Abstract
Background: WNT activation is a hallmark of colorectal cancer. BRAF mutation is present in 15% of colorectal cancers, and the role of mutations in WNT signaling regulators in this context is unclear. Here, we evaluate the mutational landscape of WNT signaling regulators in BRAF mutant cancers. Methods: we performed exome-sequencing on 24 BRAF mutant colorectal cancers and analyzed these data in combination with 175 publicly available BRAF mutant colorectal cancer exomes. We assessed the somatic mutational landscape of WNT signaling regulators, and performed hotspot and driver mutation analyses to identify potential drivers of WNT signaling. The effects of Apc and Braf mutation were modelled, in vivo, using the Apcmin/+ and BrafV637/Villin-CreERT2/+ mouse, respectively. Results: RNF43 was the most frequently mutated WNT signaling regulator (41%). Mutations in the beta-catenin destruction complex occurred in 48% of cancers. Hotspot analyses identified potential cancer driver genes in the WNT signaling cascade, including MEN1, GNG12 and WNT16. Truncating APC mutation was identified in 20.8% of cancers. Truncating APC mutation was associated with early age at diagnosis (p < 2 × 10−5), advanced stage (p < 0.01), and poor survival (p = 0.026). Apcmin/+/BrafV637 animals had more numerous and larger SI and colonic lesions (p < 0.0001 and p < 0.05, respectively), and a markedly reduced survival (median survival: 3.2 months, p = 8.8 × 10−21), compared to animals with Apc or Braf mutation alone. Conclusions: the WNT signaling axis is frequently mutated in BRAF mutant colorectal cancers. WNT16 and MEN1 may be novel drivers of aberrant WNT signaling in colorectal cancer. Co-mutation of BRAF and APC generates an extremely aggressive neoplastic phenotype that is associated with poor patient outcome.
Keywords:
colorectal cancer; WNT signaling; APC; BRAF; serrated neoplasia; genomics; driver mutations 1. Background
Colorectal cancer is a heterogeneous disease that arises through two main molecular pathways. The conventional pathway, which accounts for 75–80% of all colorectal cancer diagnoses, is initiated by the biallelic inactivation of APC and progresses to cancer via mutations in KRAS and alterations to the TP53 gene. By contrast, the serrated neoplasia pathway is initiated by activating mutations in BRAF and often progresses to malignancy via MLH1 hypermethylation, microsatellite instability and a plethora of epigenetic alterations. At the transition to dysplasia, serrated lesions usually acquire mutations that increase WNT signaling. Sessile serrated lesions (SSLs) acquire missense APC mutations [1], and truncating RNF43 mutations [2]. In traditional serrated adenomas (TSAs), common WNT pathway aberrations include RSPO3 fusions [3,4], mutations of CTNNB1 [3] and the mutation of APC [3].
In the normal enterocytes, the WNT signaling cascade exists to support stemness, differentiation and development. Appropriate levels of WNT signal are maintained intracellularly by the β-catenin destruction complex. The complex consists of AXIN, APC, GSK3β, and CK1α. The destruction complex ubiquitinates β-catenin in the cytosol, triggering its subsequent proteasomal degradation. In the absence of the destruction complex, β-catenin translocates to the nucleus and forms a complex with the TCF/LEF molecules and p300 to activate the expression of genes supporting the stem phenotype. Constitutive WNT signaling is deleterious to the cell and thus in the absence of exogenous stimuli the β-catenin destruction complex patrols the cytosol and degrades β-catenin. WNT signaling is activated by the binding of extracellular WNT ligands to frizzled receptors residing on the cell surface. This triggers the sequestering of the destruction complex to the cell membrane and facilitates the build-up of β-catenin, which enters the nucleus and activates WNT target genes.
Approximately 45–50% of BRAF mutant cancers demonstrate dysregulated WNT signaling [1], and thus the WNT signaling pathway appears to be important to serrated colorectal neoplasia. In conventional colorectal carcinogenesis, WNT signaling is dysregulated via truncating mutations of APC and the loss of 5q21, the region where the APC gene resides [5]. This dysregulation occurs very early in the evolution of conventional adenomas. However, numerous studies have indicated that mutation of BRAF is almost never identified in such APC mutated adenomas, even when they develop advanced histological features [6,7]. This suggests that BRAF and APC mutations are mutually exclusive in conventional adenomas.
In the serrated neoplastic pathway where the initiating mutation is BRAF, WNT signaling only commonly becomes dysregulated when the benign polyp transitions to malignancy. Truncating RNF43 mutations may alter WNT signaling, but these are predominantly present in mismatch repair deficient BRAF mutant cancer [2,8], and there is controversy as to whether RNF43 mutation affects canonical WNT signaling [9]. The epigenetic silencing of the WNT pathway members is another possible mechanism for altering canonical WNT pathway activity. The methylation of SFRP genes increases WNT signaling [10] and is common in colorectal cancer [11]. Similarly, DNA methylation induced the inactivation of DKK genes, which are antagonists of WNT signaling, and occur in ~20% of all colon cancers [12]. The frequency of WNT signaling dysregulation being due to APC mutation is not well established.
Here we have conducted a large-scale genomic analysis of the somatic mutations that underlie WNT signaling activation in BRAF mutant colorectal cancer. We hypothesize that WNT signaling activation in BRAF mutant cancers will be heterogeneous, and a mosaic of alterations underpin WNT signaling to achieve a “just-right” level of pathway activation.
2. Methods
This study was approved by the QIMR Berghofer Human Research Ethics Committee (P460, P773). Informed consent was obtained for all participants included in this study.
2.1. Cohorts Included in the Study
We assessed the somatic mutational landscape of 199 BRAF mutant cancers from four distinct sources. This included cancers from the Cancer Genome Atlas project (n = 51) [7,13], the Dana Faber Cancer Institute (Giannakis et al. 2016, n = 111) [14], the Clinical Proteomic Tumor Analysis Consortium (Suhas et al. 2019, n = 13) [15] and additional BRAF mutant cancers that were sequenced as part of this study (methods detailed below, n = 24). For the survival analyses, we included additional targeted sequenced data from the Memorial Sloan-Kettering Cancer Centre (Yaeger et al. 2018, n = 76) [16]. This dataset was limited to a panel of genes, and as such, was excluded from other analyses. Figure S1 shows similar tumor mutation burden across each cohort. BRAF wild type cancers (n = 512) were included for the comparison of mutational profiles between BRAF mutant and wild type cancers. Clinicopathological details of samples included in this study and mutational data are available as Supplementary Materials (Supplementary Table S1 and S2).
2.2. DNA Extraction, Library Preparation and Exome Sequencing of Local Samples
Cancer and germline samples were obtained from patients at the Royal Brisbane and Women’s Hospital, Brisbane, Australia, at the time of surgery. All participants gave their written, informed consent prior to participating in the study and the study was approved by the QIMR Berghofer Human Research Ethics Committee (P460, P773). DNA was isolated from whole blood using the salt precipitation method, as previously reported [17]. Cancer samples were snap-frozen in liquid nitrogen and DNA extracted using the AllPrep DNA/RNA/Protein mini kit (QIAGEN, Frankfurt, Germany), as previously reported [18]. Exome-sequencing libraries were generated using the Agilent SureSelect Human All Exon V4+UTR capture platform (Agilent, CA, USA). Libraries were sequenced to a target depth of 200-fold coverage on a 100 bp paired-end sequencing run using an Illumina HiSeq 2000 instrument. Sequence reads were trimmed using Cutadapt (v1.9) [19] and aligned to the GRCh37 reference with BWA-MEM (v0.7.12) [20]. Alignments were duplicate-marked with Picard (v1.129, https://broadinstitute.github.io/picard/) and coordinate-sorted using Samtools (v1.1) [21]. Single nucleotide substitution variants were detected using a dual calling strategy using qSNP (v2.0) [22] and the GATK HaplotypeCaller (v3.3-0) [23]. The HaplotypeCaller was also used to call short indels of ≤50 bp. Initial read filtering for all variants detected included: a minimum of 35 alignment matches in the CIGAR string, 3 or fewer mismatches in the MD field, and a mapping quality greater than 10. High confidence variants were selected with: a minimum coverage of 8 reads in the control data and 12 reads in the tumour data; at least 5 variant supporting reads present where the variant was not within the first or last 5 bases; at least 4 of the 5 reads with unique start positions; the variant was identified in reads of both sequencing directions; the variant was not less than 5 base pairs from a mono-nucleotide run of 7 or more bases in length. Variants were annotated with gene feature information and transcript or protein consequences using SnpEff (v4.0e) [24]. Sequencing and QC metrics are reported in Supplementary Table S3.
