Abstract
The pathogenesis of ocular adnexal marginal zone lymphomas of mucosa-associated lymphatic tissue-type (OAML) is not fully understood. We performed whole genome sequencing (WGS) and/or whole exome sequencing (WES) for 13 cases of OAML and sequenced 38 genes selected from this analysis in a large cohort of 82 OAML. Besides confirmation of frequent mutations in the genes transducin beta like 1 X-linked receptor 1 (TBL1XR1) and cAMP response element binding protein (CREBBP), we newly identifed JAK3 as a frequently mutated gene in OAML (11% of cases). In our retrospective cohort, JAK3 mutant cases had a shorter progression-free survival compared with unmutated cases. Other newly identified genes recurrently mutated in 5–10% of cases included members of the collagen family (collagen type XII alpha 1/2 (COL12A1, COL1A2)) and DOCK8. Evaluation of the WGS data of six OAML did not reveal translocations or a current infection of the lymphoma cells by viruses. Evaluation of the WGS data for copy number aberrations confirmed frequent loss of TNFAIP3, and revealed recurrent gains of the NOTCH target HES4, and of members of the CEBP transcription factor family. Overall, we identified several novel genes recurrently affected by point mutations or copy number alterations, but our study also indicated that the landscape of frequently (>10% of cases) mutated protein-coding genes in OAML is now largely known.
1. Introduction
Lymphomas of the ocular adnexa are a rare entity, accounting for about 1–2% of non-Hodgkin lymphomas (NHL) [1]. In about 80% of cases, extranodal marginal zone lymphomas of the mucosa-associated lymphatic tissue (MALT) subtype are the most frequent type of primary ocular adnexal lymphomas [2,3,4]. Ocular adnexal MALT lymphomas (OAML) have a mature B cell phenotype and derive from post-germinal center B cells [5,6]. In some geographical areas, chronic infections by Chlamydia psittaci have been linked to the pathogenesis of OAML [7,8]. Chromosomal translocations that are frequent in particular subtypes of extranodal marginal zone lymphomas—t(1;14)(p22;q32) (BCL10/IgH), t(14;18)(q32;p21) (IgH/MALT1), t(11;18)(q21;q21) (BIRC3/MALT1), and t(3;14)(p14;q32) (FOXP1/IgH)—have also been identified in OAML, but their overall frequency in OAML is low [9,10,11,12]. As these translocations promote constitutive NF-κB activity [9,10,12], which is a hallmark of MALT lymphomas [13], several studies searched for further mutations in components of the NF-κB pathway in OAML. Indeed, recurrent mutations were found in TNFAIP3 (around 30% of cases), MYD88 (around 5–20% of cases), and BCL10 (around 25% of cases in a cohort of patients from China), whereas other genes were rarely or not mutated in the cases analyzed [14,15]. We recently showed that also NFKBIA is recurrently mutated in such lymphomas [16]. Only 6% of patients in our cohort harbored BCL10 mutations, and TNIP1 was mutated in 5% of cases. Moreover, we identified NOTCH1, NOTCH2, and KMT2D, encoding an epigenetic regulator, as further recurrently mutated genes in OAML [16]. Another candidate gene analysis revealed recurrent mutations in TBL1XR1 (transducin beta like 1 X-linked receptor 1; 6% of cases), TNFRSF14, (tumor necrosis factor receptor family member 14; 5%), and TET2 (tet methycytosine dioxygenase 2; 4% of cases), besides a few less frequently mutated genes [17]. A further candidate gene mutation analysis validated several genes identified by the other studies, mainly adding KMT2C to the list of genes recurrently mutated in OAML [18].
Thus far, only one genome-wide mutation study of OAML has been published [19]. This whole genome sequencing (WGS) analysis included 10 cases. Additional cases were then investigated by targeted sequencing. Besides validation of mutations in NF-κB pathway members, and in TBL1XR1, KMT2D, and NOTCH1, this analysis newly identified mutations in cAMP response element binding protein (CREBBP; 17% of OAML in the extended cohort), encoding a histone/protein acetyltransferase, and in LRP1B (6% of cases).
