A.; Michel, J.; Schonhaar, K.; Goerdt, S.; Schledzewski, K. Differentiation and gene expression profile of tumor-associated macrophages. Semin. Cancer Biol. 2012, 22, 289–297. [Google Scholar] [CrossRef]
- Vicari, A.P.; Caux, C.; Trinchieri, G. Tumour escape from immune surveillance through dendritic cell inactivation. Semin. Cancer Biol. 2002, 12, 33–42. [Google Scholar] [CrossRef]
- Lin, K.W.; Jacek, T.; Jacket, R. Dendritic cells heterogeneity and its role in cancer immunity. Cancer Res. 2006, 2, 35–40. [Google Scholar]
- Nielsen, S.R.; Schmid, M.C. Macrophages as Key Drivers of Cancer Progression and Metastasis. Mediat. Inflamm. 2017, 2017, 9624760. [Google Scholar] [CrossRef]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, L.L.; Von Andrian, U.H. Travellers in many guises: The origins and destinations of dendritic cells. Immunol. Cell Biol 2002, 80, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Lamagna, C.; Aurrand-Lions, M.; Imhof, B.A. Dual role of macrophages in tumor growth and angiogenesis. J. Leukoc. Biol. 2006, 80, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Bronte, V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Investig. 2007, 117, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Khallou-Laschet, J.; Varthaman, A.; Fornasa, G.; Compain, C.; Gaston, A.T.; Clement, M.; Dussiot, M.; Levillain, O.; Graff-Dubois, S.; Nicoletti, A.; et al. Macrophage plasticity in experimental atherosclerosis. PLoS ONE 2010, 5, e8852. [Google Scholar] [CrossRef]
- Zeyda, M.; Farmer, D.; Todoric, J.; Aszmann, O.; Speiser, M.; Gyori, G.; Zlabinger, G.J.; Stulnig, T.M. Human adipose tissue macrophages are of an anti-inflammatory phenotype but capable of excessive pro-inflammatory mediator production. Int. J. Obes. 2007, 31, 1420–1428. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Laoui, D.; Van Overmeire, E.; Di Conza, G.; Aldeni, C.; Keirsse, J.; Morias, Y.; Movahedi, K.; Houbracken, I.; Schouppe, E.; Elkrim, Y.; et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. 2014, 74, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol. Rev. 2008, 222, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Qu, J.; Sun, Y.; Wang, J.; Liu, X.; Wang, F.; Zhang, H.; Wang, W.; Ma, X.; Gao, X.; et al. Prognostic significance of tumor-associated macrophages in breast cancer: A meta-analysis of the literature. Oncotarget 2017, 8, 30576–30586. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Liu, J.; Xu, R.; Zhu, X.; Zhao, X.; Qian, B.Z. Prognostic role of tumour-associated macrophages and macrophage scavenger receptor 1 in prostate cancer: A systematic review and meta-analysis. Oncotarget 2017, 8, 83261–83269. [Google Scholar] [CrossRef]
- Yin, S.; Huang, J.; Li, Z.; Zhang, J.; Luo, J.; Lu, C.; Xu, H.; Xu, H. The Prognostic and Clinicopathological Significance of Tumor-Associated Macrophages in Patients with Gastric Cancer: A Meta-Analysis. PLoS ONE 2017, 12, e0170042. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Xiao, Z.; Guo, C.; Pu, Q.; Ma, L.; Liu, C.; Lin, F.; Liao, H.; You, Z.; Liu, L. Prognostic impact of tumor-associated macrophage infiltration in non-small cell lung cancer: A systemic review and meta-analysis. Oncotarget 2016, 7, 34217–34228. [Google Scholar] [CrossRef]
- Guo, B.; Cen, H.; Tan, X.; Ke, Q. Meta-analysis of the prognostic and clinical value of tumor-associated macrophages in adult classical Hodgkin lymphoma. BMC Med. 2016, 14, 159. [Google Scholar] [CrossRef]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef]
- Rossi, M.; Young, J.W. Human dendritic cells: Potent antigen-presenting cells at the crossroads of innate and adaptive immunity. J. Immunol. 2005, 175, 1373–1381. [Google Scholar] [CrossRef]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef]
- Ueno, H.; Klechevsky, E.; Morita, R.; Aspord, C.; Cao, T.; Matsui, T.; Di Pucchio, T.; Connolly, J.; Fay, J.W.; Pascual, V.; et al. Dendritic cell subsets in health and disease. Immunol. Rev. 2007, 219, 118–142. [Google Scholar] [CrossRef]
- Tran Janco, J.M.; Lamichhane, P.; Karyampudi, L.; Knutson, K.L. Tumor-infiltrating dendritic cells in cancer pathogenesis. J. Immunol. 2015, 194, 2985–2991. [Google Scholar] [CrossRef]
- Lin, A.; Schildknecht, A.; Nguyen, L.T.; Ohashi, P.S. Dendritic cells integrate signals from the tumor microenvironment to modulate immunity and tumor growth. Immunol. Lett. 2010, 127, 77–84. [Google Scholar] [CrossRef]
- Krempski, J.; Karyampudi, L.; Behrens, M.D.; Erskine, C.L.; Hartmann, L.; Dong, H.; Goode, E.L.; Kalli, K.R.; Knutson, K.L. Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J. Immunol. 2011, 186, 6905–6913. [Google Scholar] [CrossRef]
- Huarte, E.; Cubillos-Ruiz, J.R.; Nesbeth, Y.C.; Scarlett, U.K.; Martinez, D.G.; Buckanovich, R.J.; Benencia, F.; Stan, R.V.; Keler, T.; Sarobe, P.; et al. Depletion of dendritic cells delays ovarian cancer progression by boosting antitumor immunity. Cancer Res. 2008, 68, 7684–7691. [Google Scholar] [CrossRef]
- Gabrilovich, D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat. Rev. Immunol. 2004, 4, 941–952. [Google Scholar] [CrossRef]
- Allavena, P.; Sica, A.; Vecchi, A.; Locati, M.; Sozzani, S.; Mantovani, A. The chemokine receptor switch paradigm and dendritic cell migration: Its significance in tumor tissues. Immunol. Rev. 2000, 177, 141–149. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef]
- Imtiyaz, H.Z.; Simon, M.C. Hypoxia-inducible factors as essential regulators of inflammation. Curr. Top. Microbiol. Immunol. 2010, 345, 105–120. [Google Scholar]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef]
- Vaupel, P.; Hockel, M. Tumor oxygenation and its relevance to tumor physiology and treatment. Adv. Exp. Med. Biol. 2003, 510, 45–49. [Google Scholar]
- Bosco, M.C.; D’Orazi, G.; Del, B.D. Targeting hypoxia in tumor: A new promising therapeutic strategy. J. Exp. Clin. Cancer Res. 2020, 39, 8. [Google Scholar] [CrossRef]