2.3. Assessing the Somatic Mutational Landscape of WNT Regulators
To assess the somatic mutational landscape of WNT signaling regulators, we downloaded mutational annotation files for each cohort from the Genome Data Commons (TCGA), cBioPortal (CPTAC), from Supplementary Materials (DFCI) or analysis of the Royal Brisbane and Women’s Hospital, Brisbane cases. MAF files concatenated to form a combined MAF file comprised of 924,366 entries, relating to 1411 samples. BRAF V600E mutant samples were subset from the larger dataset, yielding a total of 320,431 variants from 199 samples. As we sought to investigate WNT, we further selected only genes that were members of the REACTOME signaling by the WNT geneset (n = 327 genes). The final dataset was comprised of 5327 nonsynonymous variants in WNT signaling loci that corresponded to 199 samples.
An analysis of variants was performed using the MAFtools R package [25]. Cancer drivers were predicted using two orthogonal approaches (OncodriveClust [26]: default parameters as implemented in MAFtools; OncodriveFML [27]: scores: CADD v1.3, signature: computed by sample, remaining parameters: default). Driver mutation analyses were performed on the entire set of variants (not limited to the WNT signaling gene-set) to accurately model the background mutational processes and avoid statistical biases. Results from Non-WNT loci were discarded and FDR corrections were performed on the remaining p values that pertain to tests performed on genes in the WNT signaling pathway. Somatic interactions (co-mutations and mutual exclusivity) were identified by performing Fisher’s exact test on pairs of genes.
2.4. Murine Model of Apc and Braf Mutation
To model the effects of Apc and Braf mutation on colorectal neoplasia, we utilized two murine models. The ApcMin/+ mouse has a mono-allelic mutation at codon 851 and recapitulates human germline APC mutation. In both humans and mice, progression is governed by the loss of the remaining allele. Our second model, the BrafCA/CA/Villin-CreERT2/+ mouse [28,29], is an inducible model of Braf mutant colorectal neoplasia. Recombination of the mutant Braf V637E allele is induced at 2 weeks of age by a single intraperitoneal injection of tamoxifen (75mg/kg). The Braf V637E allele is the murine analogue of the BRAF V600E human mutation. To model the effects of Apc mutation and Braf mutation, we crossed ApcMin/+ mice with BrafCA/CA/Villin-CreERT2/+. Animals were monitored biweekly for signs of distress and humanely euthanized when such signs were identified, as per our approved protocol (QIMR Berghofer Animal Ethics Committee; P1208). For survival analysis, animals were deemed to be deceased if they were euthanized due to distress. If animals reached the prescribed endpoints of the experiment without any signs of distress, they were deemed to have survived, and were censored for survival analysis.
At sacrifice, the gastrointestinal tract from oesophagus to rectum was removed, cleaned and opened longitudinally. Macroscopic lesions were bisected to obtain both molecular and histological data. Matched normal hyperplastic tissue was taken minimum five centimetres from the site of the lesion. Histological assessment of lesions and lesion counts was performed on haematoxylin and eosin stained sections from formalin-fixed, paraffin-embedded blocks by specialist gastrointestinal anatomical pathologists.
2.5. Sanger Sequencing
Sanger sequencing was performed to assess the G7 repeat track of WNT16. PCR conditions were as follows: 1× GoBuffer (ProMega, USA), 2.5 mM MgCl2, 0.25 mM dNTP, 0.25 uM Forward Primer (5′ GGCAACATGACAGAGTGTTCC 3′), 0.25 uM Reverse Primer (5′ GCCATACTGGACATCATCGG 3′), 0.25 uM Syto9, 1U GoTaq DNA polymerase (ProMega, USA), 50 ng DNA; Cycing: 95 °C hold for two minutes, 40× cycles of 95 °C for 30 s, 60 °C for 30 s, 72 °C for 45 s, followed by a 72 °C hold for five minutes at the end of cycle 40. Sequencing was performed as per Fennell et al. [30]
2.6. Statistical Analysis
All statistical analyses were performed in Microsoft Open R (v3.5.1). Student’s t tests were performed for the hypothesis testing of continuous variables. Logistic regression analyses were employed to examine the probability of mutations over patient age. The likelihood–ratio test was performed to assess associations with categorical variables and Fisher’s exact test was used to examine for the mutual exclusivity of mutations in gene-pairs.
3. Results
3.1. The Somatic Mutation Landscape of WNT Signaling in BRAF Mutated Cancers
To assess the degree of variation in genetic alterations of WNT signaling pathway genes, we collated whole exome-sequencing data of BRAF mutant colorectal cancers from three previously published studies [7,14,15], combined with 24 samples that were sequenced in-house (total n = 199). We limited WXS variants to genes in the WNT signaling cascade, as identified in the REACTOME signaling by WNT gene set (n = 327 genes). The mean number of WNT pathway mutations per sample was 16.7 ± 13.6, and was highly correlated with overall tumor mutation burden (p = 4.08 × 10−85, r2 = 0.86). RNF43 was the most commonly mutated gene (41%, Figure 1). Overall, 35.3% of samples had a truncating mutation in RNF43. KMT2D, TRRAP, and APC were mutated in 33%, 30% and 28% of samples, respectively (Figure 1).
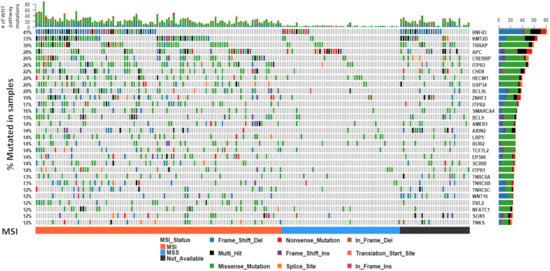
Figure 1.
The somatic mutation landscape of WNT signaling regulators in BRAF mutant colorectal cancers. The 30 most frequently mutated genes in the WNT pathway are depicted. Each column corresponds to a single cancer. The colors of bars are indicative of the type of mutation, with grey = wild-type. The barplot at the top of the figure represents the number of mutations in the WNT pathway a sample has. The vertical plot on the right of the figure represents the number of mutations in each gene, color coded by mutation type. Microsatellite instability status is indicated below the mutation plot.
The β-catenin destruction complex is an important regulator of canonical WNT signaling. It is comprised of APC, AXIN1, AXIN2, and GSK3β (Figure 2). We next evaluated how frequently mutations occur in any component of this complex or in CTNNB1 itself. Notably, 48% of all cancers had mutations in at least one of these five genes. Mutations in APC and AXIN2, but not AXIN1, are significantly mutually exclusive (p < 0.05). Missense mutations in CTNNB1 have been reported to render the molecule impervious to ubiquitin-mediated destruction. CTNNB1 mutations occur in 9% of samples. Missense CTNNB1 mutations were mutually exclusive with truncating APC mutations.
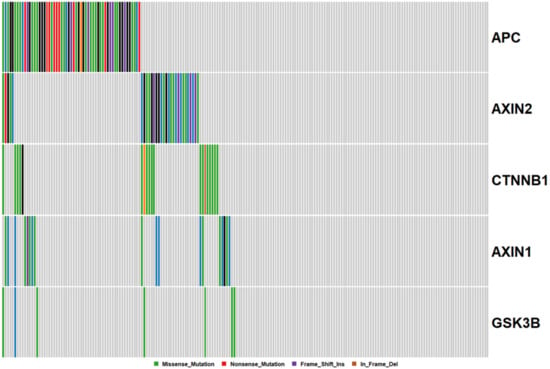
Figure 2.