In the present work, we performed WGS for six cases of OAML, and whole exome sequencing (WES) for eight cases, including one case studied by both approaches. On the basis of the results from these studies, 38 mutated genes were selected for a target gene panel, and samples from 82 OAML were studied by targeted deep sequencing for mutations in these genes. Finally, we also investigated the WGS data for translocations, gains and losses, and the presence of viral DNA.
2. Results
2.1. Whole Exome, Whole Genome, and Targeted Sequencing of OAML
We analyzed primary OAML from five patients for somatic mutations by WGS, from seven other patients by WES, and one further OAML by both sequencing approaches. Clinical data of the cohort and detailed characteristics of the patients are shown in Table 1 and Table S1. The WES analysis was revealed after filtering 139 heterozygous non-synonymous mutations in 138 genes (Table S3). Known SNPs were excluded from the analysis. The mean number of mutations per patient was 17.6 (range 1-41). The WGS analysis was revealed after filtering eight recurrently mutated genes in six patients, which carried the “impact moderate and high”, as described above (Table S2). The low number of mutations was explained by the stringent filtering criteria applied to this sequencing.

Table 1.
Patients’ characteristics.
We selected 38 genes for a targeted sequencing analysis of a large collection of OAML from 82 patients, including 11/13 index cases (Table S2). The selected genes included 36 genes mutated in at least one of the six OAML studied by WGS and/or at least in two of the eight lymphomas studied by WES, as well as two additional genes (CXCR4, ACTN4). Mutations in at least 1/38 genes analyzed by targeted sequencing were observed in 50/82 cases (61%), and 32 (39%) patients exhibited no mutations (Table S2). The internal controls used for targeted sequencing from different tissue types of the same sample revealed an overlap between paired samples of 95%, independent of tissue type. Only one of the samples per patient was therefore considered in the further analysis. A sample from a reactive hyperplasia revealed no mutations. The mean number of mutations per patient was 1.33 (range 0–15). Variant allele frequencies were between 10% and 72%, reflecting homo- and heterozygous mutations.
2.2. Detection of Numerous Recurrently Mutated Genes in OAML
Figure 1 gives an overview on the 17 genes mutated in at least two of the 82 OAML analyzed by targeted sequencing. The most frequently mutated gene (14% of cases) was TBL1XR1, an F-box-like/WD (tryptophan-aspartic acid) repeat-containing protein that is a component of both nuclear receptor corepressor (N-CoR) and histone deacetylase 3 (HDAC 3) complexes. It is required for transcriptional activation by a variety of transcription factors [20]. TBL1XR1 mutations were mostly detected with variant allele frequencies ≥ 20%, indicating a clonal representation (Figure 1).
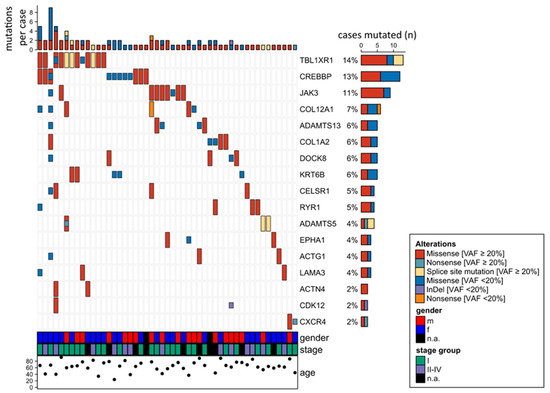
Figure 1.
Overview of 17 genes mutated in at least two cases in the ocular adnexal marginal zone lymphomas of mucosa-associated lymphatic tissue-type (OAML) cohort analyzed by targeted amplicon sequencing of 38 genes. Mutations observed in whole genome sequencing (WGS) or whole exome sequencing (WES), but not verified by targeted deep sequencing were not taken into account, considering the higher quality and sequencing depth of the latter approach. For the same reasons, mutations seen by targeted sequencing but not covered or filtered out in the WES or WGS analyses are included in this overview. Cases 101 and 102 are not considered here, because only WES and/or WGS were performed for those cases, but not targeted sequencing.