- Lu, X.; Kang, Y. Hypoxia and hypoxia-inducible factors: Master regulators of metastasis. Clin. Cancer Res. 2010, 16, 5928–5935. [Google Scholar] [CrossRef]
- Palazon, A.; Aragones, J.; Morales-Kastresana, A.; de Landazuri, M.O.; Melero, I. Molecular pathways: Hypoxia response in immune cells fighting or promoting cancer. Clin. Cancer Res. 2012, 18, 1207–1213. [Google Scholar] [CrossRef]
- Sica, A.; Melillo, G.; Varesio, L. Hypoxia: A double-edged sword of immunity. J. Mol. Med. 2011, 89, 657–665. [Google Scholar] [CrossRef]
- Sitkovsky, M.; Lukashev, D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat. Rev. Immunol. 2008, 5, 712–721. [Google Scholar] [CrossRef]
- Multhoff, G.; Vaupel, P. Hypoxia Compromises Anti-Cancer Immune Responses. Adv. Exp. Med. Biol. 2020, 1232, 131–143. [Google Scholar]
- Westendorf, A.M.; Skibbe, K.; Adamczyk, A.; Buer, J.; Geffers, R.; Hansen, W.; Pastille, E.; Jendrossek, V. Hypoxia Enhances Immunosuppression by Inhibiting CD4+ Effector T Cell Function and Promoting Treg Activity. Cell Physiol. Biochem. 2017, 41, 1271–1284. [Google Scholar] [CrossRef]
- Parodi, M.; Raggi, F.; Cangelosi, D.; Manzini, C.; Balsamo, M.; Blengio, F.; Eva, A.; Varesio, L.; Pietra, G.; Moretta, L.; et al. Hypoxia Modifies the Transcriptome of Human NK Cells, Modulates Their Immunoregulatory Profile, and Influences NK Cell Subset Migration. Front. Immunol. 2018, 9, 2358. [Google Scholar] [CrossRef]
- Terry, S.; Buart, S.; Chouaib, S. Hypoxic Stress-Induced Tumor and Immune Plasticity, Suppression, and Impact on Tumor Heterogeneity. Front. Immunol. 2017, 8, 1625. [Google Scholar] [CrossRef]
- Triner, D.; Shah, Y.M. Hypoxia-inducible factors: A central link between inflammation and cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef]
- Bosco, M.C.; Varesio, L. Dendritic cell reprogramming by the hypoxic environment. Immunobiology 2012, 217, 1241–1249. [Google Scholar] [CrossRef]
- Tripathi, C.; Tewari, B.N.; Kanchan, R.K.; Baghel, K.S.; Nautiyal, N.; Shrivastava, R.; Kaur, H.; Bhatt, M.L.; Bhadauria, S. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget 2014, 5, 5350–5368. [Google Scholar] [CrossRef]
- Escribese, M.M.; Casas, M.; Corbi, A.L. Influence of low oxygen tensions on macrophage polarization. Immunobiology 2012, 217, 1233–1240. [Google Scholar] [CrossRef]
- Bosco, M.C.; Varesio, L. Hypoxia and Gene Expression. In Hypoxia and Cancer. Biological Implications and Therapeutic Opportunities; Melillo, G., Ed.; Humana Press: Totowa, NJ, USA, 2014; pp. 91–119. [Google Scholar]
- Leblond, M.M.; Gerault, A.N.; Corroyer-Dulmont, A.; MacKenzie, E.T.; Petit, E.; Bernaudin, M.; Valable, S. Hypoxia induces macrophage polarization and re-education toward an M2 phenotype in U87 and U251 glioblastoma models. Oncoimmunology 2016, 5, e1056442. [Google Scholar] [CrossRef]
- Zhang, J.; Cao, J.; Ma, S.; Dong, R.; Meng, W.; Ying, M.; Weng, Q.; Chen, Z.; Ma, J.; Fang, Q.; et al. Tumor hypoxia enhances Non-Small Cell Lung Cancer metastasis by selectively promoting macrophage M2 polarization through the activation of ERK signaling. Oncotarget 2014, 5, 9664–9677. [Google Scholar] [CrossRef]
- Guo, X.; Xue, H.; Shao, Q.; Wang, J.; Guo, X.; Chen, X.; Zhang, J.; Xu, S.; Li, T.; Zhang, P.; et al. Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and M-CSFR. Oncotarget 2016, 7, 80521. [Google Scholar] [CrossRef]
- Zeng, Q.; Jewell, C.M. Directing toll-like receptor signaling in macrophages to enhance tumor immunotherapy. Curr. Opin. Biotechnol. 2019, 60, 138–145. [Google Scholar] [CrossRef]
- Fang, W.B.; Yao, M.; Brummer, G.; Acevedo, D.; Alhakamy, N.; Berkland, C.; Cheng, N. Targeted gene silencing of CCL2 inhibits triple negative breast cancer progression by blocking cancer stem cell renewal and M2 macrophage recruitment. Oncotarget 2016, 7, 49349–49367. [Google Scholar] [CrossRef]
- Moisan, F.; Francisco, E.B.; Brozovic, A.; Duran, G.E.; Wang, Y.C.; Chaturvedi, S.; Seetharam, S.; Snyder, L.A.; Doshi, P.; Sikic, B.I. Enhancement of paclitaxel and carboplatin therapies by CCL2 blockade in ovarian cancers. Mol. Oncol. 2014, 8, 1231–1239. [Google Scholar] [CrossRef]
- Sandhu, S.K.; Papadopoulos, K.; Fong, P.C.; Patnaik, A.; Messiou, C.; Olmos, D.; Wang, G.; Tromp, B.J.; Puchalski, T.A.; Balkwill, F.; et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 1041–1050. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Li, C.W.; Lai, Y.J.; Hsu, J.L.; Hung, M.C. Activation of phagocytosis by immune checkpoint blockade. Front. Med. 2018, 12, 473–480. [Google Scholar] [CrossRef]
- Mayoux, M.; Roller, A.; Pulko, V.; Sammicheli, S.; Chen, S.; Sum, E.; Jost, C.; Fransen, M.F.; Buser, R.B.; Kowanetz, M.; et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci. Transl. Med. 2020, 12, 473–480. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II Study of Autologous Monocyte-Derived mRNA Electroporated Dendritic Cells (TriMixDC-MEL) Plus Ipilimumab in Patients With Pretreated Advanced Melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef]
- Dietrich, J.; Nakajima, H.; Colonna, M. Human inhibitory and activating Ig-like receptors which modulate the function of myeloid cells. Microbes Infect. 2000, 2, 323–329. [Google Scholar] [CrossRef]
- Bosco, M.C.; Puppo, M.; Santangelo, C.; Anfosso, L.; Pfeffer, U.; Fardin, P.; Battaglia, F.; Varesio, L. Hypoxia modifies the transcriptome of primary human monocytes: Modulation of novel immune-related genes and identification of CC-chemokine ligand 20 as a new hypoxia-inducible gene. J. Immunol. 2006, 177, 1941–1955. [Google Scholar] [CrossRef]
- Bosco, M.C.; Pierobon, D.; Blengio, F.; Raggi, F.; Vanni, C.; Gattorno, M.; Eva, A.; Novelli, F.; Cappello, P.; Giovarelli, M.; et al. Hypoxia modulates the gene expression profile of immunoregulatory receptors in human mature dendritic cells: Identification of TREM-1 as a novel hypoxic marker in vitro and in vivo. Blood 2011, 117, 2625–2639. [Google Scholar] [CrossRef]