Mutations in the Beta-Catenin destruction complex. Each column corresponds to a single cancer, and each row, a single gene.
APC is a classical tumor suppressor gene, where the loss of both copies is required for complete abrogation of transcription. We next assessed the frequency of double, and triple hit mutational events in APC. Of cancers with truncating APC mutations, 50% also harbored a second truncating mutation in APC. One cancer had three truncating mutations in APC. When we included missense mutations in the assessment of potential double hit events, this percentage rose to 54%. These data indicate that missense mutations are not selected as second-hits at the APC locus, and raise questions regarding their functionality.
3.2. Somatic Mutation Interaction Analysis Identifies Co-Mutated WNT Signaling Loci and Mutual Exclusivity of Truncating APC and RNF43 Mutations
We performed somatic mutation interaction analyses to examine for mutations in genes that are mutually exclusive and those that tend to co-occur. We found evidence for co-occurring mutations in 222 gene pairs (Figure 3, Supplementary Table S4). As truncating mutations are more likely to influence the final protein, we next examined somatic interactions between truncating mutations in WNT pathway genes (Figure 3). We identified statistical evidence for somatic interactions between 75 gene pairs. 96% were between co-mutated gene pairs (Supplementary Table S4). Truncating APC mutation was mutually exclusive to truncating mutations in both RNF43 (P0.0003, OR: 0.20), and ZNRF3 (p = 0.001, OR: 0). AMER1 truncating mutations were mutually exclusive to RNF43 mutation (p = 0.043, OR: 0.12).
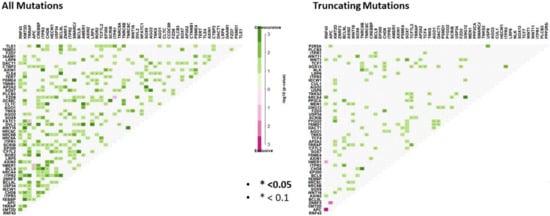
Figure 3.
Somatic interaction analysis reveals mutually exclusive mutations between gene pairs, and significant co-occurring mutations. Co-occurring mutations are indicated by green squares and mutually exclusive mutations between gene pairs in purple. The intensity of the color is proportionate the –log10 (p-value). p-values were determined using Fisher’s exact test.
3.3. The WNT Signaling Mutational Landscape of BRAF Mutant Microsatellite Unstable Cancers Differs from BRAF Mutant Microsatellite Stable Cancers
It is well established that loss of DNA mismatch repair functionality results in a hypermutable phenotype (microsatellite instability, or MSI) and that this is common in BRAF mutant colorectal cancers. We next sought to examine mutations in WNT regulators by microsatellite instability status, to identify if different regulators are targeted in these contexts. We identified fifty genes that were significantly differentially mutated according to microsatellite instability status. As expected, all genes were more frequently mutated in MSI cancers. Table 1 shows the 15 most significantly differentially mutated WNT signaling regulators between MSI and MSS cancers. This analysis reveals genes that are exclusively mutated in MSI cancers (i.e., TPR2, EP300, DVL2) and those that are mutated significantly more frequently in MSI cancers (i.e., TRRAP, RNF43, ZNRF3). The most frequently mutated genes are markedly different between MSI and MSS cancers (Figure 4). The most frequently mutated genes in MSI cancers were RNF43 (49%), KMT2D (48%), and TRRAP (41%); in contrast, APC was the most frequent mutational target in MSS cancers (21%), followed by RNF43 (20%), HECW1 (10%), and a tail of less frequently mutated genes (Figure 4).

Table 1.
WNT signaling regulators that are differentially mutated according to microsatellite instability status in BRAF mutant colorectal cancers.
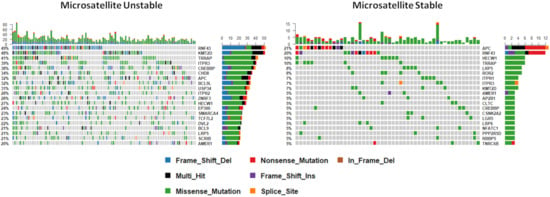
Figure 4.
The mutational landscape of WNT signaling regulators by microsatellite instability status. Note that this analysis is limited to cancers with microsatellite instability status available (n = 167).
3.4. WNT Signalling Regulators Are Differentially Mutated in BRAF Mutant Cancers in Comparison to BRAF Wild Type Cancers
To establish whether the WNT mutational landscape of colorectal cancers differs according to BRAF status, we examined the mutational profile of WNT signaling genes in 512 further colorectal cancers from the Cancer Genome Atlas that were BRAF wild type. The most frequently mutated genes are markedly different in BRAF wild type cancers (Figure 1 and Figure 5). APC was mutated in 82% of these cancers, in comparison to just 28% of BRAF mutant cancers. Of all WNT signaling regulators assessed in this study, 110 were significantly differentially mutated between BRAF mutant and wild type cancers (Table 2). Overall, 99% of these genes were more frequently mutated in BRAF mutant cancers. This is likely due to the increased frequency of MSI in BRAF mutant cancers, and the high frequency of the mutations in highly penetrant APC gene in BRAF wild type cancers, reducing the selective pressure on other WNT regulators.
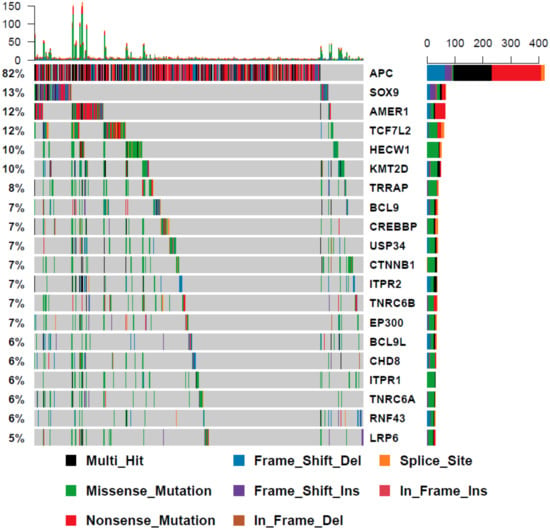
Figure 5.
The mutational landscape of WNT signaling regulators in BRAF wild type cancers.
Table 2.
Comparison of the mutational frequency of WNT signaling regulators in BRAF mutant cancers versus BRAF wild type cancers.
3.5. Mutation Clustering Analysis Reveals Mutational Hotspots in Nine WNT Signaling Genes
We next sought to identify driver genes using the OncodriveCLUST algorithm. This method identifies potential driver genes using a positional clustering method and operates on the assumption that clusters of mutations, or “mutational hotspots”, are more likely to occur in oncogenes. In keeping with previous studies [2,8,30,31] (p43), RNF43 was identified as a putative cancer driver (p = 0.07). Somatic mutations in MEN1, a gene identified as a familial cancer risk gene and as an inducer of genome wide hypermethylation, were identified as putative drivers. MEN1 was mutated in 4% (8/199, p < 0.001) of samples, and most of the identified mutations were frameshift deletions at R521. Moreover, WNT16 and GNG12 were implicated as potential cancer drivers (p = 0.06 and p = 0.0006, respectively).
We used oncodriveFML, an orthogonal computational method of predicting cancer drivers based on predictions of functionality, to identify other potential driver genes that do not necessarily harbor clusters of mutations. This analysis identified 11 potential cancer drivers in the WNT signaling cascade, three of which were identified by oncodriveCLUST (RNF43: p = 7.22 × 10−6, MEN1: p = 0.02, and GNG12: p = 0.012). Other genes that were identified include members of the beta-catenin destruction complex (APC: p = 7.22 × 10−6, AXIN1: p < 0.01, AXIN2: p = 0.0001), ZNRF3 (p = 7.22 × 10−6), SOX9 (p = 7.22 × 10−6), BCL9L (p < 0.001), PYGO2 (p < 0.001), and WNT11 (p = 0.045).
