Optimization and Characterization of Protein Nanoparticles for the Targeted and Smart Delivery of Cytochrome c to Non-Small Cell Lung Carcinoma

 ,
,
Abstract
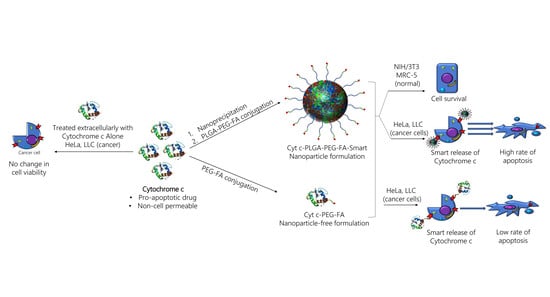
1. Introduction
2. Results
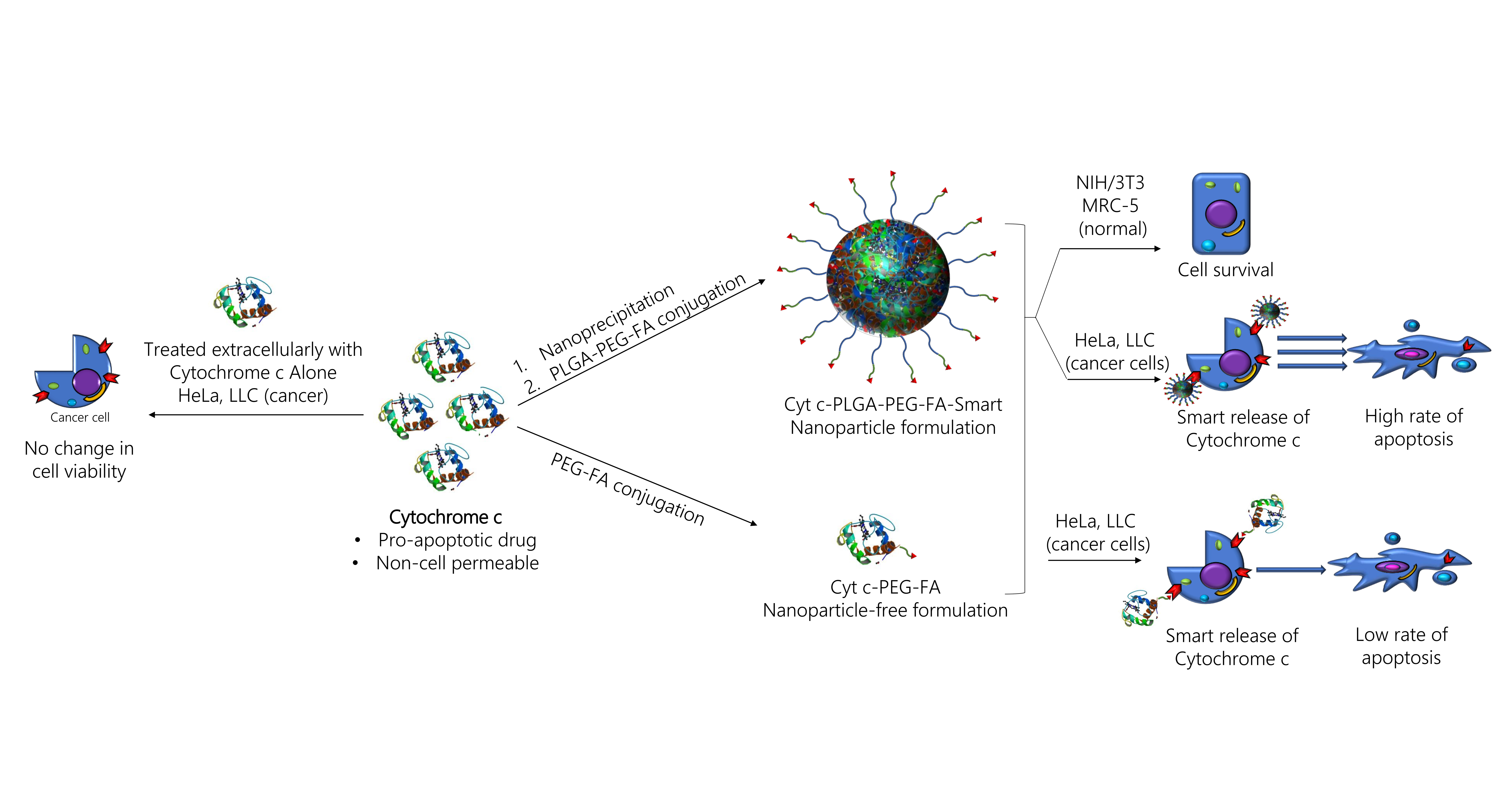
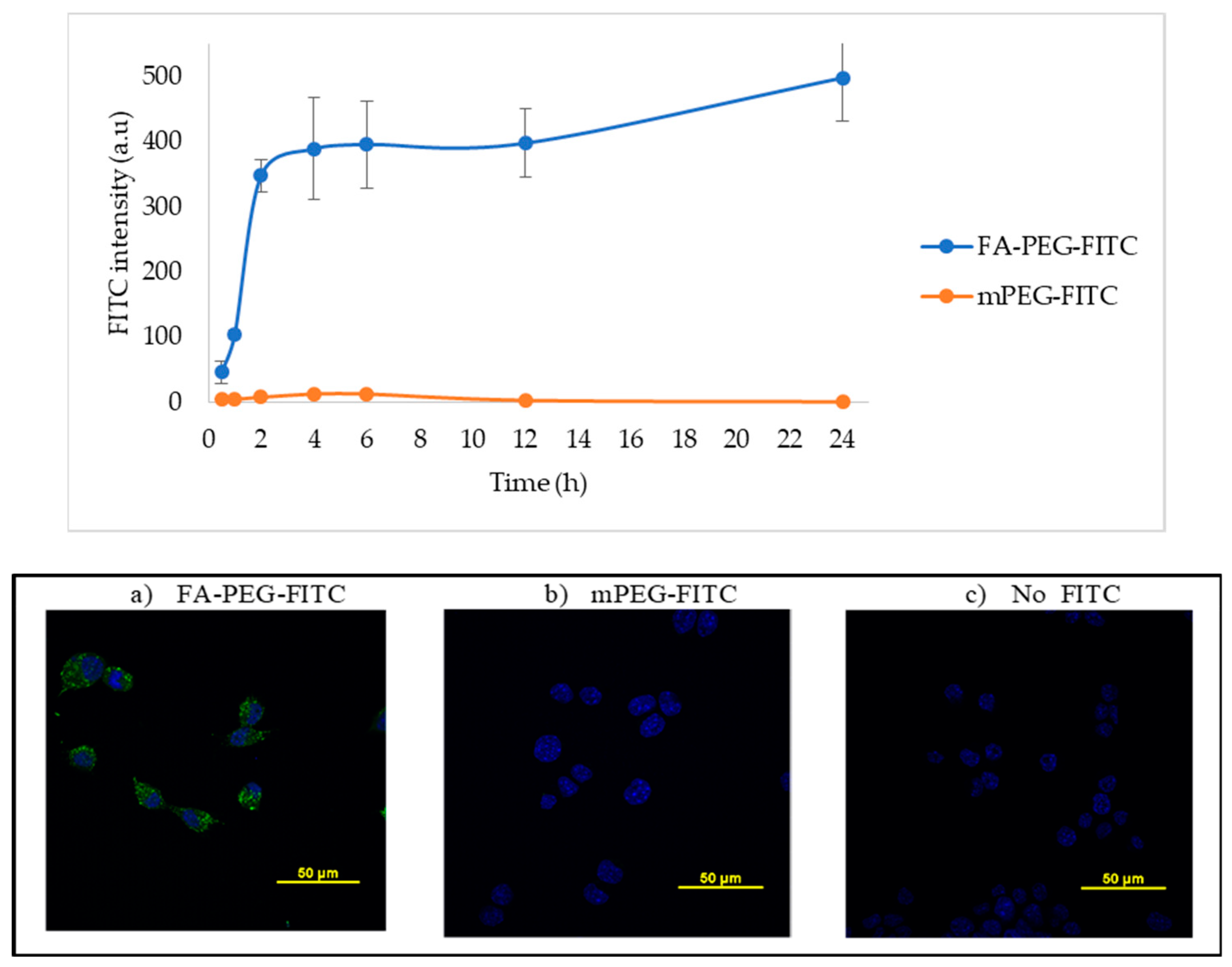
2.1. Folic Acid Uptake by LLC Cells
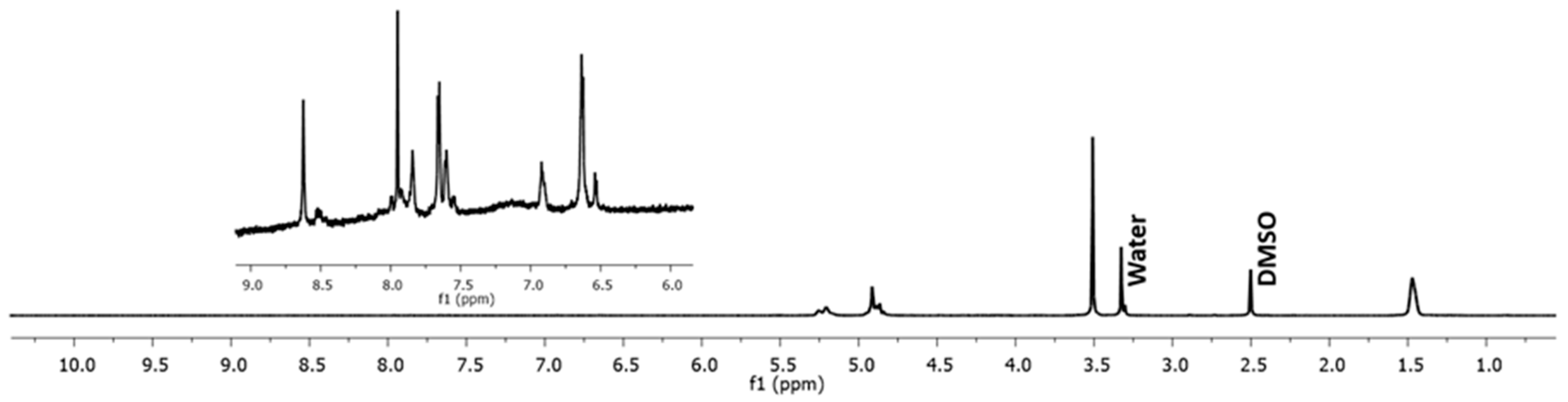
2.2. Synthesis and Characterization of Nanoparticle-Free Cyt c Formulation: Cyt c-PEG-FA
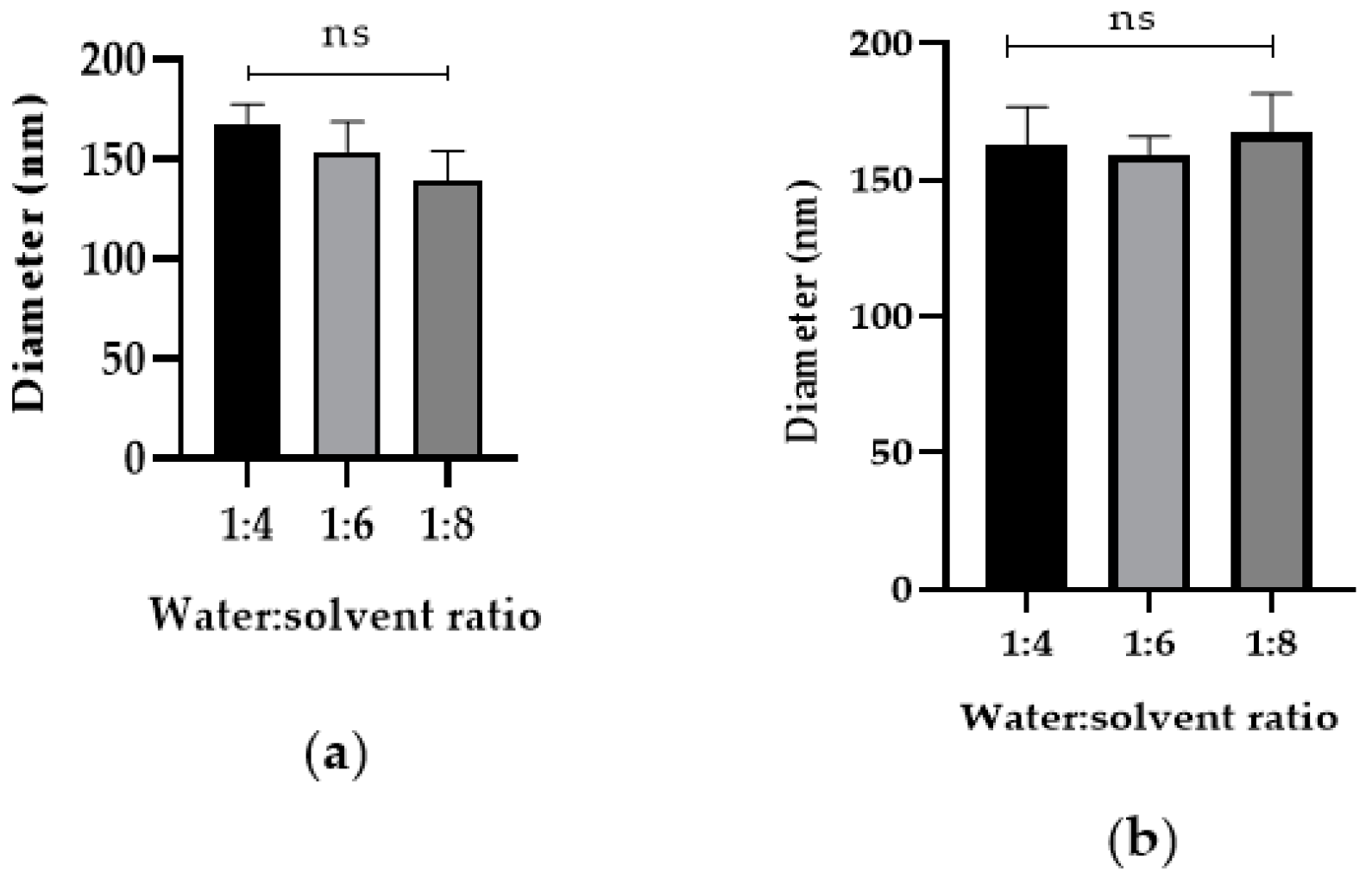
2.3. Cyt c Nanoprecipitation Optimization and Characterization (Synthesis of Cyt c Nanoparticles)
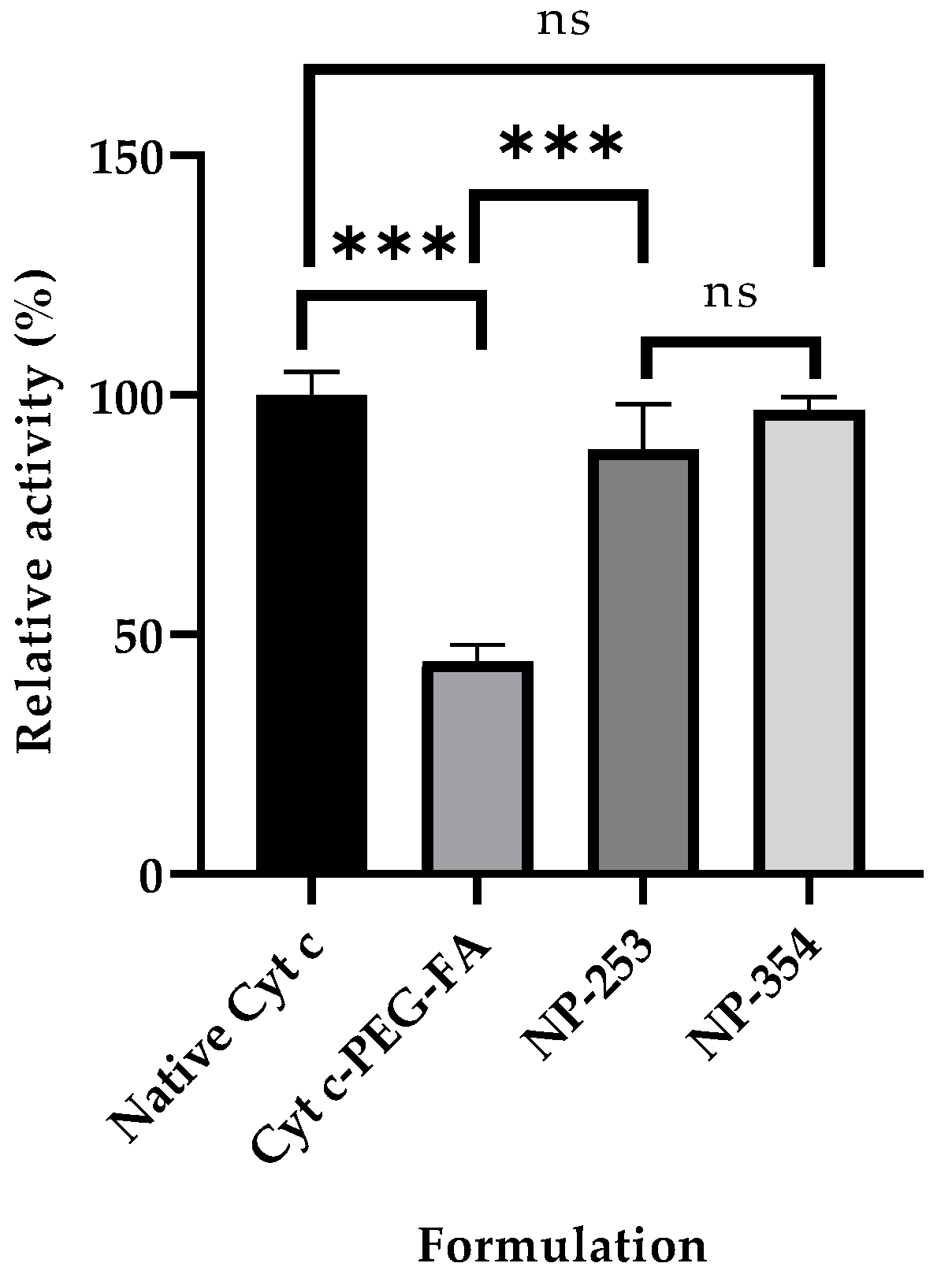
2.4. Cyt c-PLGA-PEG-FA NP Optimization and Characterization
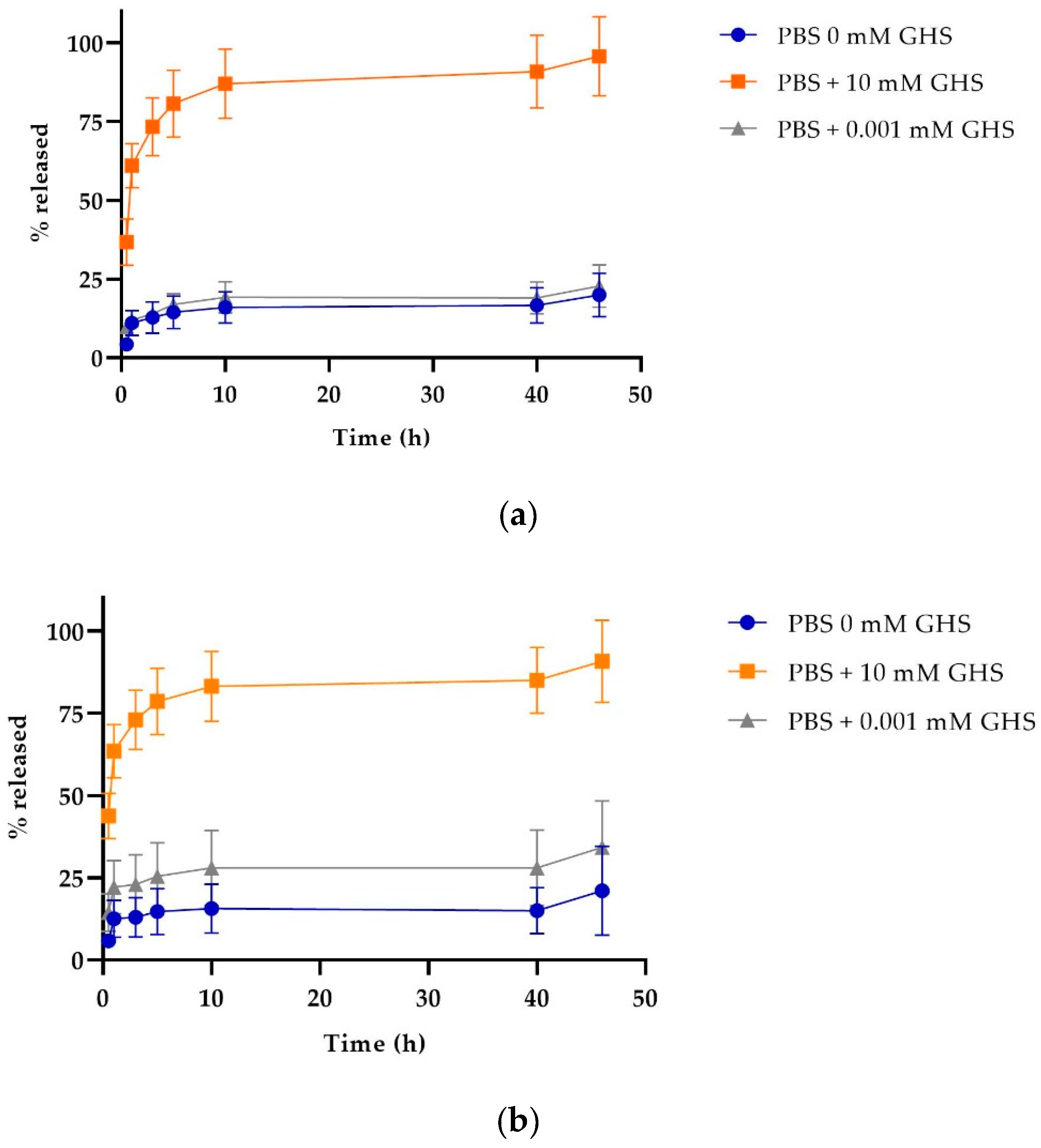
2.5. Release Profile of Nanoparticles
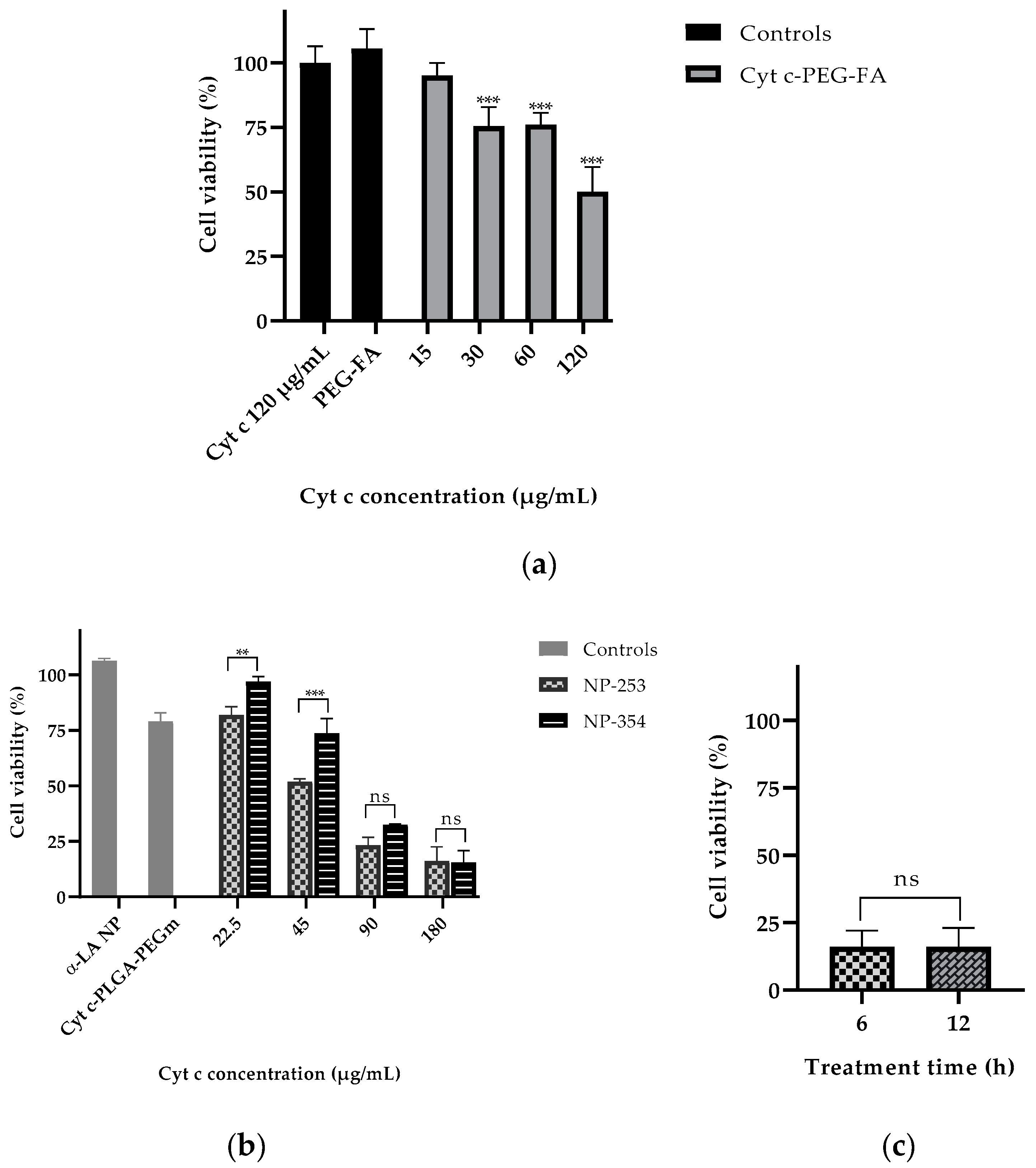
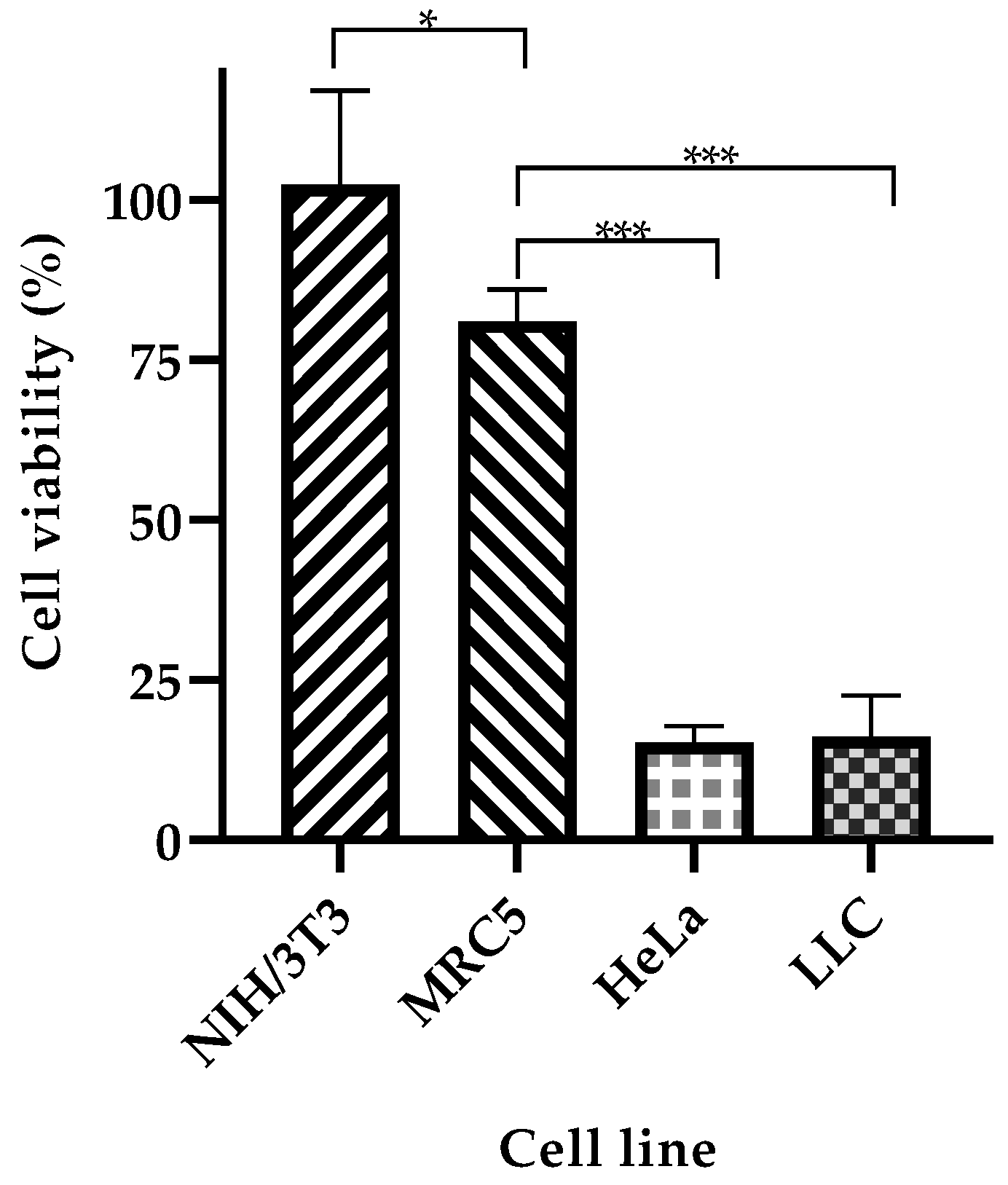
2.6. Cell Viability and Target Specificity Studies
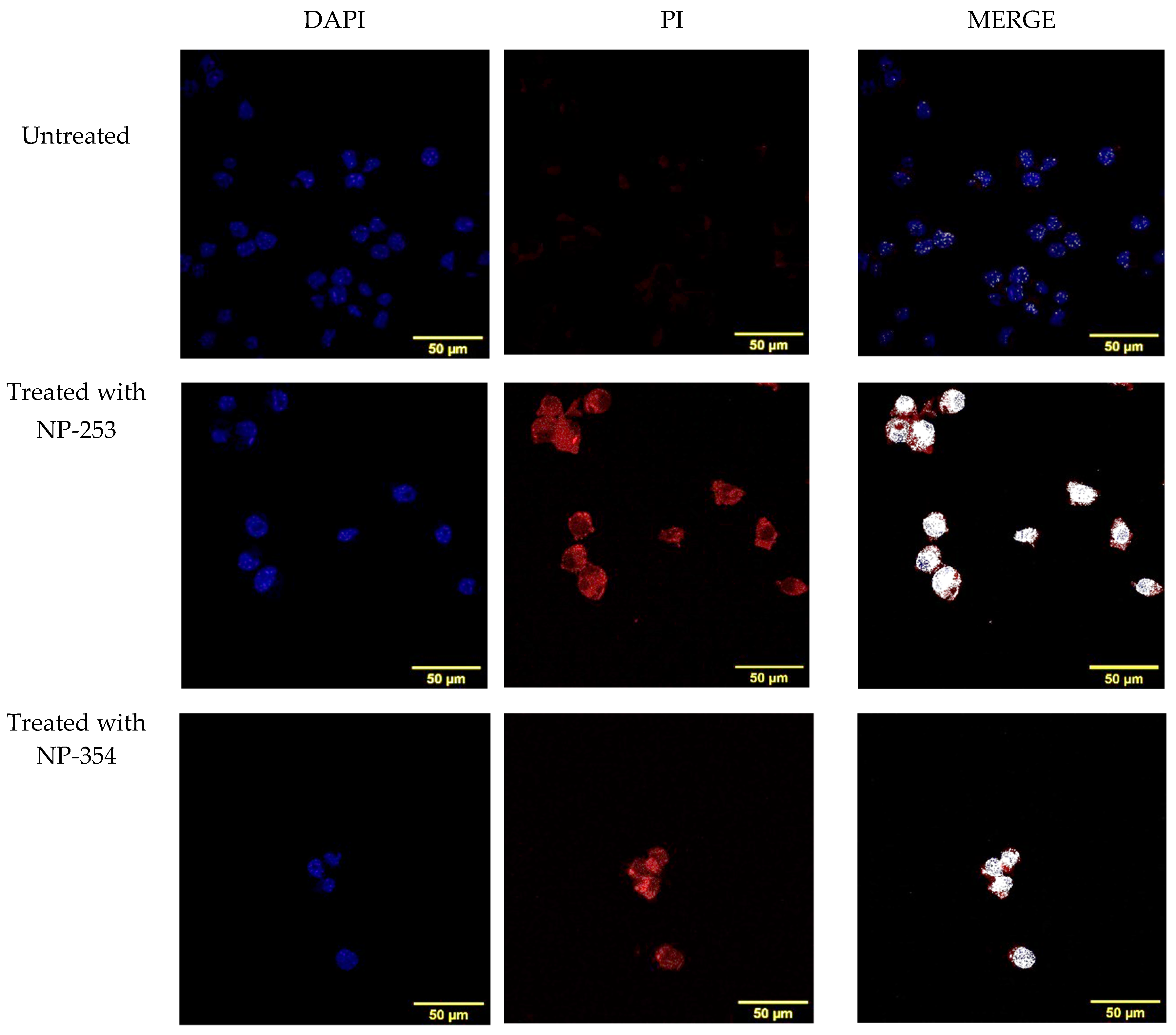
2.7. Study of Cell Death Induction Using DAPI and Propidium Iodide Co-Localization
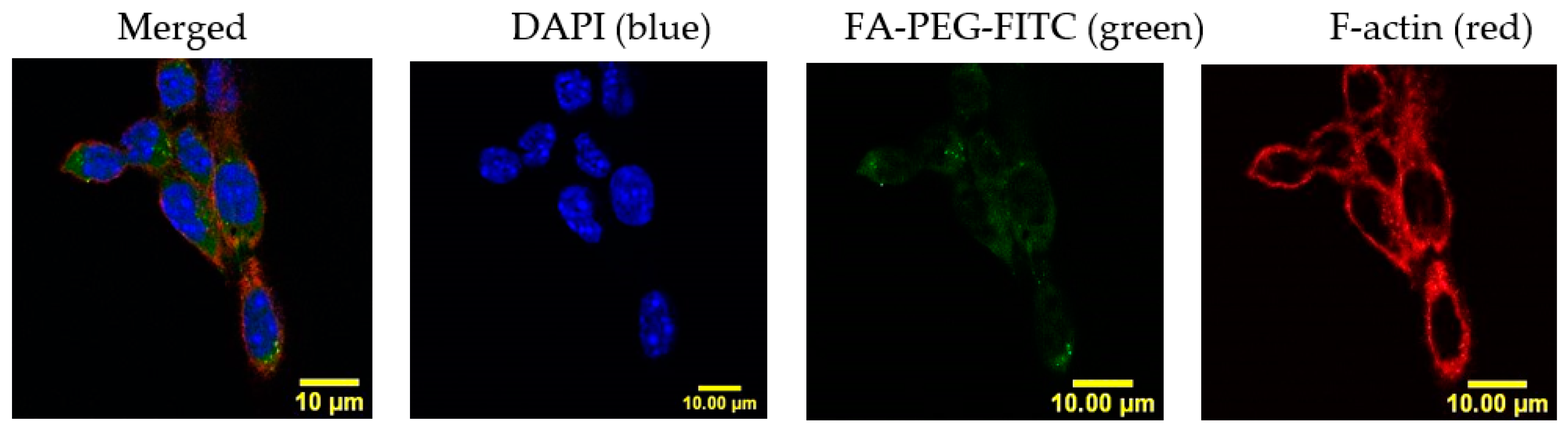
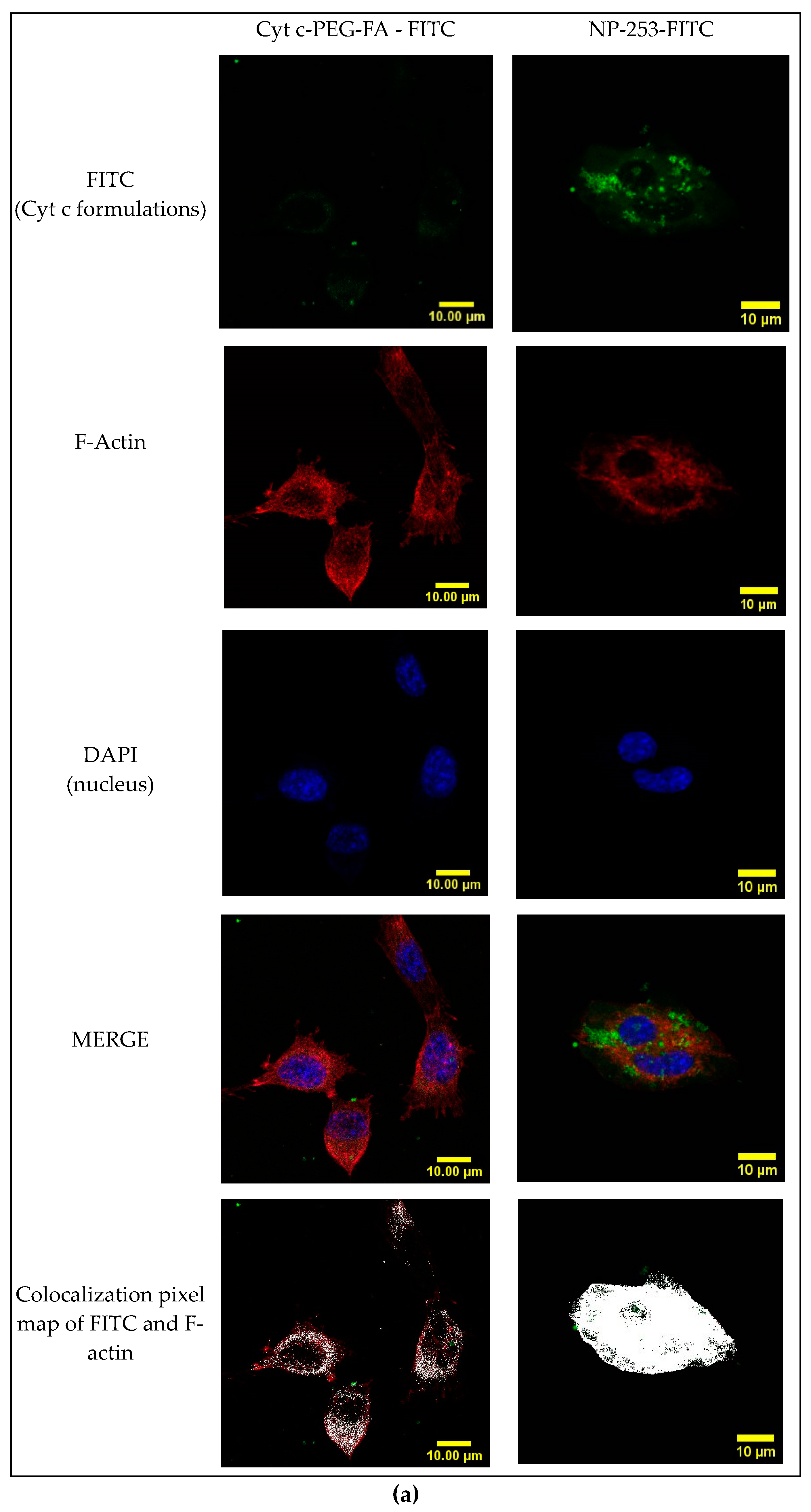
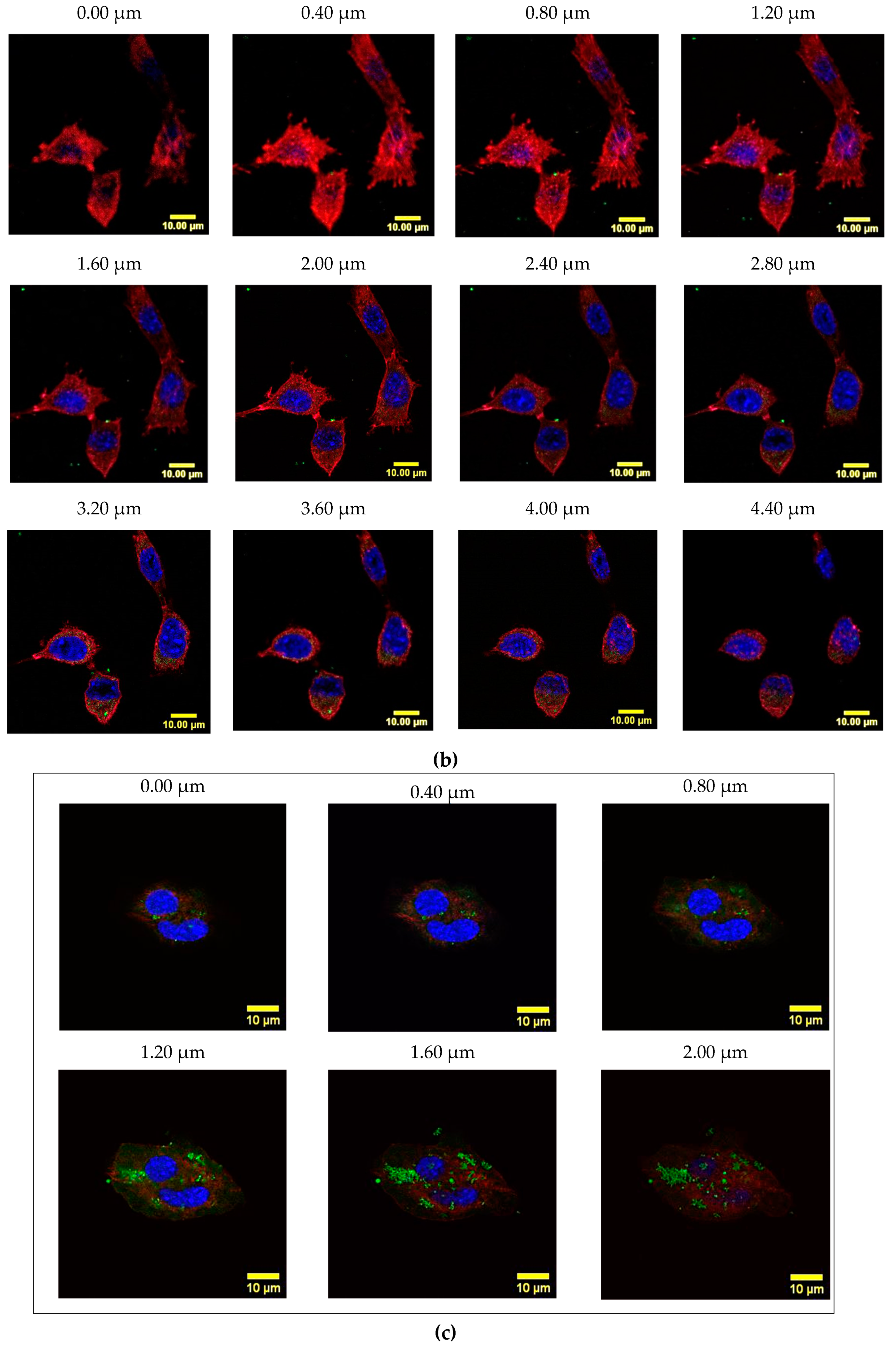
2.8. Cyt c-PEG-FA and NP-253 Internalization
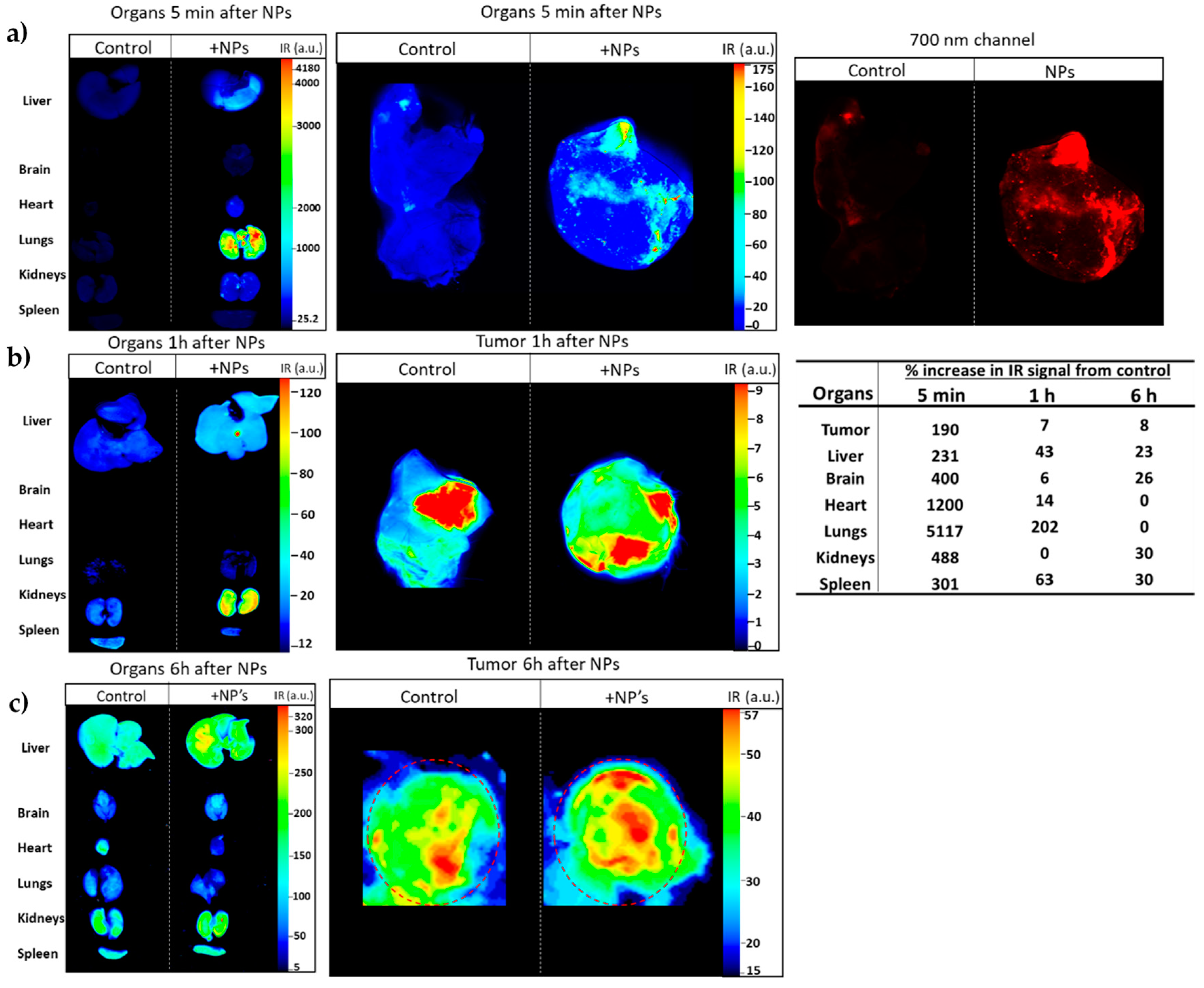
2.9. In Vivo Imaging Studies of NP Biodistribution in a Lung Carcinoma Mouse Model
3. Discussion
4. Materials and Methods
4.1. Materials
4.1.1. Constructs and Nanoparticle Synthesis and Decoration
4.1.2. In Vitro Immunofluorescence Studies and Cell Uptake of FITC-Labeled NPs
4.1.3. Caspase Activity and Cell Viability Assays
4.1.4. In Vivo Studies
4.2. Constructs and Nanoparticle Synthesis and Decoration
4.2.1. Synthesis of Cyt c-PEG-FA (Nanoparticle-Free Formulation)
4.2.2. Protein Nanoprecipitation
4.2.3. Determination of Precipitation Efficiency
4.2.4. Synthesis of SH-PLGA-PEG-FA