- Pierobon, D.; Bosco, M.C.; Blengio, F.; Raggi, F.; Eva, A.; Filippi, M.; Musso, T.; Novelli, F.; Cappello, P.; Varesio, L.; et al. Chronic hypoxia reprograms human immature dendritic cells by inducing a proinflammatory phenotype and TREM-1 expression. Eur. J. Immunol. 2013, 43, 949–966. [Google Scholar] [CrossRef]
- Clark, G.J.; Ju, X.; Tate, C.; Hart, D.N. The CD300 family of molecules are evolutionarily significant regulators of leukocyte functions. Trends Immunol. 2009, 30, 209–217. [Google Scholar] [CrossRef]
- Kanazawa, N. Dendritic cell immunoreceptors: C-type lectin receptors for pattern-recognition and signaling on antigen-presenting cells. J. Dermatol. Sci. 2007, 45, 77–86. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef]
- Melillo, G.; Cox, G.; Biragyn, A.; Sheffler, L.; Varesio, L. Regulation of nitric oxide synthase mRNA expression by interferon-gamma and picolinic acid. J. Biol. Chem. 1994, 269, 8128. [Google Scholar]
- Arredouani, M.S.; Palecanda, A.; Koziel, H.; Huang, Y.C.; Imrich, A.; Sulahian, T.H.; Ning, Y.Y.; Yang, Z.; Pikkarainen, T.; Sankala, M.; et al. MARCO is the major binding receptor for unopsonized particles and bacteria on human alveolar macrophages. J. Immunol. 2005, 175, 6058–6064. [Google Scholar] [CrossRef]
- Nakamura, K.; Funakoshi, H.; Miyamoto, K.; Tokunaga, F.; Nakamura, T. Molecular cloning and functional characterization of a human scavenger receptor with C-type lectin (SRCL), a novel member of a scavenger receptor family. Biochem. Biophys. Res. Commun. 2001, 280, 1028–1035. [Google Scholar] [CrossRef]
- Pasare, C.; Medzhitov, R. Toll-like receptors: Linking innate and adaptive immunity. Adv. Exp. Med. Biol. 2005, 560, 11–18. [Google Scholar]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat. Immunol. 2001, 2, 675–680. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Wenink, M.H.; van den Berg, W.B.; van Riel, P.L.; Radstake, T.R. Fc gamma receptor mediated modulation of dendritic cells as a potential strategy in the battle against rheumatoid arthritis. Neth. J. Med. 2006, 64, 103–108. [Google Scholar]
- Javaid, N.; Choi, S. Toll-like Receptors from the Perspective of Cancer Treatment. Cancers 2020, 12, 297. [Google Scholar] [CrossRef] [PubMed]
- Georgoudaki, A.M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Ostling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef] [PubMed]
- La, F.L.; Boura, V.F.; Alexeyenko, A.; Berglund, A.; Ponten, V.; Mattsson, J.S.M.; Djureinovic, D.; Persson, J.; Brunnstrom, H.; Isaksson, J.; et al. Expression of scavenger receptor MARCO defines a targetable tumor-associated macrophage subset in non-small cell lung cancer. Int. J. Cancer 2018, 143, 1741–1752. [Google Scholar]
- LeMaoult, J.; Zafaranloo, K.; Le Danff, C.; Carosella, E.D. HLA-G up-regulates ILT2, ILT3, ILT4, and KIR2DL4 in antigen presenting cells, NK cells, and T cells. FASEB J. 2005, 19, 662–664. [Google Scholar] [CrossRef]
- Weiskopf, K. Cancer immunotherapy targeting the CD47/SIRPalpha axis. Eur. J. Cancer 2017, 76, 100–109. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef]
- Curiel, T.J.; Wei, S.; Dong, H.; Alvarez, X.; Cheng, P.; Mottram, P.; Krzysiek, R.; Knutson, K.L.; Daniel, B.; Zimmermann, M.C.; et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med. 2003, 9, 562–567. [Google Scholar] [CrossRef]
- Han, G.; Chen, G.; Shen, B.; Li, Y. Tim-3: An activation marker and activation limiter of innate immune cells. Front. Immunol. 2013, 4, 449. [Google Scholar] [CrossRef]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef]
- Fang, H.Y.; Hughes, R.; Murdoch, C.; Coffelt, S.B.; Biswas, S.K.; Harris, A.L.; Johnson, R.S.; Imityaz, H.Z.; Simon, M.C.; Fredlund, E.; et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood 2009, 114, 844–859. [Google Scholar] [CrossRef]
- Yang, M.; Ma, C.; Liu, S.; Sun, J.; Shao, Q.; Gao, W.; Zhang, Y.; Li, Z.; Xie, Q.; Dong, Z.; et al. Hypoxia skews dendritic cells to a T helper type 2-stimulating phenotype and promotes tumour cell migration by dendritic cell-derived osteopontin. Immunology 2009, 128, e237–e249. [Google Scholar] [CrossRef] [PubMed]
- Raggi, F.; Blengio, F.; Eva, A.; Pende, D.; Varesio, L.; Bosco, M.C. Identification of CD300a as a new hypoxia-inducible gene and a regulator of CCL20 and VEGF production by human monocytes and macrophages. Innate Immun. 2014, 20, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Kuhlicke, J.; Frick, J.S.; Morote-Garcia, J.C.; Rosenberger, P.; Eltzschig, H.K. Hypoxia inducible factor (HIF)-1 coordinates induction of Toll-like receptors TLR2 and TLR6 during hypoxia. PLoS ONE 2007, 2, e1364. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.; Vonderheide, R.H. Anti-CD40 agonist antibodies: Preclinical and clinical experience. Update Cancer Ther. 2007, 2, 61–65. [Google Scholar] [CrossRef]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef]
- Perry, C.J.; Munoz-Rojas, A.R.; Meeth, K.M.; Kellman, L.N.; Amezquita, R.A.; Thakral, D.; Du, V.Y.; Wang, J.X.; Damsky, W.; Kuhlmann, A.L.; et al. Myeloid-targeted immunotherapies act in synergy to induce inflammation and antitumor immunity. J. Exp. Med. 2018, 215, 877–893. [Google Scholar] [CrossRef]
- Hoves, S.; Ooi, C.H.; Wolter, C.; Sade, H.; Bissinger, S.; Schmittnaegel, M.; Ast, O.; Giusti, A.M.; Wartha, K.; Runza, V.; et al. Rapid activation of tumor-associated macrophages boosts preexisting tumor immunity. J. Exp. Med. 2018, 215, 859–876. [Google Scholar] [CrossRef]
- Li, F.; Ravetch, J.V. Inhibitory Fcgamma receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science 2011, 333, 1030–1034. [Google Scholar] [CrossRef]
- Beatty, G.L.; Torigian, D.A.; Chiorean, E.G.; Saboury, B.; Brothers, A.; Alavi, A.; Troxel, A.B.; Sun, W.; Teitelbaum, U.R.; Vonderheide, R.H.; et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2013, 19, 6286–6295. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef]