WNT16 harbors a mutational hotspot at G165. This codon resides in a G7 repeat track that was the subject of frameshift indels in 15 cancers. We used Sanger sequencing to orthogonally validate the presence of WNT16 hotspot mutations in BRAF mutant cancers (n = 79) and identified frameshift mutations in 20.2% (16/79) of cancers (Figure S2). Clinicopathological analyses of WNT16 mutant cancers revealed an association with MSI (mutant: 100% vs. Wt: 61.9%, p = 0.0054), right sided tumors (mutant: 100% vs. Wt: 86.8%, p = 0.038) and earlier stage at diagnosis (Mutant: 87.6% vs. Wt: 63.2%, p = 0.029). When corrected for covariates that may influence cancer stage (MSI, tumour side, sex, age, CIMP), WNT16 mutations were marginally associated with an earlier stage at diagnosis (p = 0.05).
3.6. Mutations in WNT Signaling Regulators Confer Prognostic Implications
To determine whether frequently mutated WNT regulators, or those identified as putative cancer drivers, is relevant to patient outcome, we performed survival analysis on patients with cancers bearing mutations in the five most frequently mutated WNT signaling regulators (RNF43, KMT2D, TRRAP, APC, CREBBP) and those identified by driver analysis (MEN1, GNG12, WNT16, AXIN1, AXIN2, ZNRF3, SOX9, BCL9L, PYGO2 and WNT11). Four genes (APC, RNF43, CREBBP and WNT16) were associated with survival (Table 3). Mutations in WNT16 and APC were significantly associated with shorter overall survival (Table 3). By contrast, mutations of CREBBP and RNF43 were associated with prolonged survival (Table 3).
Table 3.
Univariate survival analysis of the most frequently mutated WNT signaling regulators, and WNT signaling regulators identified as potential drivers by computational analysis. Differences in overall survival were assessed using the log-rank test.
3.7. Co-Mutation of APC and BRAF Represents a Unique and Aggressive Subtype of BRAF Mutant Cancers
We next evaluated the relationship between BRAF mutation and APC mutation in further detail, to characterize the clinical and molecular correlates of this subtype of cancers. We supplemented the 199 BRAF mutant exomes assessed earlier in the manuscript with 76 BRAF mutant cancers that were subjected to targeted sequencing as part of Yaeger et al. 2018 [16]. Truncating mutation was present in 20% of BRAF mutant cancers. We examined whether there was a relationship between age at diagnosis and APC mutation by logistic regression analysis. The probability of truncating APC mutation occurring in a BRAF mutant cancer decreases markedly with age from ~60% in patients diagnosed at age 40, to <10% of patients diagnosed at >90 years of age (Logistic Regression p = 3.74 × 10−7). The average age of patients with a BRAFV600E/APCTruncated cancer was significantly lower than both patients with a BRAF V600E/APCMissense tumour (61 vs. 72, p = 2.03 × 10−5, Table 4) and a patient with a BRAF V600E/APCWild-type cancer (61 vs. 71, p = 9.3 × 10−6). BRAFV600E/APCTruncated cancers were more likely to be left sided when compared with BRAFV600E/APCMissense cancer (24.5% vs. 4.2%, p = 0.02, Table 4). There was no difference in the frequency of CIMP versus either missense or wild-type APC cancers. Overall, 42.3% of BRAFV600E/APCTruncated cancers were microsatellite unstable. MSI is less frequent than both BRAF V600E/APCMissense (91.3%, p = 5.3 × 10−5, Table 4) and BRAF V600E/APCWild-type (53.8%, p = 0.14) cancers.
Table 4.
Clinical and molecular characteristics of BRAF mutant cancers with APC mutation.
BRAFV600E/APCTruncated cancers were aggressive cancers, with 67.3% of patients presenting with metastatic disease. In contrast, only 36.4% and 45.7% of BRAF V600E/APCMissense and BRAF V600E/APCWild-type cancers presented at stage III/IV (p = 0.01 and 0.002 versus BRAFV600E/APCTruncated, respectively). BRAFV600E/APCTruncated cancers that were also microsatellite stable were enriched further for late-stage disease, with 100% of these patients presenting with metastatic disease (Stage III or IV), and 88% with distant metastases (Stage IV).
Furthermore, the univariable analysis of survival indicated that BRAFV600E/APCTruncated cancers have a significantly poorer median survival (504 days vs. 1390 days, log-rank p = 0.026, n = 32 and n = 78 for truncating mutant and wild-type, respectively; Figure 6A). The five-year survival of BRAFV600E/APCTruncated patients was 12%. By contrast, the five year survival of BRAFV600E/APCWild-Type patients was 42%. This effect was most pronounced in microsatellite stable cancers (Figure S3A) compared with microsatellite unstable cancers (Figure S3B,C). We performed a multivariate survival analysis, including age at diagnoses, gender, stage and microsatellite instability as potential prognosticators. Using the Cox-proportional hazard method, microsatellite instability status, and gender are significantly independently associated with survival. Truncating APC mutation trends toward conferring independent negative prognostic implications, however this failed to reach the threshold for significance (Table 5, p = 0.17). Collectively, these data indicate that the activating mutation of BRAF and the truncating mutation of APC represent an aggressive subtype of colorectal cancers that occur at a relatively young age in comparison to BRAF mutant cancers more generally.
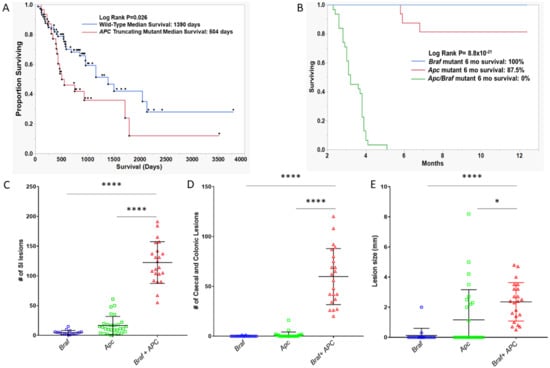
Figure 6.
Survival analysis of (A) BRAF mutant human cancers by the presence or absence of truncating APC mutation. (B) Apc, Braf, and Apc/Braf mutant murine models. p-values are univariate and derived from the log-rank test. (C–E) Assessment of the number and size of lesions in Apc, Braf, and Apc/Braf mutant mouse models. (C) Total lesions in the small intestine. (D) Total number of lesions in the colon and caecum. E: Mean size of lesions in the colon and caecum.
Table 5.
Cox-proportional Hazard analysis of survival of BRAF mutant cancers.
3.8. Mutation of Braf in APCmin/+ Mouse Results in Massive Polyp Load, Rapid Disease Progression and Poor Survival
To determine whether we could recapitulate the apparently aggressive phenotype of the co-mutation of BRAF and APC, we crossed inducible BrafV637 mutant mice with Apcmin/+ mice. The Braf mutation was induced at wean in ApcMin/+ mice and we compared the number of lesions per animal and survival to mice with just the mutant Braf allele or the mutant Apc allele.
We next assessed differences in survival between ApcMin/+ (n = 29), BrafV63 7(n = 15), and APCMin/+/ BrafV637 mice (n = 22). Animals were regarded as having survived and were censored if they were healthy at the time of sacrifice; animals were regarded as deceased if the animal had to be euthanized due to illness. Notably, 100% of Braf mutant animals survived to 12 months, as did 81.25% of Apc mutant animals. Mutation of both Braf and Apc significantly reduced the survival of the animals (p = 8.8 × 10−21, Figure 6B). The median survival of animals with both Apc and Braf mutation was 3.2 months. No animal with both mutations survived longer than six months.