4.2.5. Synthesis of Cyt c-PLGA-PEG-FA Nanoparticles
4.3. Physicochemical Characterization of Nanoparticles
4.3.1. 1H NMR
4.3.2. Dynamic Light Scattering
4.3.3. Scanning Electron Microscopy
4.3.4. Determination of Encapsulation Efficiency and Actual Loading
4.3.5. Study of the Release Profile of Cyt c-PLGA-PEG-FA NPs
4.4. In Vitro Cell Studies to Characterize Constructs and NPs
4.4.1. Study of the Kinetics of Folic Acid Uptake by LLC Cells
4.4.2. Cell-Free Caspase 3, 7, and 10 Assay
4.4.3. In Vitro Cell Viability Assay
4.4.4. Calculation of IC50 for NP-Free Cyt c-PEG-FA Formulation and Surface-Decorated NP-253 and NP-354 Formulations
4.4.5. Study of Apoptosis Induction Using Propidium Iodide
4.4.6. Study of Formulations for NP Internalization
4.5. In Vivo Studies to Detect Near-Infrared NPs in Mice
4.5.1. Cell Growth and Induction of Lewis Lung Carcinoma in Mice
4.5.2. Labeling of Nanoparticles with a Near-Infrared Dye
4.5.3. Nanoparticle Optimization for In Vivo Tumor Targeting
4.5.4. Detection of Infrared-Labeled Nanoparticles In Vivo
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fouad:, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Lopez, J.; Tait, S.W.G. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, P.K.; Goel, A.; Mittal, R.D. Survivin: A molecular biomarker in cancer. Indian. J. Med. Res. 2015, 141, 389–397. [Google Scholar] [PubMed]
- Pandey, M.K.; Prasad, S.; Tyagi, A.K.; Deb, L.; Huang, J.; Karelia, D.N.; Amin, S.G.; Aggarwal, B.B. Targeting Cell Survival Proteins for Cancer Cell Death. Pharmaceuticals 2016, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.; Singh, A. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, X. Cytochrome C-mediated apoptosis. Annu. Rev. Biochem. 2004, 73, 87–106. [Google Scholar] [CrossRef]