- Ring, N.G.; Herndler-Brandstetter, D.; Weiskopf, K.; Shan, L.; Volkmer, J.P.; George, B.M.; Lietzenmayer, M.; McKenna, K.M.; Naik, T.J.; McCarty, A.; et al. Anti-SIRPalpha antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E10578–E10585. [Google Scholar] [CrossRef] [PubMed]
- Sikic, B.I.; Lakhani, N.; Patnaik, A.; Shah, S.A.; Chandana, S.R.; Rasco, D.; Colevas, A.D.; O’Rourke, T.; Narayanan, S.; Papadopoulos, K.; et al. First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients With Advanced Cancers. J. Clin. Oncol. 2019, 37, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Barkal, A.A.; Weiskopf, K.; Kao, K.S.; Gordon, S.R.; Rosental, B.; Yiu, Y.Y.; George, B.M.; Markovic, M.; Ring, N.G.; Tsai, J.M.; et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat. Immunol. 2018, 19, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Gubin, M.M.; Esaulova, E.; Ward, J.P.; Malkova, O.N.; Runci, D.; Wong, P.; Noguchi, T.; Arthur, C.D.; Meng, W.; Alspach, E.; et al. High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint Cancer Therapy. Cell 2018, 175, 1443. [Google Scholar] [CrossRef] [PubMed]
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng 2018, 2, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Kaczanowska, S.; Joseph, A.M.; Davila, E. TLR agonists: Our best frenemy in cancer immunotherapy. J. Leukoc. Biol. 2013, 93, 847–863. [Google Scholar] [CrossRef]
- Vidyarthi, A.; Khan, N.; Agnihotri, T.; Negi, S.; Das, D.K.; Aqdas, M.; Chatterjee, D.; Colegio, O.R.; Tewari, M.K.; Agrewala, J.N. TLR-3 Stimulation Skews M2 Macrophages to M1 Through IFN-alphabeta Signaling and Restricts Tumor Progression. Front. Immunol. 2018, 9, 1650. [Google Scholar] [CrossRef]
- Sato-Kaneko, F.; Yao, S.; Ahmadi, A.; Zhang, S.S.; Hosoya, T.; Kaneda, M.M.; Varner, J.A.; Pu, M.; Messer, K.S.; Guiducci, C.; et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight 2017, 2, e93397. [Google Scholar] [CrossRef]
- Solinas, C.; De Silva, P.; Bron, D.; Willard-Gallo, K.; Sangiolo, D. Significance of TIM3 expression in cancer: From biology to the clinic. Semin. Oncol. 2019, 46, 372–379. [Google Scholar] [CrossRef]
- Scarlett, U.K.; Cubillos-Ruiz, J.R.; Nesbeth, Y.C.; Martinez, D.G.; Engle, X.; Gewirtz, A.T.; Ahonen, C.L.; Conejo-Garcia, J.R. In situ stimulation of CD40 and Toll-like receptor 3 transforms ovarian cancer-infiltrating dendritic cells from immunosuppressive to immunostimulatory cells. Cancer Res. 2009, 69, 7329–7337. [Google Scholar] [CrossRef]
- Colonna, M. TREMs in the immune system and beyond. Nat. Rev. Immunol. 2003, 3, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.W.; McVicar, D.W. TREM and TREM-like receptors in inflammation and disease. Curr. Opin. Immunol. 2009, 21, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Sharif, O.; Knapp, S. From expression to signaling: Roles of TREM-1 and TREM-2 in innate immunity and bacterial infection. Immunobiology 2008, 213, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Gingras, M.C.; Lapillonne, H.; Margolin, J.F. TREM-1, MDL-1, and DAP12 expression is associated with a mature stage of myeloid development. Mol. Immunol. 2002, 38, 817–824. [Google Scholar] [CrossRef]
- Tessarz, A.S.; Cerwenka, A. The TREM-1/DAP12 pathway. Immunol. Lett. 2008, 116, 111–116. [Google Scholar] [CrossRef]
- Pelham, C.J.; Pandya, A.N.; Agrawal, D.K. Triggering receptor expressed on myeloid cells receptor family modulators: A patent review. Expert Opin. Ther. Pat. 2014, 24, 1383–1395. [Google Scholar] [CrossRef]
- Allcock, R.J.; Barrow, A.D.; Forbes, S.; Beck, S.; Trowsdale, J. The human TREM gene cluster at 6p21.1 encodes both activating and inhibitory single IgV domain receptors and includes NKp44. Eur. J. Immunol. 2003, 33, 567–577. [Google Scholar] [CrossRef]
- Chung, D.H.; Seaman, W.E.; Daws, M.R. Characterization of TREM-3, an activating receptor on mouse macrophages: Definition of a family of single Ig domain receptors on mouse chromosome 17. Eur. J. Immunol. 2002, 32, 59–66. [Google Scholar] [CrossRef]
- Nguyen, A.H.; Berim, I.G.; Agrawal, D.K. Chronic inflammation and cancer: Emerging roles of triggering receptors expressed on myeloid cells. Expert. Rev. Clin. Immunol. 2015, 11, 849–857. [Google Scholar] [CrossRef]
- Colonna, M.; Facchetti, F. TREM-1 (triggering receptor expressed on myeloid cells): A new player in acute inflammatory responses. J. Infect. Dis. 2003, 187 (Suppl. S2), S397–S401. [Google Scholar] [CrossRef]
- Bouchon, A.; Facchetti, F.; Weigand, M.A.; Colonna, M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature 2001, 410, 1103–1107. [Google Scholar] [CrossRef] [PubMed]
- Radaev, S.; Kattah, M.; Rostro, B.; Colonna, M.; Sun, P.D. Crystal structure of the human myeloid cell activating receptor TREM-1. Structure 2003, 11, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Kelker, M.S.; Foss, T.R.; Peti, W.; Teyton, L.; Kelly, J.W.; Wuthrich, K.; Wilson, I.A. Crystal structure of human triggering receptor expressed on myeloid cells 1 (TREM-1) at 1.47 A. J. Mol. Biol. 2004, 342, 1237–1248. [Google Scholar] [CrossRef] [PubMed]
- Kelker, M.S.; Debler, E.W.; Wilson, I.A. Crystal structure of mouse triggering receptor expressed on myeloid cells 1 (TREM-1) at 1.76 A. J. Mol. Biol. 2004, 344, 1175–1181. [Google Scholar] [CrossRef]
- Baruah, S.; Keck, K.; Vrenios, M.; Pope, M.R.; Pearl, M.; Doerschug, K.; Klesney-Tait, J. Identification of a Novel Splice Variant Isoform of TREM-1 in Human Neutrophil Granules. J. Immunol. 2015, 195, 5725–5731. [Google Scholar] [CrossRef]
- Gomez-Pina, V.; Soares-Schanoski, A.; Rodriguez-Rojas, A.; Del Fresno, C.; Garcia, F.; Vallejo-Cremades, M.T.; Fernandez-Ruiz, I.; Arnalich, F.; Fuentes-Prior, P.; Lopez-Collazo, E. Metalloproteinases shed TREM-1 ectodomain from lipopolysaccharide-stimulated human monocytes. J. Immunol. 2007, 179, 4065–4073. [Google Scholar] [CrossRef]
- Gibot, S.; Kolopp-Sarda, M.N.; Bene, M.C.; Bollaert, P.E.; Lozniewski, A.; Mory, F.; Levy, B.; Faure, G.C. A soluble form of the triggering receptor expressed on myeloid cells-1 modulates the inflammatory response in murine sepsis. J. Exp. Med. 2004, 200, 1419–1426. [Google Scholar] [CrossRef]