We assessed polyp load by microscopic enumeration. Animals with Braf and Apc mutations alone develop an average of 4.6 and 16.55 polyps in the small intestine, respectively. Animals with both Braf and Apc mutation simultaneously develop significantly more lesions in the SI (p < 0.0001, Figure 6C). Animals with Braf or Apc mutation rarely developed colonic or caecal lesions (mean lesions per mouse: 0.11 and 1.1, respectively, Figure 6D). In contrast, the dual mutation of Apc and Braf resulted in the accumulation of an average of 59.82 colonic/caecal lesions per animal (p < 0.0001, Figure 6D). We did not observe a significant increase in lesion size in the small intestine between groups, however we did observe significantly larger lesions in the colon and caecum of animals bearing both Apc and Braf mutation (p < 0.0001, Figure 6E). Lesions had a morphology that was reminiscent of human conventional adenomas, rather than dysplastic serrated lesions.
4. Discussion
Here, we have investigated the role of somatic mutation in shaping the WNT signaling landscape of colorectal cancers bearing the BRAF mutation. We have shown that 48% of BRAF mutant cancers mutate at least one member of the B-catenin destruction complex. Other common modes of activation include the mutation of RNF43 and ZNRF3. We have identified a number of novel mutations that may alter the WNT signaling landscape of cancers. These include MEN1, a known WNT pathway tumor suppressor, and WNT16, a WNT ligand that may act as an antagonist of ligand mediated WNT activation. Both MEN1 and WNT16 harbor hotspot frameshift mutations, that were identified as potential drivers by computational analysis. Mutation of RNF43 was mutually exclusive to the mutation of APC. We examined the clinical and molecular correlates of BRAF mutant cancers bearing truncating mutations of APC, which occurred in 20% of samples. These cancers were predominantly microsatellite stable, and late stage. Cancers with a truncating APC mutation occurred at an average age that was >10 years lower than the wider cohort of BRAF mutant cancers. Survival analysis revealed a significantly poorer prognosis for this subtype of patients. Using the murine models of Apc and Braf mutations, we showed that mutating both genes results in an extensive phenotype with a massive lesion burden. Animals had a median survival of 3.2 months, and no animal bearing both mutations survived longer than six months. Collectively, these data indicate that the mutation of both BRAF and APC results in an aggressive and rapidly progressing cancer phenotype and confers a poor prognosis.
WNT signaling underpins colorectal carcinogenesis. In the conventional pathway, WNT signaling is usually activated via bi-allelic inactivation of the APC tumor suppressor gene at the beginning of the tumorigenic process. However, the mechanisms governing the WNT pathway activation in the serrated neoplasia pathway, which is uniquely marked by BRAF mutation, is less clear. In the present study, we sought to identify WNT signaling genes that are mutated in the context of BRAF mutant serrated colorectal neoplasia. We obtained exome sequencing data from 175 BRAF mutant colorectal cancers from four previously published studies [7,14,15,32] and sequenced a further 24 BRAF mutant samples collected locally. Our analyses revealed a mosaic of mutations in WNT signaling regulators, including well-known WNT regulators such as RNF43, APC, AXIN2 and ZNRF3. Our analysis identified significant mutual exclusivity between truncating mutations of RNF43 and APC. The mutual exclusivity of mutations in these genes has been previously reported, [8] however it has not been clear whether this mutual exclusivity was, in part, due to the abundance of RNF43 mutations in BRAF mutant cancers, and the relative rarity of APC mutations in this context. Here, we have shown that within cancers that bear BRAF mutation, the mutually exclusive nature of mutations in these genes remain, indicating that the mutual exclusivity is likely between APC and RNF43 mutations, rather than between BRAF mutations and APC mutations. Likewise, ZNRF3 mutation was mutually exclusive to truncating mutations of APC. This association was present only when missense mutations were not included. It is possible that the addition of a truncating APC mutation in this context is disadvantageous to tumor progression. Therefore, the mutation of RNF43/ZNRF3 may create a genetic dependency on APC. If true, exploiting the dependency on APC, a canonical tumor suppressor gene, may be a novel therapeutic treatment for patients with an RNF43 mutated cancer.
We next examined the exome sequencing data to identify potential novel drivers of WNT signaling activation in colorectal cancer. We adopted a mutational clustering-based approach to identify potential cancer drivers based on the presence of mutational hotspots, as implemented in the OncodriveCLUST algorithm [26]. Reassuringly, RNF43, which has two mutational hotspots [2,8,30], was successfully identified as a cancer driver. RSPO fusions, which have been implicated in the WNT dysregulation of serrated lesions and cancers [4,33], were not identified due to technological limitations. It is likely that some cancers in this cohort harbored such fusions, given the frequency of RSPO fusions previously reported. We identified eleven other potential cancer driver genes in the WNT signaling cascade. MEN1 was mutated in eight samples and most mutations were frameshift alterations at codon R521. Germline MEN1 mutations result in multiple endocrine neoplasia type 1, a tumor predisposition syndrome. It has also been identified as a tumor suppressor gene in a number of different cancer types, including tumors of the parathyroid [34] (p1), entero-pancreatic neuroendocrine cancers [35] (p1), and carcinoids [36]. Interestingly, especially in the context of highly methylated BRAF mutant cancers, the loss of MEN1 has been associated with aberrant DNMT1 activity and an altered DNA methylation landscape. To our knowledge, MEN1 alterations have not been previously reported in colorectal cancer, nor is colorectal cancer a typical presentation of MEN1 syndrome. It is possible that these patients had an underlying germline mutation in MEN1, and the mutations identified in this studied were the second hit at the locus.
WNT16 was also identified as a potential cancer driver gene. WNT16 is a WNT ligand, a seemingly unlikely candidate tumor suppressor. However, Nalesso et al. [37] showed that while WNT16 was capable of binding Fzd receptors and activating canonical WNT signaling, the degree of activation was significantly lower when compared with the more abundant WNT3A. TOPFlash assays showed that co-stimulation with both WNT3A and WNT16 resulted in significantly less canonical WNT activation when compared with stimulation using WNT3A alone [37]. Thus, it appears WNT16 acts as a competitive inhibitor of Fzd and acts to ensure the homeostasis of WNT signaling. In cancer, the loss of WNT16 may facilitate excessive canonical WNT activation by failure to compete with more potent WNT ligands, such as WNT3A and WNT8. In combination with data indicating poorer prognosis for patients harboring WNT16 mutant cancers, it is likely that WNT16 acts as a tumor suppressor. Inhibitors of WNT ligand secretion, such as porcupine inhibitors, are currently being trialed in colorectal and other solid tumors [38]. Cancers that lack WNT16 are prone to excessive ligand-dependent WNT activation [37] (p16) and may represent a subset of patients that could benefit from this therapy. Indeed, as much of WNT16 mutations occur on a background of RNF43 alterations, which has been shown to confer sensitivity to porcupine inhibitors [31], the mutation of both genes could further sensitize cells to this class of drugs. Although we do not present data in support of this hypothesis in this study, the frequency in which WNT16 is mutated in BRAF mutant colorectal cancers may provide an impetus for investigating whether WNT16 has a role in determining sensitivity to WNT-ligand inhibitors.
We recently assessed a series of 80 BRAF mutant cancers [1] and identified truncating APC mutation in 11% of these cancers. It has been postulated that truncating APC mutation is uncommon in the context of pre-existing BRAF mutation, because the dysregulation of the WNT signal is too profound in this cellular context. This is consistent with the model proposed by Albuquerque et al. [39] and indicates that mutations such as RNF43/ZNRF3 provide a “just-right” level of WNT signaling to confer a selective advantage. However, the present study has confirmed that a minority of BRAF mutant cancers do carry a truncating APC mutation. The higher proportion of cancers bearing both APC and BRAF mutation in the present study may be due to selection bias in the present series, with a higher proportion of late stage microsatellite stable cancer included.