- Kumar, R.; Bhat, T.A.; Walsh, E.M.; Chaudhary, A.K.; O’Malley, J.; Rhim, J.S.; Wang, J.; Morrison, C.D.; Attwood, K.; Bshara, W.; et al. Cytochrome c deficiency confers apoptosome and mitochondrial dysfunction in African-American men with prostate cancer. Cancer Res. 2019, 79, 1353–1368. [Google Scholar] [CrossRef]
- Srinivasan, S.; Guha, M.; Dong, D.W.; Whelan, K.A.; Ruthel, G.; Uchikado, Y.; Natsugoe, S.; Nakagawa, H.; Avadhani, N.G. Disruption of cytochrome c oxidase function induces the Warburg effect and metabolic reprogramming. Oncogene 2016, 35, 1585–1595. [Google Scholar] [CrossRef]
- Li, F.; Srinivasan, A.; Wang, Y.; Armstrong, R.C.; Tomaselli, K.J.; Fritz, L.C. Cell-specific induction of apoptosis by microinjection of cytochrome c. Bcl-xL has activity independent of cytochrome c release. J. Biol. Chem. 1997, 272, 30299–30305. [Google Scholar] [CrossRef]
- Zhivotovsky, B.; Orrenius, S.; Brustugun, O.T.; Døskeland, S.O. Injected cytochrome c induces apoptosis. Nature 1998, 391, 449–450. [Google Scholar] [CrossRef]
- Santra, S.; Kaittanis, C.; Perez, J.M. Cytochrome c encapsulating theranostic nanoparticles: A novel bifunctional system for targeted delivery of therapeutic membrane-impermeable proteins to tumors and imaging of cancer therapy. Mol. Pharm. 2010, 7, 1209–1222. [Google Scholar] [CrossRef] [PubMed]
- Morales-Cruz, M.; Cruz-Montañez, A.; Figueroa, C.M.; González-Robles, T.; Davila, J.; Inyushin, M.; Loza-Rosas, S.A.; Molina, A.M.; Muñoz-Perez, L.; Kucheryavykh, L.Y.; et al. Combining stimulus-triggered release and active targeting strategies improves cytotoxicity of cytochrome c nanoparticles in tumor cells. Mol. Pharm. 2016, 13, 2844–2854. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, C.M.; Suarez, B.; Molina, A.; Mendez, J.; Torres, Z.; Griebenow, K. Smart release nano-formulation of cytochrome c and hyaluronic acid induces apoptosis in cancer cells. J. Nanomed. Nanotechnol. 2017, 8, 427. [Google Scholar]