The second most frequently mutated gene was cAMP response element binding protein (CREBBP), with 13% mutated cases. CREBBP belongs to the KAT3 family of histone/protein lysine acetyltransferases and is a coactivator of many transcription factors.
JAK3 was identified as the third most frequently mutated gene. Mutations were observed in 9/82 patients (11%), two samples harbored subclonal mutations with a variant allele frequency below 20%. All mutations were missense mutations, mostly located in the FERM domain. Other mutations were distributed in the pseudokinase and kinase domain (Figure 2). Analyzing the survival data in this patient cohort, we observed that JAK3 mutant cases had a shorter progression free survival (PFS) compared to unmutated cases (Figure 3). No differences in overall survival were observed. For the two other most frequently mutated genes, TBL1XR1 and CREBBP, no significant association of the presence of mutations with progression-free or overall survival was obtained.
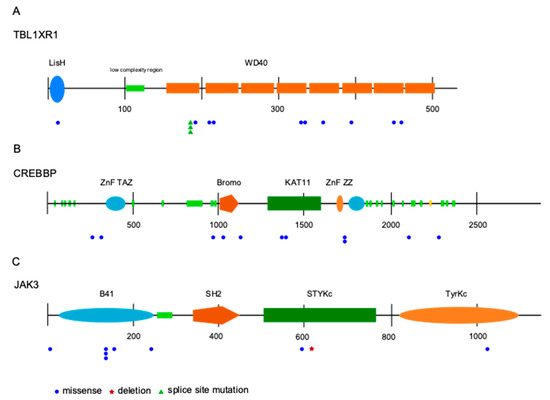
Figure 2.
Distribution and type of mutations in the three most recurrently mutated genes identified by targeted sequencing, transducin beta like 1 X-linked receptor 1 (TBL1XR1) (A), cAMP response element binding protein (CREBBP) (B), and JAK3 (C). The coding region on mRNA level is shown. Numbers indicate basepairs. Abbreviations: LisH—lis homology domain; WD40—beta-transducin repeats; ZnF—zinc finger domain; TAZ—transcription adaptor putative zinc finger; Bromo—Bromo domain; KAT11—histone acetylation protein domain; ZZ—ZZ-type zinc finger domain; B41—band 4.1 homologues; SH2—Src homology 2 domain; STYKc—protein kinase, unclassified specificity; TyrKc—tyrosine kinase, catalytic domain.
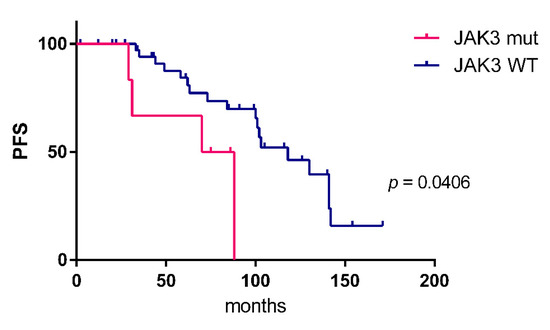
Figure 3.
Kaplan–Meier estimates (progression-free survival, PFS) of patients with (n = 6) and without JAK3 mutation (n = 39). The Log-rank (Mantel–Cox) test was applied. No differences in overall survival were observed.
2.3. Analysis of WGS Data for CNVs, Chromosomal Translocations, and Viral Sequences
Somatic CNV calling revealed 167 loss and 3437 gain regions (overview displayed in Figure S1). Most frequent losses were observed on chromosome 6 at the locus 6q23.3, including the TNFAIP3 gene in three of six patients (Table S4). Numerous genes showed recurrent gains in the six cases (Figure 4, Table S4). Although not all gains in a cancer have pathogenetic roles, several genes with recurrent gains in our collection deserve mentioning. HES4 showed gains in four of the cases. HES4 is a downstream target and effector of NOTCH activity [21]. CEBPB, which showed gains in four OAML, functions as transcription factor in myeloid cells, but it also has a pathogenetic role in multiple myelomas, and positively regulates BCL2 in B cells [22,23]. CEBPD, encoding another member of the CEBP family, was also recurrently affected by gains (4/6 cases). Little is known about the role of CEBPD in B cells, but it is mutated in a Burkitt lymphoma cell line, pointing to a potential role in B cell malignancies [24].