- Haselmayer, P.; Grosse-Hovest, L.; von Landenberg, P.; Schild, H.; Radsak, M.P. TREM-1 ligand expression on platelets enhances neutrophil activation. Blood 2007, 110, 1029–1035. [Google Scholar] [CrossRef]
- Dorner, B.G.; Scheffold, A.; Rolph, M.S.; Huser, M.B.; Kaufmann, S.H.; Radbruch, A.; Flesch, I.E.; Kroczek, R.A. MIP-1alpha, MIP-1beta, RANTES, and ATAC/lymphotactin function together with IFN-gamma as type 1 cytokines. Proc. Natl. Acad. Sci. USA 2002, 99, 6181–6186. [Google Scholar] [CrossRef]
- Bleharski, J.R.; Kiessler, V.; Buonsanti, C.; Sieling, P.A.; Stenger, S.; Colonna, M.; Modlin, R.L. A role for triggering receptor expressed on myeloid cells-1 in host defense during the early-induced and adaptive phases of the immune response. J. Immunol. 2003, 170, 3812–3818. [Google Scholar] [CrossRef]
- Fortin, C.F.; Lesur, O.; Fulop, T., Jr. Effects of TREM-1 activation in human neutrophils: Activation of signaling pathways, recruitment into lipid rafts and association with TLR4. Int. Immunol. 2007, 19, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Radsak, M.P.; Salih, H.R.; Rammensee, H.G.; Schild, H. Triggering receptor expressed on myeloid cells-1 in neutrophil inflammatory responses: Differential regulation of activation and survival. J. Immunol. 2004, 172, 4956–4963. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Heiderscheidt, C.A.; Joo, M.; Gao, X.; Knezevic, N.; Mehta, D.; Sadikot, R.T. MYD88-dependent and -independent activation of TREM-1 via specific TLR ligands. Eur. J. Immunol. 2010, 40, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Azam, T.; Ferwerda, G.; Girardin, S.E.; Kim, S.H.; Dinarello, C.A. Triggering receptor expressed on myeloid cells-1 (TREM-1) amplifies the signals induced by the NACHT-LRR (NLR) pattern recognition receptors. J. Leukoc. Biol. 2006, 80, 1454–1461. [Google Scholar] [CrossRef]
- Rolli, J.; Loukili, N.; Levrand, S.; Rosenblatt-Velin, N.; Rignault-Clerc, S.; Waeber, B.; Feihl, F.; Pacher, P.; Liaudet, L. Bacterial flagellin elicits widespread innate immune defense mechanisms, apoptotic signaling, and a sepsis-like systemic inflammatory response in mice. Crit. Care 2010, 14, R160. [Google Scholar] [CrossRef]
- Molad, Y.; Pokroy-Shapira, E.; Carmon, V. CpG-oligodeoxynucleotide-induced TLR9 activation regulates macrophage TREM-1 expression and shedding. Innate Immun. 2013, 19, 623–630. [Google Scholar] [CrossRef]
- Derive, M.; Massin, F.; Gibot, S. Triggering receptor expressed on myeloid cells-1 as a new therapeutic target during inflammatory diseases. Self/Nonself 2010, 1, 225–230. [Google Scholar] [CrossRef]
- Arts, R.J.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G. TREM-1: Intracellular signaling pathways and interaction with pattern recognition receptors. J. Leukoc. Biol. 2013, 93, 209–215. [Google Scholar] [CrossRef]
- Roe, K.; Gibot, S.; Verma, S. Triggering receptor expressed on myeloid cells-1 (TREM-1): A new player in antiviral immunity? Front. Microbiol. 2014, 5, 627. [Google Scholar] [CrossRef]
- Hyun, J.; McMahon, R.S.; Lang, A.L.; Edwards, J.S.; Badilla, A.D.; Greene, M.E.; Stone, G.W.; Pallikkuth, S.; Stevenson, M.; Dykxhoorn, D.M.; et al. HIV and HCV augments inflammatory responses through increased TREM-1 expression and signaling in Kupffer and Myeloid cells. PLoS Pathog. 2019, 15, e1007883. [Google Scholar] [CrossRef]
- Zanzinger, K.; Schellack, C.; Nausch, N.; Cerwenka, A. Regulation of triggering receptor expressed on myeloid cells 1 expression on mouse inflammatory monocytes. Immunology 2009, 128, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Ornatowska, M.; Joo, M.S.; Sadikot, R.T. TREM-1 expression in macrophages is regulated at transcriptional level by NF-kappaB and PU.1. Eur. J. Immunol. 2007, 37, 2300–2308. [Google Scholar] [CrossRef] [PubMed]
- Gibot, S.; Buonsanti, C.; Massin, F.; Romano, M.; Kolopp-Sarda, M.N.; Benigni, F.; Faure, G.C.; Bene, M.C.; Panina-Bordignon, P.; Passini, N.; et al. Modulation of the triggering receptor expressed on the myeloid cell type 1 pathway in murine septic shock. Infect. Immun. 2006, 74, 2823–2830. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Peng, A.; Sun, M.; Deng, Q.; Hazlett, L.D.; Yuan, J.; Liu, X.; Gao, Q.; Feng, L.; He, J.; et al. TREM-1 amplifies corneal inflammation after Pseudomonas aeruginosa infection by modulating Toll-like receptor signaling and Th1/Th2-type immune responses. Infect. Immun. 2011, 79, 2709–2716. [Google Scholar] [CrossRef]
- Gibot, S.; Alauzet, C.; Massin, F.; Sennoune, N.; Faure, G.C.; Bene, M.C.; Lozniewski, A.; Bollaert, P.E.; Levy, B. Modulation of the triggering receptor expressed on myeloid cells-1 pathway during pneumonia in rats. J. Infect. Dis. 2006, 194, 975–983. [Google Scholar] [CrossRef]
- Buckland, K.F.; Ramaprakash, H.; Murray, L.A.; Carpenter, K.J.; Choi, E.S.; Kunkel, S.L.; Lukacs, N.W.; Xing, Z.; Aoki, N.; Hartl, D.; et al. Triggering receptor expressed on myeloid cells-1 (TREM-1) modulates immune responses to Aspergillus fumigatus during fungal asthma in mice. Immunol. Investig. 2011, 40, 692–722. [Google Scholar] [CrossRef]
- Hu, L.T.; Du, Z.D.; Zhao, G.Q.; Jiang, N.; Lin, J.; Wang, Q.; Xu, Q.; Cong, L.; Qiu, S. Role of TREM-1 in response to Aspergillus fumigatus infection in corneal epithelial cells. Int. Immunopharmacol. 2014, 23, 288–293. [Google Scholar] [CrossRef]
- DePaula-Silva, A.B.; Gorbea, C.; Doty, D.J.; Libbey, J.E.; Sanchez, J.M.S.; Hanak, T.J.; Cazalla, D.; Fujinami, R.S. Differential transcriptional profiles identify microglial- and macrophage-specific gene markers expressed during virus-induced neuroinflammation. J. Neuroinflamm. 2019, 16, 152. [Google Scholar] [CrossRef]
- Gibot, S.; Cravoisy, A.; Levy, B.; Bene, M.C.; Faure, G.; Bollaert, P.E. Soluble triggering receptor expressed on myeloid cells and the diagnosis of pneumonia. N. Engl. J. Med. 2004, 350, 451–458. [Google Scholar] [CrossRef]