- Saxena, M.; Delgado, Y.; Sharma, R.K.; Sharma, S.; Guzmán, S.L.P.D.L.; Tinoco, A.D.; Griebenow, K. Inducing cell death in vitro in cancer cells by targeted delivery of cytochrome c via a transferrin conjugate. PLoS ONE 2018, 13, e0195542. [Google Scholar] [CrossRef] [PubMed]
- Kucheryavykh, Y.V.; Davila, J.; Ortiz-Rivera, J.; Inyushin, M.; Almodovar, L.; Mayol, M.; Morales-Cruz, M.; Cruz-Montañez, A.; Barcelo-Bovea, V.; Griebenow, K.; et al. Targeted delivery of nanoparticulate Cytochrome C into glioma cells through the proton-coupled folate transporter. Biomolecules 2019, 9, 154. [Google Scholar] [CrossRef] [PubMed]
- Morales-Cruz, M.; Flores-Fernández, G.M.; Morales-Cruz, M.; Orellano, E.A.; Rodriguez-Martinez, J.A.; Ruiz, M.; Griebenow, K. Two-step nanoprecipitation for the production of protein-loaded PLGA nanospheres. Res. Pharm. Sci. 2012, 2, 79–85. [Google Scholar] [CrossRef]
- Morales-Cruz, M.; Figueroa, C.M.; González-Robles, T.; Delgado, Y.; Molina, A.; Méndez, J.; Morales, M.; Griebenow, K. Activation of caspase-dependent apoptosis by intracellular delivery of cytochrome c-based nanoparticles. J. Nanobiotechnol. 2014, 12, 33. [Google Scholar] [CrossRef]
- Cheng, R.; Feng, F.; Meng, F.; Deng, C.; Feijen, J.; Zhong, Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J. Control. Release 2011, 152, 2–12. [Google Scholar] [CrossRef]
- Fernández, M.; Javaid, F.; Chudasama, V. Advances in targeting the folate receptor in the treatment/imaging of cancers. Chem. Sci. 2018, 9, 790–810. [Google Scholar] [CrossRef]
- Grodzinski, P.; Kircher, M.; Goldberg, M.; Gabizon, A. Integrating Nanotechnology into Cancer Care. ACS Nano 2019, 13, 7370–7376. [Google Scholar] [CrossRef]