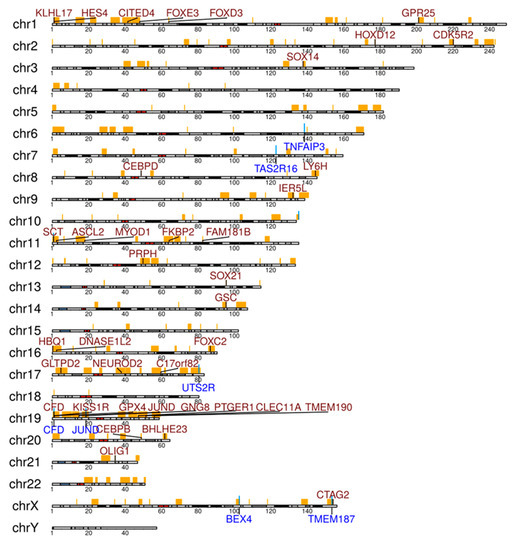
Figure 4.
Selected genes in significantly enriched regions of gains (genes written in red) and losses (genes written in blue) analyzed by copy number analysis from the WGS analysis of six OAML. Regions with false discovery rate (FDR)-corrected p-values below 0.05 from Fisher’s exact tests were considered as being significantly enriched in patients. Regions with gains or losses in at least one patient are marked at the respective chromosome in orange.
In the translocation analysis, none of the translocations described in MALT lymphomas (i.e., t(1;14) (p22;q32) (BCL10/IgH), t(14;18)(q32;p21) (IgH/MALT1), t(11;18)(q21;q21) (BIRC3/MALT1), and t(3;14)(p14;q32) (FOXP1/IgH)) were observed in the WGS data. We also did not find any convincing indication of other novel translocation events.
In a search for viral sequences, no conclusive hits against eukaryotic viruses were obtained.
2.4. Analysis for Bacterial Sequences in the OAML Tissues
Because many MZL subtypes develop by triggering certain bacteria, such as Helicobacter pylori in gastric MALT or Chlamydophila psittaci in OAML (at least in some geographical regions), we performed a metagenomic analysis of bacterial DNA in 17 samples. We also did not identify DNA of C. psittaci in any of the six lymphomas studied by WGS (data not shown). No other bacteria were recurrently detected in these lymphomas.
3. Discussion
Previous studies on somatic mutations and chromosomal abnormalities in OAML mostly focused on candidate gene approaches, interrogating genetic lesions that were known from other types of B cell lymphomas [10,11,14,15,16,25,26,27]. This led to major insights into recurrent gene mutations, chromosomal translocations, and copy number gains and losses in OAML. A major finding was that somatic mutations affecting genes of the NF-κB pathway are a hallmark of OAML, but that translocations deregulating NF-κB pathway members are relatively rare in comparison to other MALT lymphomas [9,10,11,12,28]. However, the question remained as to whether the landscape of frequently mutated genes in OAML is now known, or whether numerous other main drivers have not been detected in the prior focused mutation studies. We therefore followed the strategy to first perform unbiased WGS and/or WES of 13 cases of OAML and then select candidate genes from these genome-wide mutation screens to study a large cohort of cases for recurrence of these mutations. We selected 38 candidate genes and studied them for mutations in 82 OAML by deep sequencing. Furthermore, the WGS data were also analyzed for chromosomal translocations, CNVs, and viral sequences.
The absence of translocations affecting the genes BCL10, MALT1, BIRC3, or FOXP1, as they are frequently seen in other types of MALT lymphomas, in six OAML analyzed by WGS is in line with published data [29]. We also did not detect other novel translocations. Thus, chromosomal translocations apparently have only a minor role in the pathogenesis of OAML.
The presence of Chlamydophila psittaci in OAML as a potential bacterial trigger for this lymphoma is described only in some geographical areas [7,8]. We performed a bacterial metagenomic analysis of 17 OAML and did not identify C. psittaci in our cohort. Other bacteria were not recurrently detected in that analysis either. Furthermore, we analyzed the WGS data of six OAML for the occurrence of pathogenic viruses infecting humans, but did not observe any viral sequences. Hence, neither bacteria nor known viruses appear to play a substantial or any role in the pathogenesis of OAML, at least in patients from Germany.