APC mutations were much more common in a minority of BRAF mutant cancers diagnosed at a younger age. The average age of BRAF mutant cancers harboring APC mutation was 12 years less than APC wild-type. These cancers were more likely to be microsatellite stable, and present with metastatic disease. The median survival of patients with BRAF mutation and APC mutation was 64% lower than patients with BRAF mutation alone, and patients with both mutations had a five-year survival rate of 12%. BRAF mutant microsatellite stable cancers are known to confer a particularly poor prognosis [40], in part due to the low neoantigen burden and poor immunogenicity of the cancers [41]. Although APC mutation in BRAF mutant cancers was not significantly associated with poorer prognosis on multivariate analysis, we did identify APC mutations as prognosticators on univariate analysis. Furthermore, there was a strong and borderline significant trend to a worse still prognosis for BRAF mutant MSS cancers with APC mutations, compared to those without. Hence, cancers appear to be highly aggressive and occur earlier in life. We generated a murine model to recapitulate mutation of APC and BRAF to examine interactions between these mutations and the consequences of mutating both genes on polyp development and overall survival. We observed massive polyp loads in animals bearing both mutations, and a markedly reduced survival. Moreover, 100% and 81.25% of Braf mutant and Apc mutant animals survived to 12 months. When we mutated both genes, no animals survived past six months and the median survival was reduced to a mere 3.2 months. While animals did not develop invasive cancer, and instead died due to polyp load, these data indicate that the commutation of APC and BRAF in an enterocyte induces rapid neoplastic alterations and an overt proliferative phenotype. Collectively, these data provide strong evidence that the mutation of both APC and BRAF, whilst uncommon in humans, generates a remarkably aggressive neoplastic phenotype.
It is difficult to resolve how these particular cancers have evolved. Both APC and BRAF mutations are tumor-initiating events [28,42,43] and give rise to different precursor lesions [44]. Moreover, the cell of origin for APC initiated polyps and BRAF initiated polyps is hypothesized to differ. APC initiated lesions adhere to the “top-down” model [45], whereas BRAF mutant lesions are initiated in the stem compartment [46]. Methylation profiling of BRAF mutant and APC mutant cancers confirmed this model, and showed that BRAF mutant cancers had a methylation profile reminiscent of the intestinal stem cell [46] (p). In our study, we observed no difference in the frequency of CIMP between BRAF mutants with APC mutation and those without, suggesting that these dual-mutant cancers may have arisen in the stem component, and therefore may have been initiated by BRAF and acquired an APC mutation at a later stage. However, it is difficult to reconcile this with the rarity to which APC is mutated in BRAF mutant precursor lesions [1]. It is possible that, upon acquiring an APC mutation, progression to cancer is rapid and, as a result, identifying lesions in a transitional state is rare. This fits with the aggressive phenotype of these cancers. An alternative hypothesis, supporting an age at diagnosis that is similar to conventional pathway cancers [18] and the morphology of our murine adenomas, is that polyps are initiated by APC, acquiring a BRAF mutation. If this is the case, such lesions must progress extremely rapidly to cancer, as they are very rarely identified in large series of conventional adenomas.
The mechanisms associated with APC mutation induced aggressiveness in the context of BRAF mutant cancers are not clear. Aberrant WNT signaling appears to be a prerequisite for the acquisition of dysplasia in traditional serrated adenomas [3] and sessile serrated lesion [29,47]. In traditional serrated adenomas, which are by nature dysplastic, are thought to have transitioned from hyperplastic polyps or sessile serrated lesions, and this transition coincides with the acquisition of WNT pathway alterations, such as RSPO fusions [4] and RNF43 mutations [3]. In sessile serrated lesions that acquire dysplasia, RNF43 mutations are extremely common (>50%) [47], however they are rare in lesions that are not yet dysplastic. This is an important distinction, as lesions with dysplasia are thought to progress within 12 months to invasive cancer [48]. Alterations to RNF43 are dependent on DNA methylation induced loss of MLH1 and mismatch repair function. In sessile serrated lesions, DNA methylation may develop over a protracted period of time, increasing the sojourn of these lesions to dysplasia and ultimately malignancy [49]. BRAF mutation has been reported to induce senescence, which can be overcome through silencing of the CDKN2A loci [50]. It has been previously reported that APC loss in the intestine does require loss of p21 to generate adenomas and does not induce senescence [51]. It is possible that APC mutations, in the context of BRAF mutation, facilitate an exit to oncogene induced senescence and while simultaneously satisfying the requirement for WNT signaling, facilitating a rapid transition to dysplasia and invasive cancer. This may explain why precancerous lesions with both APC mutation and BRAF mutation are rarely identified, and why the cancers with these mutations present at a younger age.
Our study has comprehensively assessed the somatic mutation landscape of WNT signaling regulators in a large series of BRAF mutant colorectal cancers, however several limitations remain. As we assessed publicly accessible sequencing data from various sources, the depth of sequencing, bioinformatic pipeline and variant filtering methods was not uniform across cohorts. This may result in the under (or over) reporting of certain variants. Our study was also limited in the ability to assess the role of the CpG island methylator phenotype in WNT signaling. CIMP was assessed in some samples in our study cohorts, however the method of assessment was not uniform across samples. For example, cancers from TCGA assessed CIMP via microarray based clustering; in contrast, other studies used qPCR based marker panels. Moreover, many WNT signaling genes are putative methylation targets. Further study is necessary to assess the role of CIMP in shaping WNT signaling in the context of BRAF mutant colorectal cancer. Although our study is the largest to date to assess WNT regulators in BRAF mutant cancers, we did not have a sufficiently large enough sample size to draw conclusions as to the prognostic implications of genes that are mutated more rarely, such as MEN1 and GNG12. Mutations in hotspots, or in relatively small genes, could be assessed in much larger cohorts using less expensive genotyping technologies, to determine whether mutations in these genes are relevant to patient outcome.
5. Conclusions
In conclusion, here we have conducted a comprehensive survey of the somatic mutational landscape shaping WNT signaling in BRAF mutant serrated colorectal neoplasia. The mutational landscape of WNT signaling regulators is a mosaic that is underpinned by mutations in key driver genes, such as RNF43 and APC. The mutations of RNF43 and APC are mutually exclusive. We identified potential cancer driver genes in the WNT signaling axis. MEN1 has previously been implicated in cancers of endocrine origin, but has not been identified as a tumor suppressor gene in colorectal cancer. We have identified a hotspot mutation in MEN1 that effects 4% of BRAF mutant cancers. We have identified WNT16 as a potential driver gene by mutational hotspot analysis. WNT16 is a competitive inhibitor of canonical WNT and the mutation of WNT16 is common in BRAF mutant cancers. BRAF mutant colorectal cancers with truncating APC mutation tended to arise earlier in life, and presented at a significantly later stage. These cancers are extremely aggressive and the survival of patients with both BRAF and APC mutation is poor (12% 5-year survival). The in vivo modelling of Apc and Braf mutation revealed a dramatically increased tumor burden, with a median survival of 3.2 months for animals with both mutations. Therefore, we conclude that the co-mutation of BRAF and APC in colorectal cancers is conducive to an aggressive phenotype.
Supplementary Materials
The following are available online at https://www.mdpi.com/2072-6694/12/5/1171/s1, Figure S1: Tumor mutation burden (nsSNV/mb) by MSI status in each cohort, Figure S2: Sanger sequencing of WNT16 G7 repeat confirms mutational hotspot, Figure S3: Kaplan-Meir survival analysis of BRAF mutant stratified by APC mutation, Table S1: Clinicopathological details of patients included in the study, Table S2: All mutations detected in the study, Table S3: Sequencing metrics in the RBWH cohort, Table S4: Mutual Exclusivity analyses by pair-wise fishers exact tests.