- Lagler, H.; Sharif, O.; Haslinger, I.; Matt, U.; Stich, K.; Furtner, T.; Doninger, B.; Schmid, K.; Gattringer, R.; de Vos, A.F.; et al. TREM-1 activation alters the dynamics of pulmonary IRAK-M expression in vivo and improves host defense during pneumococcal pneumonia. J. Immunol. 2009, 183, 2027–2036. [Google Scholar] [CrossRef]
- de Oliveira, M.A.; Dos Santos Dantas, P.H.; Figueira, M.S.-S.; Sales-Campos, H. The role of the triggering receptor expressed on myeloid cells-1 (TREM-1) in non-bacterial infections. Crit. Rev. Microbiol. 2020, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Schenk, M.; Bouchon, A.; Birrer, S.; Colonna, M.; Mueller, C. Macrophages expressing triggering receptor expressed on myeloid cells-1 are underrepresented in the human intestine. J. Immunol. 2005, 174, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Mehta, H.J.; Mohammed, K.; Nasreen, N.; Roman, R.; Brantly, M.; Sadikot, R.T. TREM-1 is induced in tumor associated macrophages by cyclo-oxygenase pathway in human non-small cell lung cancer. PLoS ONE 2014, 9, e94241. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Kohsaka, H.; Kitasato, H.; Akahoshi, T. Lipopolysaccharide-induced up-regulation of triggering receptor expressed on myeloid cells-1 expression on macrophages is regulated by endogenous prostaglandin E2. J. Immunol. 2007, 178, 1144–1150. [Google Scholar] [CrossRef]
- Hosoda, H.; Tamura, H.; Kida, S.; Nagaoka, I. Transcriptional regulation of mouse TREM-1 gene in RAW264.7 macrophage-like cells. Life Sci. 2011, 89, 115–122. [Google Scholar] [CrossRef]
- Syed, M.A.; Joo, M.; Abbas, Z.; Rodger, D.; Christman, J.W.; Mehta, D.; Sadikot, R.T. Expression of TREM-1 is inhibited by PGD2 and PGJ2 in macrophages. Exp. Cell Res. 2010, 316, 3140–3149. [Google Scholar] [CrossRef]
- Hosoda, H.; Tamura, H.; Nagaoka, I. Evaluation of the lipopolysaccharide-induced transcription of the human TREM-1 gene in vitamin D3-matured THP-1 macrophage-like cells. Int. J. Mol. Med. 2015, 36, 1300–1310. [Google Scholar] [CrossRef]
- Kim, T.H.; Lee, B.; Kwon, E.; Choi, S.J.; Lee, Y.H.; Song, G.G.; Sohn, J.; Ji, J.D. Regulation of TREM-1 expression by 1,25-dihydroxyvitamin D3 in human monocytes/macrophages. Immunol. Lett. 2013, 154, 80–85. [Google Scholar] [CrossRef]
- Rigo, I.; McMahon, L.; Dhawan, P.; Christakos, S.; Yim, S.; Ryan, L.K.; Diamond, G. Induction of triggering receptor expressed on myeloid cells (TREM-1) in airway epithelial cells by 1,25(OH)(2) vitamin D(3). Innate Immun. 2012, 18, 250–257. [Google Scholar] [CrossRef]
- Pierobon, D.; Raggi, F.; Cambieri, I.; Pelassa, S.; Occhipinti, S.; Cappello, P.; Novelli, F.; Musso, T.; Eva, A.; Castagnoli, C.; et al. Regulation of Langerhans cell functions in a hypoxic environment. J. Mol. Med. 2016, 94, 943–955. [Google Scholar] [CrossRef]
- Murat, A.; Migliavacca, E.; Hussain, S.F.; Heimberger, A.B.; Desbaillets, I.; Hamou, M.F.; Ruegg, C.; Stupp, R.; Delorenzi, M.; Hegi, M.E. Modulation of angiogenic and inflammatory response in glioblastoma by hypoxia. PLoS ONE 2009, 4, e5947. [Google Scholar] [CrossRef] [PubMed]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005, 2005, re12. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhou, W.; Yin, S.; Zhou, Y.; Chen, T.; Qian, J.; Su, R.; Hong, L.; Lu, H.; Zhang, F.; et al. Blocking Triggering Receptor Expressed on Myeloid Cells-1-Positive Tumor-Associated Macrophages Induced by Hypoxia Reverses Immunosuppression and Anti-Programmed Cell Death Ligand 1 Resistance in Liver Cancer. Hepatology 2019, 70, 198–214. [Google Scholar] [CrossRef]
- Wong-Baeza, I.; Gonzalez-Roldan, N.; Ferat-Osorio, E.; Esquivel-Callejas, N.; Aduna-Vicente, R.; Arriaga-Pizano, L.; Astudillo-de la Vega, H.; Villasis-Keever, M.A.; Torres-Gonzalez, R.; Estrada-Garcia, I.; et al. Triggering receptor expressed on myeloid cells (TREM-1) is regulated post-transcriptionally and its ligand is present in the sera of some septic patients. Clin. Exp. Immunol. 2006, 145, 448–455. [Google Scholar] [CrossRef]
- El Mezayen, R.; El Gazzar, M.; Seeds, M.C.; McCall, C.E.; Dreskin, S.C.; Nicolls, M.R. Endogenous signals released from necrotic cells augment inflammatory responses to bacterial endotoxin. Immunol. Lett. 2007, 111, 36–44. [Google Scholar] [CrossRef]
- Wu, J.; Li, J.; Salcedo, R.; Mivechi, N.F.; Trinchieri, G.; Horuzsko, A. The proinflammatory myeloid cell receptor TREM-1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res. 2012, 72, 3977–3986. [Google Scholar] [CrossRef]
- Read, C.B.; Kuijper, J.L.; Hjorth, S.A.; Heipel, M.D.; Tang, X.; Fleetwood, A.J.; Dantzler, J.L.; Grell, S.N.; Kastrup, J.; Wang, C.; et al. Cutting Edge: Identification of neutrophil PGLYRP1 as a ligand for TREM-1. J. Immunol. 2015, 194, 1417–1421. [Google Scholar] [CrossRef]
- Mohamadzadeh, M.; Coberley, S.S.; Olinger, G.G.; Kalina, W.V.; Ruthel, G.; Fuller, C.L.; Swenson, D.L.; Pratt, W.D.; Kuhns, D.B.; Schmaljohn, A.L. Activation of triggering receptor expressed on myeloid cells-1 on human neutrophils by marburg and ebola viruses. J. Virol. 2006, 80, 7235–7244. [Google Scholar] [CrossRef]
- Dower, K.; Ellis, D.K.; Saraf, K.; Jelinsky, S.A.; Lin, L.L. Innate immune responses to TREM-1 activation: Overlap, divergence, and positive and negative cross-talk with bacterial lipopolysaccharide. J. Immunol. 2008, 180, 3520–3534. [Google Scholar] [CrossRef]
- Barraud, D.; Gibot, S. Triggering receptor expressed on myeloid cell 1. Crit. Care Clin. 2011, 27, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Lemarie, J.; Barraud, D.; Gibot, S. Host response biomarkers in sepsis: Overview on sTREM-1 detection. Methods Mol. Biol. 2015, 1237, 225–239. [Google Scholar] [PubMed]