The copy number variation analyses performed on WGS data revealed overall and recurrently more gains than losses in this cohort (Table S4). Among genes in areas with losses, TNFAIP3 as a major inhibitor of the NF-κB signaling pathway is affected by chromosomal copy number loss, validating prior studies [10,13,26,27,30]. Among genes with recurrent gains in our cohort, HES4 and two members of the CEBP transcription factor family may be particularly of interest. The gains in HES4 fit to the indications that NOTCH pathway activation is important in OAML. We previously reported recurrent activating mutations of NOTCH1 and NOTCH2 in OAML [22,23]. Thus, copy number gains of HES4 may be a further mechanism for enforced NOTCH pathway activation in OAML. Although CEBP family members are mainly known for their function in myeloid cells, there are indications that CEBPB and CEBPD may have roles in B lineage malignancies [21,22,23].
Regarding the mutational analysis of 38 candidate genes selected from the WES and WGS studies in 82 cases of OAML, 9 of the 38 genes were not mutated, which is explained by applying different filtering criteria for targeted sequencing compared to filtering criteria applied for WGS and WES due to different pipelines for analysis. Moreover, the tumor cell content in formalin-fixed paraffin-embedded (FFPE) material used for targeted sequencing was lower compared to the tumor cell content of sorted tumor cells for WGS and WES, resulting in a lower variant allele frequency in targeted sequencing. Additionally, tumor-derived mutations were compared against non-tumor mutations in WGS and WES. In targeted sequencing and WES, the mutations were filtered for variant allele frequency and quality score; in WGS, the mutations were required to occur in both read directions. We focus in the following discussion on those of the 38 genes that were mutated in at least two of the cases (Figure 1). The most frequently mutated gene was TBL1XR1, with mutations in 14% of the cases, validating two prior studies that detected frequent TBL1XR1 mutations in OAML [17,19]. This gene can activate the transcription of transcription factors such as NF-κB and JUN, and hence may contribute to the strong NF-κB activity in OAML. TBL1XR1 is mutated in various tumors, promoting tumor cell survival, and has been linked to a poor prognosis [31,32]. In one study, TBL1XR1 mutant cells exhibited a 10–15% higher proliferation rate than wild-type TBL1XR1 cells [19], suggesting a survival advantage for lymphoma cells carrying this mutation.
The second most frequently mutated gene in our cohort was CREBBP (13% of cases). Thus far, only one prior study had detected CREBBP mutations in OAML [19]. We therefore provided a valuable validation of frequent mutations in this gene in OAML. CREBBP, which is also mutated in other types of B cell lymphomas [33], is an epigenetic regulator, belonging to the KAT3 family of histone/protein lysine acetyltransferases. Mutations were also reported in other studies for the epigenetic regulators KMT2D and KMT2C [16,18]. Therefore, epigenetic dysregulation seems to be involved in the pathogenesis of OAML.
The JAK/STAT pathway has not yet been reported as playing a role in the pathogenesis of OAML. We here identified for the first time JAK3 mutations in OAML. With 11% of patients harbouring JAK3 mutations, it is the third most frequently mutated gene in our cohort. All mutations were located at positions known to alter the protein function (Figure 2) and leading to activation of the JAK/STAT signaling pathway by causing a gain of function of JAK3 [34,35,36]. Analyzing the survival data in this cohort, we observed that JAK3 mutant cases have a shorter PFS compared to unmutated cases (Figure 3). Because OAML have a high relapse rate of up to 50% after several years including systemic relapses [37,38], the information on mutations leading to a shorter PFS could be used to modify initial treatment and/or monitoring these patients more closely. Identification of activating JAK3 mutations in OAML also represent a possible therapeutic approach by targeting the JAK/STAT pathway with JAK inhibitors [39]. However, as this is a retrospective analysis, validation of the link of JAK3 mutation to clinical behaviour needs to be validated in future studies.