Author Contributions
L.J.F. conceptualized the study, bioinformatic analysis, statistical analysis, and drafted the manuscript. A.K. contributed to animal work. C.L. carried out the histopathological analyses, D.M. contributed to animal work. W.F. contributed to animal work. C.S. contributed to the molecular analyses. C.B. contributed to the molecular analyses and animal work. S.J. contributed to the molecular analyses. T.D. contributed to the molecular analyses. A.-M.P. contributed to the bioinformatic analyses. S.H.K. contributed to the bioinformatic analyses. J.V.P. contributed to the bioinformatic analyses. N.W. contributed to the bioinformatic analyses. B.L. conceptualized and supervised the study. V.L.J.W. conceptualized and supervised the study, and obtained funding. All authors contributed to editing the manuscript and approved the submitted version. All authors have read and agree to the published version of the manuscript.
Funding
This work was supported through funding from the National Health and Medical Research Council (Grant #: 1050455, 1063105), the Cancer Council Queensland (1160923), the RBWH Research Foundation and Pathology Queensland. VW is the recipient of a Senior Research Fellowship from the Gastroenterological Society of Australia. LF was supported by a Research Training Program Living Scholarship from the Australia Government, a Top-Up award from QIMR Berghofer and Australian Rotary Health. NW is funded by a National Health and Medical Research Council fellowship (Grant #: APP1139071).
Acknowledgments
We acknowledge the contributions of the reviewers and their insightful comments that helped shape the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Borowsky, J.; Dumenil, T.; Bettington, M.; Pearson, S.-A.; Bond, C.; Fennell, L.; Liu, C.; McKeone, D.; Rosty, C.; Brown, I.; et al. The role of APC in WNT pathway activation in serrated neoplasia. Mod. Pathol. 2018, 31, 495–504. [Google Scholar] [CrossRef]
- Bond, C.E.; McKeone, D.M.; Kalimutho, M.; Bettington, M.L.; Pearson, S.-A.; Dumenil, T.D.; Wockner, L.F.; Burge, M.; Leggett, B.A.; Whitehall, V.L.J. RNF43 and ZNRF3 are commonly altered in serrated pathway colorectal tumorigenesis. Oncotarget 2016, 7, 70589–70600. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Ogawa, R.; Yoshida, H.; Taniguchi, H.; Kojima, M.; Saito, Y.; Sekine, S. Acquisition of WNT Pathway Gene Alterations Coincides with the Transition From Precursor Polyps to Traditional Serrated Adenomas. Am. J. Surg. Pathol. 2019, 43, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Yamashita, S.; Tanabe, T.; Hashimoto, T.; Yoshida, H.; Taniguchi, H.; Kojima, M.; Shinmura, K.; Saito, Y.; Hiraoka, N.; et al. Frequent PTPRK-RSPO3 fusions and RNF43 mutations in colorectal traditional serrated adenoma. J. Pathol. 2016, 239, 133–138. [Google Scholar] [CrossRef]
- Rowan, A.J.; Lamlum, H.; Ilyas, M.; Wheeler, J.; Straub, J.; Papadopoulou, A.; Bicknell, D.; Bodmer, W.F.; Tomlinson, I.P.M. APC mutations in sporadic colorectal tumors: A mutational “hotspot” and interdependence of the “two hits”. Proc. Natl. Acad. Sci. USA 2000, 97, 3352–3357. [Google Scholar] [CrossRef]
- Kambara, T.; Matsubara, N.; Nagao, A.; Uchida, T.; Nakagawa, H.; Tanaka, N.; Nagasaka, T.; Young, J.; Leggett, B.; Jass, J. Mtations in BRAF, KRAS, and APC, and CpG island methylation: Alternative pathways to colorectal cancer. Cancer Res. 2006, 66, 81. [Google Scholar]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef]
- Tu, J.; Park, S.; Yu, W.; Zhang, S.; Wu, L.; Carmon, K.; Liu, Q.J. The most common RNF43 mutant G659Vfs*41 is fully functional in inhibiting Wnt signaling and unlikely to play a role in tumorigenesis. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Suzuki, H.; Watkins, D.N.; Jair, K.-W.; Schuebel, K.E.; Markowitz, S.D.; Chen, W.D.; Pretlow, T.P.; Yang, B.; Akiyama, Y.; van Engeland, M.; et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat. Genet. 2004, 36, 417–422. [Google Scholar] [CrossRef]
- Suzuki, H.; Gabrielson, E.; Chen, W.; Anbazhagan, R.; van Engeland, M.; Weijenberg, M.P.; Herman, J.G.; Baylin, S.B. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat. Genet. 2002, 31, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, O.; Fraga, M.F.; Ballestar, E.; Paz, M.F.; Herranz, M.; Espada, J.; García, J.M.; Muñoz, A.; Esteller, M.; González-Sancho, J.M. Epigenetic inactivation of the Wnt antagonist DICKKOPF-1 (DKK-1) gene in human colorectal cancer. Oncogene 2006, 25, 4116–4121. [Google Scholar] [CrossRef]
- Liu, Y.; Sethi, N.S.; Hinoue, T.; Schneider, B.G.; Cherniack, A.D.; Sanchez-Vega, F.; Seoane, J.A.; Farshidfar, F.; Bowlby, R.; Islam, M.; et al. Comparative Molecular Analysis of Gastrointestinal Adenocarcinomas. Available online: https://pubmed.ncbi.nlm.nih.gov/29622466-comparative-molecular-analysis-of-gastrointestinal-adenocarcinomas/ (accessed on 20 January 2020).
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Vasaikar, S.; Huang, C.; Wang, X.; Petyuk, V.A.; Savage, S.R.; Wen, B.; Dou, Y.; Zhang, Y.; Shi, Z.; Arshad, O.A.; et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019, 177, 1035–1049. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Available online: https://pubmed.ncbi.nlm.nih.gov/29316426/ (accessed on 16 January 2020).
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef]
- Fennell, L.; Dumenil, T.; Wockner, L.; Hartel, G.; Nones, K.; Bond, C.; Borowsky, J.; Liu, C.; McKeone, D.; Bowdler, L.; et al. Integrative Genome-Scale DNA Methylation Analysis of a Large and Unselected Cohort Reveals Five Distinct Subtypes of Colorectal Adenocarcinomas. Cell. Mol. Gastroenterol. Hepatol. 2019. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:13033997. Q-Bio. Available online: http://arxiv.org/abs/1303.3997 (accessed on 6 February 2020).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinforma. Oxf. Engl. 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Kassahn, K.S.; Holmes, O.; Nones, K.; Patch, A.-M.; Miller, D.K.; Christ, A.N.; Harliwong, I.; Bruxner, T.J.; Xu, Q.; Anderson, M.; et al. Somatic Point Mutation Calling in Low Cellularity Tumors. PLoS ONE 2013, 8, e74380. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Tamborero, D.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveCLUST: Exploiting the Positional Clustering of Somatic Mutations to Identify Cancer Genes. Available online: https://pubmed.ncbi.nlm.nih.gov/23884480/ (accessed on 20 January 2020).
- Mularoni, L.; Sabarinathan, R.; Deu-Pons, J.; Gonzalez-Perez, A.; López-Bigas, N. OncodriveFML: A general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol. 2016, 17, 128. [Google Scholar] [CrossRef]
- Bond, C.E.; Liu, C.; Kawamata, F.; McKeone, D.M.; Fernando, W.; Jamieson, S.; Pearson, S.-A.; Kane, A.; Woods, S.L.; Lannagan, T.R.M.; et al. Oncogenic BRAF mutation induces DNA methylation changes in a murine model for human serrated colorectal neoplasia. Epigenetics 2018, 13, 40–48. [Google Scholar] [CrossRef]
- Kane, A.M.; Fennell, L.J.; Liu, C.; Borowsky, J.; McKeone, D.M.; Bond, C.E.; Kazakoff, S.; Patch, A.-M.; Koufariotis, L.T.; Pearson, J.; et al. Alterations in signaling pathways that accompany spontaneous transition to malignancy in a mouse model of BRAF mutant microsatellite stable colorectal cancer. Neoplasia 2020, 22, 120–128. [Google Scholar] [CrossRef]
- Fennell, L.J.; Clendenning, M.; McKeone, D.M.; Jamieson, S.H.; Balachandran, S.; Borowsky, J.; Liu, J.; Kawamata, F.; Bond, C.E.; Rosty, C.; et al. RNF43 is mutated less frequently in Lynch Syndrome compared with sporadic microsatellite unstable colorectal cancers. Fam. Cancer 2018, 17, 63–69. [Google Scholar] [CrossRef]
- Jiang, X.; Hao, H.X.; Growney, J.D.; Woolfenden, S.; Bottiglio, C.; Ng, N.; Lu, B.; Hsieh, M.H.; Bagdasarian, L.; Meyer, R.; et al. Inactivating Mutations of RNF43 Confer Wnt Dependency in Pancreatic Ductal Adenocarcinoma. Available online: https://pubmed.ncbi.nlm.nih.gov/23847203/ (accessed on 20 January 2020).