- Derive, M.; Boufenzer, A.; Gibot, S. Attenuation of responses to endotoxin by the triggering receptor expressed on myeloid cells-1 inhibitor LR12 in nonhuman primate. Anesthesiology 2014, 120, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Derive, M.; Boufenzer, A.; Bouazza, Y.; Groubatch, F.; Alauzet, C.; Barraud, D.; Lozniewski, A.; Leroy, P.; Tran, N.; Gibot, S. Effects of a TREM-like transcript 1-derived peptide during hypodynamic septic shock in pigs. Shock 2013, 39, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Gibot, S.; Massin, F.; Marcou, M.; Taylor, V.; Stidwill, R.; Wilson, P.; Singer, M.; Bellingan, G. TREM-1 promotes survival during septic shock in mice. Eur. J. Immunol. 2007, 37, 456–466. [Google Scholar] [CrossRef]
- Cavaillon, J.M. Monocyte TREM-1 membrane expression in non-infectious inflammation. Crit. Care 2009, 13, 152. [Google Scholar] [CrossRef]
- Collins, C.E.; La, D.T.; Yang, H.T.; Massin, F.; Gibot, S.; Faure, G.; Stohl, W. Elevated synovial expression of triggering receptor expressed on myeloid cells 1 in patients with septic arthritis or rheumatoid arthritis. Ann. Rheum. Dis. 2009, 68, 1768–1774. [Google Scholar] [CrossRef]
- Kuai, J.; Gregory, B.; Hill, A.; Pittman, D.D.; Feldman, J.L.; Brown, T.; Carito, B.; O’Toole, M.; Ramsey, R.; Adolfsson, O.; et al. TREM-1 expression is increased in the synovium of rheumatoid arthritis patients and induces the expression of pro-inflammatory cytokines. Rheumatology 2009, 48, 1352–1358. [Google Scholar] [CrossRef]
- Murakami, Y.; Akahoshi, T.; Aoki, N.; Toyomoto, M.; Miyasaka, N.; Kohsaka, H. Intervention of an inflammation amplifier, triggering receptor expressed on myeloid cells 1, for treatment of autoimmune arthritis. Arthritis Rheum. 2009, 60, 1615–1623. [Google Scholar] [CrossRef]
- Ho, C.C.; Liao, W.Y.; Wang, C.Y.; Lu, Y.H.; Huang, H.Y.; Chen, H.Y.; Chan, W.K.; Chen, H.W.; Yang, P.C. TREM-1 expression in tumor-associated macrophages and clinical outcome in lung cancer. Am. J. Respir. Crit. Care Med. 2008, 177, 763–770. [Google Scholar] [CrossRef]
- Kuemmel, A.; Alflen, A.; Schmidt, L.H.; Sebastian, M.; Wiewrodt, R.; Schulze, A.B.; Buhl, R.; Radsak, M. Soluble Triggering Receptor Expressed on Myeloid Cells 1 in lung cancer. Sci. Rep. 2018, 8, 10766. [Google Scholar] [CrossRef] [PubMed]
- Fontana, R.; Raccosta, L.; Rovati, L.; Steffensen, K.R.; Paniccia, A.; Jakobsson, T.; Melloni, G.; Bandiera, A.; Mangili, G.; Bergamini, A.; et al. Nuclear receptor ligands induce TREM-1 expression on dendritic cells: Analysis of their role in tumors. Oncoimmunology 2019, 8, 1554967. [Google Scholar] [CrossRef] [PubMed]
- Schenk, M.; Bouchon, A.; Seibold, F.; Mueller, C. TREM-1—Expressing intestinal macrophages crucially amplify chronic inflammation in experimental colitis and inflammatory bowel diseases. J. Clin. Investig. 2007, 117, 3097–3106. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.; Cheon, J.H.; Kim, B.Y.; Kim, D.H.; Kim, E.S.; Kim, T.I.; Lee, K.R.; Kim, W.H. Correlation of serum-soluble triggering receptor expressed on myeloid cells-1 with clinical disease activity in inflammatory bowel disease. Dig. Dis. Sci. 2009, 54, 1525–1531. [Google Scholar] [CrossRef]
- Weber, B.; Schuster, S.; Zysset, D.; Rihs, S.; Dickgreber, N.; Schurch, C.; Riether, C.; Siegrist, M.; Schneider, C.; Pawelski, H.; et al. TREM-1 deficiency can attenuate disease severity without affecting pathogen clearance. PLoS Pathog. 2014, 10, e1003900. [Google Scholar] [CrossRef]
- Tzivras, M.; Koussoulas, V.; Giamarellos-Bourboulis, E.J.; Tzivras, D.; Tsaganos, T.; Koutoukas, P.; Giamarellou, H.; Archimandritis, A. Role of soluble triggering receptor expressed on myeloid cells in inflammatory bowel disease. World J. Gastroenterol. 2006, 12, 3416–3419. [Google Scholar] [CrossRef]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef]
- Liao, R.; Sun, T.W.; Yi, Y.; Wu, H.; Li, Y.W.; Wang, J.X.; Zhou, J.; Shi, Y.H.; Cheng, Y.F.; Qiu, S.J.; et al. Expression of TREM-1 in hepatic stellate cells and prognostic value in hepatitis B-related hepatocellular carcinoma. Cancer Sci. 2012, 103, 984–992. [Google Scholar] [CrossRef]
- Duan, M.; Wang, Z.C.; Wang, X.Y.; Shi, J.Y.; Yang, L.X.; Ding, Z.B.; Gao, Q.; Zhou, J.; Fan, J. TREM-1, an inflammatory modulator, is expressed in hepatocellular carcinoma cells and significantly promotes tumor progression. Ann. Surg. Oncol. 2015, 22, 3121–3129. [Google Scholar] [CrossRef]
- Jung, K.H.; Yoo, W.; Stevenson, H.L.; Deshpande, D.; Shen, H.; Gagea, M.; Yoo, S.Y.; Wang, J.; Eckols, T.K.; Bharadwaj, U.; et al. Multifunctional Effects of a Small-Molecule STAT3 Inhibitor on NASH and Hepatocellular Carcinoma in Mice. Clin. Cancer Res. 2017, 23, 5537–5546. [Google Scholar] [CrossRef]
- Shen, Z.T.; Sigalov, A.B. Novel TREM-1 Inhibitors Attenuate Tumor Growth and Prolong Survival in Experimental Pancreatic Cancer. Mol. Pharm. 2017, 14, 4572–4582. [Google Scholar] [CrossRef] [PubMed]
- Anaya-Prado, R.; Norzgaray-Ibarra, F.G.; Bravo-Cuellar, A.; Perez-Avila, C.E.; Schadegg-Pena, D.; Anaya-Fernandez, M.M. [Expression of TREM-1 in patients with invasive cervical cancer and precursor lesions]. Rev. Med. Inst. Mex. Seguro Soc. 2015, 53, 722–727. [Google Scholar] [PubMed]
- Rubin, D.C.; Shaker, A.; Levin, M.S. Chronic intestinal inflammation: Inflammatory bowel disease and colitis-associated colon cancer. Front. Immunol. 2012, 3, 107. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chai, F.; Lu, G.; Hang, G.; Chen, C.; Chen, X.; Shi, J. TREM-1 inhibition attenuates inflammation and tumor within the colon. Int. Immunopharmacol. 2013, 17, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Dantas, P.H.D.S.; Matos, A.O.; da Silva, F.E.; Silva-Sales, M.; Sales-Campos, H. Triggering receptor expressed on myeloid cells-1 (TREM-1) as a therapeutic target in infectious and noninfectious disease: A critical review. Int. Rev. Immunol. 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sigalov, A.B. A novel ligand-independent peptide inhibitor of TREM-1 suppresses tumor growth in human lung cancer xenografts and prolongs survival of mice with lipopolysaccharide-induced septic shock. Int. Immunopharmacol. 2014, 21, 208–219. [Google Scholar] [CrossRef]