Further recurrently mutated genes include COL12A1, collagen type XII alpha 1 chain, which plays a role in cell adhesion. Different splicing variants occur in colorectal carcinoma [40], and mutations are reported for resistant small-cell lung cancers [41] and papillary renal cell carcinoma [42]. Notably, recurrent deletions of COL12A1 have been identified in primary central nervous system lymphoma [43], and thus we now have identified a second B cell lymphoma with recurrent genetic alterations of this gene. The gene of a second collagen family member, COL1A2, was mutated in 6% of cases. Although no mutations of COL1A2 have previously been reported for lymphomas, a potential pathogenetic role of these mutations is indicated from the finding that it is frequently epigenetically silenced in medulloblastoma and colorectal cancer [44,45]. A further recurrently mutated gene in OAML (6% of cases) is DOCK8. The fact that DOCK8 is involved in RhoGTPase signaling, that germline loss of DOCK8 causes an immunodeficiency, that it has an important role in murine B cell differentiation, and that mutations of this gene have previously been reported in other cancers [46,47], point to a potential involvement of DOCK8 mutations in the pathogenesis of OAML.
4. Materials and Methods
4.1. Patients and Samples
Patient samples were obtained from fresh and FFPE material of patients treated at the University Hospital Essen and the Kliniken Essen-Mitte. A total of 84 samples were selected for analysis with approval of the ethical review committee of the University of Duisburg-Essen. This research was approved by the Ethics Committee of the Medical Faculty of the University Duisburg-Essen on 04.12.2012 (ethic code: 12-5001-BO). Clinical data of these patients are summarized in Table 1. Samples used for sequencing analyses were collected from untreated patients. Patients were diagnosed between 2005 and 2016 in accordance with the WHO 2016 classification [48]. Only patients with primary OAML were included in this study. Samples obtained from fresh biopsies were either immediately processed or suspensions were prepared and cryopreserved. Samples were only further analyzed if the diagnosis of OAML was verified. For diagnosis, the corresponding immunohistochemical stainings were evaluated on FFPE sections with antibodies against CD3, CD5, CD10, CD20, CD23, CD79a, CXCR4, immunoglobulin light chains, cyclin D1, and Ki-67 (details not shown). For cases in which monoclonality could not be determined by light chain restriction, the diagnosis was complemented by flow cytometry analysis of the respective cell suspension and/or a molecular analysis for clonal immunoglobulin heavy chain gene rearrangements. WGS was performed in six cases with paired tumor and non-tumor samples. WES was performed for eight cases with paired tumor and non-tumor samples available (Table S1).
4.2. Tumor and Non-Tumor Cell Enrichment
For WGS analyses, tumor cells were enriched by fluorescence-activated cell sorting (FACS; Aria III, BD Biosciences; Heidelberg, Germany) after staining with antibodies against CD19, and immunoglobulin kappa and lambda light chains (BD Biosciences). For cases without light chain restriction, cells were sorted for positivity of CD19, CD27, and IgD (BD Biosciences). To obtain non-tumor controls, we performed density gradient centrifugation of heparinized peripheral blood samples of the respective patients (Pancoll human, PAN-biotech, Aidenbach, Germany). Peripheral blood mononuclear cells (PBMCs) depleted for CD19+ B cells by magnetic cell separation with a bead-coupled anti-CD19 antibody (MACS, Miltenyi Biotech, Bergisch Gladbach, Germany) served as non-tumor controls. For WES, tumor cells were obtained from fresh biopsies, as described above. As non-tumor controls, T cells were enriched from PBMCs by MACS with an anti-CD3 antibody. After tumor cell enrichment, the purity of all samples for WGS and WES was at least 95%, as determined by flow cytometry.
4.3. DNA Isolation
DNA was extracted from the isolated cells with the QIAamp Blood mini kit (Qiagen, Hilden, Germany). For FFPE material DNA was extracted from up to eight 10 µm thick sections per sample using the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany). DNA concentrations were measured with a Qubit Fluorometer 2.0 (Life Technologies, Darmstadt, Germany). Between 175 ng and 74 µg genomic DNA was isolated from the samples.