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-Spondin Fusions in Colon Cancer. Available online: https://pubmed.ncbi.nlm.nih.gov/22895193/ (accessed on 15 January 2020).
- Sekine, S.; Ogawa, R.; Hashimoto, T.; Motohiro, K.; Yoshida, H.; Taniguchi, H.; Saito, Y.; Yasuhiro, O.; Ochiai, A.; Hiraoka, N. Comprehensive characterization of RSPO fusions in colorectal traditional serrated adenomas. Histopathology 2017, 71, 601–609. [Google Scholar] [CrossRef]
- Yuan, Z.; Sánchez Claros, C.; Suzuki, M.; Maggi, E.C.; Kaner, J.D.; Kinstlinger, N.; Gorecka, J.; Quinn, T.J.; Geha, R.; Corn, A.; et al. Loss of MEN1 Activates DNMT1 Implicating DNA Hypermethylation as a Driver of MEN1 Tumorigenesis. Available online: https://pubmed.ncbi.nlm.nih.gov/26871472/ (accessed on 20 January 2020).
- Agarwal, S.K. The future: Genetics advances in MEN1 therapeutic approaches and management strategies. Endocr. Relat. Cancer 2017, 24, T119–T134. [Google Scholar] [CrossRef]
- Marini, F.; Falchetti, A.; Luzi, E.; Tonelli, F.; Maria Luisa, B. Multiple Endocrine Neoplasia Type 1 (MEN1) Syndrome. In Cancer Syndromes; Riegert-Johnson, D.L., Boardman, L.A., Hefferon, T., Roberts, M., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2009. Available online: http://www.ncbi.nlm.nih.gov/books/NBK7029/ (accessed on 20 January 2020).
- Nalesso, G.; Thomas, B.L.; Sherwood, J.C.; Yu, J.; Addimanda, O.; Eldridge, S.E.; Thorup, A.-S.; Dale, L.; Schett, G.; Zwerina, J.; et al. WNT16 antagonises excessive canonical WNT activation and protects cartilage in osteoarthritis. Ann. Rheum. Dis. 2017, 76, 218–226. [Google Scholar] [CrossRef]
- Rodon, J.; Argilés, G.; Connolly, R.M.; Vaishampayan, U.; de Jonge, M.; Garralda, E.; Giannakis, M.; Smith, D.C.; Dobson, J.R.; McLaughlin, M.; et al. Abstract CT175: Biomarker analyses from a phase I study of WNT974, a first-in-class Porcupine inhibitor, in patients (pts) with advanced solid tumors. Cancer Res. 2018, 78, CT175. [Google Scholar] [CrossRef]
- Albuquerque, C.; Breukel, C.; van der Luijt, R.; Fidalgo, P.; Lage, P.; Slors, F.J.M.; Leitão, C.N.; Fodde, R.; Smits, R. The ‘just-right’ signaling model: APC somatic mutations are selected based on a specific level of activation of the β-catenin signaling cascade. Hum. Mol. Genet. 2002, 11, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Pai, R.K.; Jayachandran, P.; Koong, A.C.; Chang, D.T.; Kwok, S.; Ma, L.; Arber, D.A.; Balise, R.R.; Tubbs, R.R.; Shadrach, B.; et al. BRAF-mutated, Microsatellite-stable Adenocarcinoma of the Proximal Colon: An Aggressive Adenocarcinoma With Poor Survival, Mucinous Differentiation, and Adverse Morphologic Features. Am. J. Surg. Pathol. 2012, 36, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Bolzacchini, E.; Cerutti, R.; Digiacomo, N.; Sahnane, N.; Pinotti, G.; Bregni, M.; Artale, S.; Verusio, C.; Crivelli, F.; Capella, C.; et al. Difference in immune infiltration in MSI and MSS BRAF mutant colorectal cancer. J. Clin. Oncol. 2018, 36, e15624. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Rad, R.; Cadiñanos, J.; Rad, L.; Varela, I.; Strong, A.; Kriegl, L.; Constantino-Casas, F.; Eser, S.; Hieber, M.; Seidler, B.; et al. A Genetic Progression Model of BrafV600E-Induced Intestinal Tumorigenesis Reveals Targets for Therapeutic Intervention. Cancer Cell 2013, 24, 15–29. [Google Scholar] [CrossRef]
- Pai, R.K.; Bettington, M.; Srivastava, A.; Rosty, C. An Update on the Morphology and Molecular Pathology of Serrated Colorectal Polyps and Associated Carcinomas. Available online: https://pubmed.ncbi.nlm.nih.gov/31028362/ (accessed on 20 January 2020).
- Shih, I.-M.; Wang, T.-L.; Traverso, G.; Romans, K.; Hamilton, S.R.; Ben-Sasson, S.; Kinzler, K.W.; Vogelstein, B. Top-down morphogenesis of colorectal tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 2640–2645. [Google Scholar] [CrossRef]
- Bormann, F.; Rodríguez-Paredes, M.; Lasitschka, F.; Edelmann, D.; Musch, T.; Benner, A.; Bergman, Y.; Dieter, S.M.; Ball, C.R.; Glimm, H.; et al. Cell-of-Origin DNA Methylation Signatures Are Maintained during Colorectal Carcinogenesis. Cell Rep. 2018, 23, 3407–3418. [Google Scholar] [CrossRef]
- Hashimoto, T.; Yamashita, S.; Yoshida, H.; Taniguchi, H.; Ushijima, T.; Yamada, T.; Saito, Y.; Ochiai, A.; Sekine, S.; Hiraoka, N. WNT Pathway Gene Mutations Are Associated With the Presence of Dysplasia in Colorectal Sessile Serrated Adenoma/Polyps. Am. J. Surg. Pathol. 2017, 41, 1188–1197. [Google Scholar] [CrossRef]
- Bettington, M.; Walker, N.; Rosty, C.; Brown, I.; Clouston, A.; McKeone, D.; Pearson, S.-A.; Leggett, B.; Whitehall, V. Clinicopathological and molecular features of sessile serrated adenomas with dysplasia or carcinoma. Gut 2017, 66, 97–106. [Google Scholar] [CrossRef]
- Liu, C.; Bettington, M.L.; Walker, N.I.; Dwine, J.; Hartel, G.F.; Leggett, B.A.; Whitehall, V.L.J. CpG Island Methylation in Sessile Serrated Adenomas Increases with Age, Indicating Lower Risk of Malignancy in Young Patients. Gastroenterology 2018, 155, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Bennecke, M.; Kriegl, L.; Bajbouj, M.; Retzlaff, K.; Robine, S.; Jung, A.; Arkan, M.C.; Kirchner, T.; Greten, F.R. Ink4a/Arf and Oncogene-Induced Senescence Prevent Tumor Progression during Alternative Colorectal Tumorigenesis. Cancer Cell 2010, 18, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.M.; Ridgway, R.A.; Derkits, S.E.; Parry, L.; Barker, N.; Clevers, H.; Clarke, A.R.; Sansom, O.J. p21 loss blocks senescence following Apc loss and provokes tumourigenesis in the renal but not the intestinal epithelium. EMBO Mol. Med. 2010, 2, 472–486. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).