- Derive, M.; Bouazza, Y.; Sennoun, N.; Marchionni, S.; Quigley, L.; Washington, V.; Massin, F.; Max, J.P.; Ford, J.; Alauzet, C.; et al. Soluble TREM-like transcript-1 regulates leukocyte activation and controls microbial sepsis. J. Immunol. 2012, 188, 5585–5592. [Google Scholar] [CrossRef]
- Cuvier, V.; Lorch, U.; Witte, S.; Olivier, A.; Gibot, S.; Delor, I.; Garaud, J.J.; Derive, M.; Salcedo-Magguilli, M. A first-in-man safety and pharmacokinetics study of nangibotide, a new modulator of innate immune response through TREM-1 receptor inhibition. Br. J. Clin. Pharmacol. 2018, 84, 2270–2279. [Google Scholar] [CrossRef]
- Horst, S.A.; Linner, A.; Beineke, A.; Lehne, S.; Holtje, C.; Hecht, A.; Norrby-Teglund, A.; Medina, E.; Goldmann, O. Prognostic value and therapeutic potential of TREM-1 in Streptococcus pyogenes- induced sepsis. J. Innate Immun. 2013, 5, 581–590. [Google Scholar] [CrossRef]
- Wang, F.; Liu, S.; Wu, S.; Zhu, Q.; Ou, G.; Liu, C.; Wang, Y.; Liao, Y.; Sun, Z. Blocking TREM-1 signaling prolongs survival of mice with Pseudomonas aeruginosa induced sepsis. Cell Immunol. 2012, 272, 251–258. [Google Scholar] [CrossRef]
- Hyder, L.A.; Gonzalez, J.; Harden, J.L.; Johnson-Huang, L.M.; Zaba, L.C.; Pierson, K.C.; Eungdamrong, N.J.; Lentini, T.; Gulati, N.; Fuentes-Duculan, J.; et al. TREM-1 as a potential therapeutic target in psoriasis. J. Investig. Dermatol. 2013, 133, 1742–1751. [Google Scholar] [CrossRef] [PubMed]
- Nochi, H.; Aoki, N.; Oikawa, K.; Yanai, M.; Takiyama, Y.; Atsuta, Y.; Kobayashi, H.; Sato, K.; Tateno, M.; Matsuno, T.; et al. Modulation of hepatic granulomatous responses by transgene expression of DAP12 or TREM-1-Ig molecules. Am. J. Pathol. 2003, 162, 1191–1201. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, H.; Huang, J.; Chen, S.; Pan, X.; Huang, H.; Wang, L. TREM-1low is a novel characteristic for tumor-associated macrophages in lung cancer. Oncotarget 2016, 7, 40508–40517. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, Y.S.; Yeo, I.J.; Kim, K.C.; Han, S.B.; Hong, J.T. Inhibition of Lung Tumor Development in ApoE Knockout Mice via Enhancement of TREM-1 Dependent NK Cell Cytotoxicity. Front. Immunol. 2019, 10, 1379. [Google Scholar] [CrossRef] [PubMed]
- Onnis, B.; Rapisarda, A.; Melillo, G. Development of HIF-1 Inhibitors for Cancer Therapy. J. Cell Mol. Med. 2009, 13, 2780–2786. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 inhibitors for cancer therapy: From gene expression to drug discovery. Curr. Pharm. Des. 2009, 15, 3839–3843. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cancer Type | MP Type | Biological Fluid | Effects | References |
|---|---|---|---|---|
| NSCLC | Macrophages in tumors and pleural effusions | Pleural effusions | Correlates with aggressive tumor behavior, recurrence, poor disease-free, and overall patient survival | [194,221,222] |
| Lung adenocarcinoma | DCs in pleural effusions | ND | Associates with disease aggressiveness and bad prognosis | [223] |
| Colon carcinoma | Intestinal macrophages | Serum/plasma | Amplifies macrophage- mediated inflammation and intestinal tissue damage; correlates with aggressive tumor behavior and recurrence | [224,225,226,227] |
| HCC | Kupffer cells; HSCs; macrophages in tumor specimens | Plasma | Increases inflammatory responses, hepatic injury, tumor development and aggressive behavior, and poor patient survival; prognostic predictor for both early tumor recurrence and low patient overall and relapse-free survival after resection; mediates immunosupression | [205,208,228,229,230,231] |
| Pancreatic cancer | TAMs | ND | Correlates with cancer progression | [232] |
| T-cell lymphoma | MDSCs; TAMs | ND | Correlates with increased tumor volume | [182] |
| Cervical cancer | Monocytes | ND | Correlates with high-grade, invasive cancer | [233] |
| Glioblastoma | TAMs | ND | Associates with low outcome of chemotherapy-treated patients | [202] |
| Cancer Type | TREM-1 Peptide | Therapeutic Effects | References |
|---|---|---|---|
| Colon Cancer | LP-17 | Reduces pro-inflammatory mediator secretion by intestinal macrophages; attenuates intestinal inflammation, permeability, and epithelial damage; decreases epithelial histopathological alterations, proliferative activity, and progression to colon carcinoma | [235] |
| NSCLC | GF9 | Decreases cytokine production and delays tumor growth | [237] |
| Pancreatic Cancer | GF9 | Reduces TAMs infiltration, pro-inflammatory cytokine serum levels, and tumor growth, increasing animal survival | [232] |
| HCC | GF9 | Reduces tumor development and growth; abrogates TREM-1+TAM-mediated immunosuppressive effect by reducing Treg recruitment and CD8+T cell apoptosis/dysfunction; improves mouse survival; attenuates resistance to PD-L1 blockade improving its therapeutic efficacy | [205] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raggi, F.; Bosco, M.C. Targeting Mononuclear Phagocyte Receptors in Cancer Immunotherapy: New Perspectives of the Triggering Receptor Expressed on Myeloid Cells (TREM-1). Cancers 2020, 12, 1337. https://doi.org/10.3390/cancers12051337
Raggi F, Bosco MC. Targeting Mononuclear Phagocyte Receptors in Cancer Immunotherapy: New Perspectives of the Triggering Receptor Expressed on Myeloid Cells (TREM-1). Cancers. 2020; 12(5):1337. https://doi.org/10.3390/cancers12051337
Chicago/Turabian StyleRaggi, Federica, and Maria Carla Bosco. 2020. "Targeting Mononuclear Phagocyte Receptors in Cancer Immunotherapy: New Perspectives of the Triggering Receptor Expressed on Myeloid Cells (TREM-1)" Cancers 12, no. 5: 1337. https://doi.org/10.3390/cancers12051337
APA StyleRaggi, F., & Bosco, M. C. (2020). Targeting Mononuclear Phagocyte Receptors in Cancer Immunotherapy: New Perspectives of the Triggering Receptor Expressed on Myeloid Cells (TREM-1). Cancers, 12(5), 1337. https://doi.org/10.3390/cancers12051337