4.4. Whole Genome Sequencing
WGS was performed for six cases with paired tumor cells and non-B cell PBMC samples. Genomic DNA was sheared to a size of 300 bp with a Covaris S220 ultrasonicator (Covaris Inc., Woburn, MA, USA). Libraries were prepared with NexteraXT DNA sample preparation kit with 1 ng DNA. Whole genome paired-end sequencing was carried out on a HiSeq 2500 apparatus (Illumina, San Diego, CA, USA). Sequencing reads were trimmed on the basis of quality and adapter sequences with the tool Trim Galore! (Babraham Bioinformatics, Babraham Institute, Cambridge, U.K.). Reads passing quality control were mapped to human reference genome hg38 with BWA [49]. The average sequence coverage was 21 (range 6 to 59). The SNV-caller Platypus [50] generated a list of putative SNVs on the basis of the mapping files, and we added annotations with SnpEff [51]. The annotated list was further shortened as follows: SNVs predicted with impact less than moderate by SnpEff were filtered out, reads with low coverage for the variant (threshold: < 5 variant reads) were removed, the variant must occur in at least two forward and two reverse reads, no significant strand bias was allowed to be detected, variants must occur in at least one of the tumor samples and not in the non-tumor samples, and variants annotated in dbSNP (v132) were removed. Genes affected by SNVs were analyzed for an enrichment of known cancer signaling pathways. Pathway annotations were taken from ConsensusPathDB [52]. Numbers of affected genes per pathway were counted and pathway enrichment was determined by Fisher’s exact test. Data are available under accession number xxx (to be provided later) in the NCBI Sequence Read Archive.
4.5. Gene Copy Number Analysis
Gene copy number alterations were determined on the basis of WGS data. Sequencing reads were trimmed on the basis of quality and adapter sequences with Trim Galore! (Babraham Bioinformatics, Babraham Institute, Cambridge, UK). Reads that passed quality control were mapped to the human reference genome hg19 with BWA-MEM (https://arxiv.org/abs/1303.3997). Read coverage of protein-coding genes for tumor and normal samples were determined with the Genome Analysis Tool Kit (GATK 3.5) [53] using the tool DepthOfCoverage. Mean coverages were computed for both samples of each tumor-normal pair and used to shift the mean coverage of the sample with the lower coverage to the mean coverage of the sample with the higher coverage by adding the difference of both means to the coverage of each individual gene in the sample with the lower coverage. This ensures that both samples are comparable under the assumption that the majority of genes do not differ in their copy number between tumor and normal samples. Finally, log2-ratios of tumor to normal were computed for each gene to obtain a log-ratio profile for each tumor-normal pair. Gains or losses of genes with a false discovery rate (FDR)-corrected p-value below 0.05 from Fisher’s exact tests were considered as being significantly enriched in patients.
4.6. Search for Viral Sequences
WGS data from six OAML were analyzed for sequences with a detectable degree of sequence homology to known viruses. The sequencing reads were aligned to the human reference genome using BWA [49], and sequencing reads not aligning to the human reference were retained. The number of these unmapped reads ranged from 4–50 × 106 between the tumor samples. In a first search, the unmapped reads were assembled using SPAdes [54], and the resulting contigs were submitted to a BLAST [55] search at the nucleotide level (BLASTN) against a database of viral sequences obtained from the RINS package [56]. This viral database contained 32,102 genome sequences of all known viruses. In a second search, the unassembled reads, such as raw sequencing reads, were submitted to a BLAST search at the protein level (tblastx) against the same virus database.
4.7. Translocations Analysis
Translocation analysis was performed on the basis of WGS data that were trimmed and mapped to the human reference genome, as described for the gene copy number analysis. BreakDancer [57] was used to search for trans-chromosomal rearrangements with a minimum alternative mapping quality of 10. The results were reviewed manually for already known translocations occurring in MALT lymphomas.