- Kellar, A.; Egan, C.; Morris, D. Preclinical murine models for lung cancer: Clinical trial applications. BioMed Res. Int. 2015, 2015, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, R.; Tosi, G.; Grabrucker, A. Emerging use of nanotechnology in the treatment of neurological disorders. Curr. Pharm. 2015, 21, 3111–3130. [Google Scholar] [CrossRef] [PubMed]
- Mendez, J.; Morales-Cruz, M.; Delgado, Y.; Figueroa, C.M.; Orellano, E.A.; Morales, M.; Monteagudo, A.; Griebenow, K. Delivery of chemically glycosylated cytochrome c immobilized in mesoporous silica nanoparticles induces apoptosis in HeLa cancer cells. Mol. Pharm. 2014, 11, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef]
- Wu, G.; Fang, Y.-Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef]
- Chenthamara, D.; Subramaniam, S.; Ramakrishnan, S.G.; Krishnaswamy, S.; Essa, M.M.; Lin, F.H.; Qoronfleh, M.W. Therapeutic efficacy of nanoparticles and routes of administration. Biomater. Res. 2019, 23, 20. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Gonda, A.; Zhao, N.; Shah, J.V.; Calvelli, H.R.; Kantamneni, H.; Francis, N.L.; Ganapathy, V. Engineering Tumor-Targeting Nanoparticles as Vehicles for Precision Nanomedicine. Med. One 2019, 4, e190021. [Google Scholar]
- Baumann, A.; Tuerck, D.; Prabhu, S.; Dickmann, L.; Sims, J. Pharmacokinetics, metabolism and distribution of PEGs and PEGylated proteins: Quo vadis? Drug Discov. Today 2014, 19, 1623–1631. [Google Scholar] [CrossRef]
- Kang, B.; Okwieka, P.; Schöttler, S.; Winzen, S.; Langhanki, J.; Mohr, K.; Opatz, T.; Mailänder, V.; Landfester, K.; Wurm, F.R. Carbohydrate-Based Nanocarriers Exhibiting Specific Cell Targeting with Minimum Influence from the Protein Corona. Angew. Chem. Int. Ed. Engl. 2015, 54, 7436–7440. [Google Scholar] [CrossRef]
- Dai, Q.; Walkey, C.; Chan, W.C. Polyethylene glycol backfilling mitigates the negative impact of the protein corona on nanoparticle cell targeting. Angew. Chem. Int. Ed. 2014, 53, 5093–5096. [Google Scholar]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Huennekens, F.M. Folic acid coenzymes in the biosynthesis of purines and pyrimidines. Vitam. Horm. 1968, 26, 375–394. [Google Scholar] [PubMed]
- Yu, Y.; Wang, J.; Kaul, S.C.; Wadhwa, R.; Miyako, E. Folic Acid Receptor-Mediated Targeting Enhances the Cytotoxicity, Efficacy, and Selectivity of Withania somnifera Leaf Extract: In vitro and in vivo Evidence. Front. Oncol. 2019, 9, 602. [Google Scholar] [CrossRef]
- Elkhodiry, M.A.; Husseini, G.A.; Velluto, D. Targeting the Folate Receptor: Effects of Conjugating Folic Acid to DOX Loaded Polymeric Micelles. Anti-Cancer Agent Med. 2016, 16, 1275–1280. [Google Scholar] [CrossRef]
- Tang, L.; Fan, T.M.; Borst, L.B.; Cheng, J. Synthesis and biological response of size-specific, monodisperse drug–silica nanoconjugates. ACS Nano 2012, 6, 3954–3966. [Google Scholar] [CrossRef]
- Morales-Cruz, M.; Delgado, Y.; Castillo, B.; Figueroa, C.M.; Molina, A.M.; Torres, A.; Milián, M.; Griebenow, K. Smart Targeting to Improve Cancer Therapeutics. Drug Des. Dev. Ther. 2019, 13, 3753–3772. [Google Scholar] [CrossRef]
- Kluck, R.M.; Ellerby, L.M.; Ellerby, H.M.; Naiem, S.; Yaffe, M.P.; Margoliash, E.; Bredesen, D.; Mauk, A.G.; Sherman, F.; Newmeyer, D.D. Determinants of cytochrome c pro-apoptotic activity. The role of lysine 72 trimethylation. J. Biol. Chem. 2000, 275, 16127–16133. [Google Scholar] [CrossRef]
- Yu, T.; Wang, X.; Purring-Koch, C.; Wei, Y.; McLendon, G.L. A mutational epitope for Cytochrome c binding to the apoptosis protease activation Factor-1. J. Biol. Chem. 2001, 276, 13034–13038. [Google Scholar] [CrossRef]
- Hao, Z.; Duncan, G.S.; Chang, C.-C.; Elia, A.; Fang, M.; Wakeham, A.; Okada, H.; Calzascia, T.; Jang, Y.; You-Ten, A.; et al. Specific ablation of the apoptotic functions of Cytochrome c reveals a differential requirement for Cytochrome c and Apaf-1 in apoptosis. Cell 2005, 121, 579–591. [Google Scholar] [CrossRef]
- Langer, K.; Balthasar, S.; Vogel, V.; Dinauer, N.; von Briesen, H.; Schubert, D. Optimization of the preparation process for human serum albumin (HSA) nanoparticles. Int. J. Pharm. 2003, 257, 169–180. [Google Scholar] [CrossRef]
- Santucci, R.; Sinibaldi, F.; Cozza, P.; Polticelli, F.; Fiorucci, L. Cytochrome c: An extreme multifunctional protein with a key role in cell fate. Int. J. Biol. Macromol. 2019, 136, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, K.; Sundarrajan, P. A Study of Phylogenetic Relationships and Homology of Cytochrome C using Bioinformatics. Int. Res. J. Sci. Eng. 2016, 4, 11. [Google Scholar]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molar Ratio Cyt c to SPDP | Moles of SPDP Per Mole of Cyt c |
|---|---|
| 1 to 2 | 0 |
| 1 to 4 | 0.93 ± 0.06 |
| 1 to 8 | 1.95 ± 0.34 |
| Solvent | Particle Diameter (nm) |
|---|---|
| Acetonitrile | 163 ± 13 |
| THF | 219 ± 9 |
| Acetone | No Cyt c precipitation |
| Ethanol | No Cyt c precipitation |
| Cyt c Concentration (mg/mL) | Diameter (nm) b | PDI b | Precipitation Efficiency (%) b | Relative Caspase Activation (%) b |
|---|---|---|---|---|
| 2.5 | 139 ± 15 | 0.041 ± 0.03 | 33.0 ± 7.2 | 104 ± 10 |
| 5 | 163 ± 13 | 0.064 ± 0.03 | 71.9 ± 8.0 | 96 ± 3 |
| 10 | 251 ± 7 | 0.13 ± 0.08 | 84.5 ± 12.3 | 88 ± 2 |
| Nanoparticle Name | Cyt c NP a | NP-253 | NP-354 | |
|---|---|---|---|---|
| Parameter | ||||
| Cyt c used during nanoprecipitation (mg/mL) | 2.5 | 5 | 10 | |
| Diameter before coating with PLGA-PEG-FA (nm) | 139 ± 15 | 163 ± 13 | 251 ± 7 | |
| Diameter after coating with PLGA-PEG-FA (nm) | - | 253 ± 55 | 354 ± 11 | |
| PDI | - | 0.07 ± 0.06 | 0.13 ± 0.03 | |
| Encapsulation efficiency (%) | 0 | 45 ± 7 | 48 ± 9 | |
| Actual loading (%) | - | 62 ± 10 | 66 ± 10 | |
| Relative caspase activity (%) | - | 89 ± 9 | 99 ± 6 | |
| Zeta potential (mV) | - | 26.9 ± 5.03 | 22.4 ± 6.36 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barcelo-Bovea, V.; Dominguez-Martinez, I.; Joaquin-Ovalle, F.; Amador, L.A.; Castro-Rivera, E.; Medina-Álvarez, K.; McGoron, A.; Griebenow, K.; Ferrer-Acosta, Y. Optimization and Characterization of Protein Nanoparticles for the Targeted and Smart Delivery of Cytochrome c to Non-Small Cell Lung Carcinoma. Cancers 2020, 12, 1215. https://doi.org/10.3390/cancers12051215
Barcelo-Bovea V, Dominguez-Martinez I, Joaquin-Ovalle F, Amador LA, Castro-Rivera E, Medina-Álvarez K, McGoron A, Griebenow K, Ferrer-Acosta Y. Optimization and Characterization of Protein Nanoparticles for the Targeted and Smart Delivery of Cytochrome c to Non-Small Cell Lung Carcinoma. Cancers. 2020; 12(5):1215. https://doi.org/10.3390/cancers12051215
Chicago/Turabian StyleBarcelo-Bovea, Vanessa, Irivette Dominguez-Martinez, Freisa Joaquin-Ovalle, Luis A. Amador, Elizabeth Castro-Rivera, Kristofer Medina-Álvarez, Anthony McGoron, Kai Griebenow, and Yancy Ferrer-Acosta. 2020. "Optimization and Characterization of Protein Nanoparticles for the Targeted and Smart Delivery of Cytochrome c to Non-Small Cell Lung Carcinoma" Cancers 12, no. 5: 1215. https://doi.org/10.3390/cancers12051215
APA StyleBarcelo-Bovea, V., Dominguez-Martinez, I., Joaquin-Ovalle, F., Amador, L. A., Castro-Rivera, E., Medina-Álvarez, K., McGoron, A., Griebenow, K., & Ferrer-Acosta, Y. (2020). Optimization and Characterization of Protein Nanoparticles for the Targeted and Smart Delivery of Cytochrome c to Non-Small Cell Lung Carcinoma. Cancers, 12(5), 1215. https://doi.org/10.3390/cancers12051215