4.8. Whole Exome Sequencing
WES was performed for eight cases with paired tumor and non-tumor T cell samples. A total of 50 ng of DNA was sheared to a size of 300 bp with a S220 ultrasonicator (Covaris). Libraries were prepared from isolated tumor cells and controls with the NEBNext Ultra DNA library prep kit (New England Biolabs, Ipswich, MA, USA). WES was carried out after capturing with NimbleGen SeqCap EZ enrichment kit v3 (Roche NimbleGen, Madison, WI, USA) on the HiSeq 2500 system with 2 × 101 bp paired-end reads (Illumina). More than 90% of the targeted exome had a minimum coverage of 20. For de-multiplexing and adapter trimming CASAVA v1.7 (Illumina) and Trimmomatic [58] were used. Read duplicates were removed with SAMtools rmdup [59]. The alignment was performed with BWA [49] against GRCh37 (hg19). Strand Avadis NGS software (Strand Genomics Inc.: San Francisco, CA, USA) was used for variant calling. Genes affected by SNVs were analyzed for enrichment of known cancer signaling pathways, as described for the WGS analysis.
4.9. Determination of the Coding Sequence of Bacterial 16S Ribosomal RNA
For 17 samples, we performed amplicon-based DNA sequencing of the prokaryotic variable regions V3 and V4 of the 16S ribosomal RNA. DNA was extracted from suspensions prepared from whole tissue biopsies using the QIAamp Blood mini kit (Qiagen). A positive control (Escherichia coli) and a no-template control were included. A total of 50 ng genomic DNA was used for the PCR, and 25 cycles of PCR were performed as published in the Illumina 16S Library Preparation Workflow. Primers [60] were combined with Illumina adapters (see Illumina 16S Library Preparation Workflow). After magnetic bead-cleanup (Agencourt AMPure XP, Beckman Coulter, Krefeld, Germany), eight further cycles of PCR were performed to add NexteraXT indices (Illumina) for multiplexing, followed by a second clean-up. Sequencing was carried out on an Illumina MiSeq apparatus.
4.10. Targeted Re-Sequencing
Genes for targeted re-sequencing were selected on the basis of the following criteria. In the WES analysis, genes had to be recurrently mutated in at least two patients. A high quality score > 500 was set as determined by Strand Avadis NGS software. In the WGS analysis, genes mutated in at least one patient were selected when the filtering criteria according to SnpEff [51] tool were set to “impact moderate and high”. We further added CXCR4 to the candidate list, although it was neither mutated in the WGS nor the WES analysis, as its high expression in OAML [61] raised our interest in this gene. ACTN4 was included in the list of target genes, although it was mutated only in one of the lymphomas studied by WES, as it can function as a co-activator of NF-κB [62].
After selection of 37 mutated genes plus CXCR4 (Table S2), a custom panel was designed for the coding exons of those 38 genes using the IonApliSeq platform (Thermo Fisher Scientific, Waltham, MA, USA). Sequencing was carried out on an IonProton sequencing apparatus (Thermo Fisher). For 82/84 patients, enough material was available for targeted re-sequencing. A total of 91 samples from those 82 patients were used, including 11/13 index patients (seven from the WES analysis, four from the WGS analysis). For three lymphoma samples, the sequencing quality was not sufficient, and therefore these samples were removed. Six samples were included as additional internal controls, which were one patient exhibiting a reactive lymphoid hyperplasia as precursor lesion with no evidence for a lymphoma, and four patients for whom one or two additional sample types of the same biopsy were available. These were two patients with a paired sample of FFPE and a suspension; one patient with a suspension and sorted tumor cells; and one patient with FFPE material, sorted tumor cells, and a cell suspension. The distinct samples from the same patient were compared to each other and revealed the same mutations in most cases. The samples additionally included for internal control were then excluded from further analyses, and hence only one sample per patient was kept for the final analysis. The samples finally included in the evaluation were 63 FFPE samples, 10 cell suspensions, and 11 samples of sorted tumor cells.
Filtering criteria were set for non-synonymous mutations only and novel SNVs only. Frameshift mutations were manually reviewed and removed, if located in homopolymer stretches or repeats. Mutations with a strand bias ≥ 100 according to Strand Avadis NGS were removed. The variant allele frequency was set to a minimum of 10%.
5. Conclusions
We conclude from our WGS, WES, and targeted deep sequencing analysis of the 38 candidate genes that the majority of frequently mutated protein-coding genes involved in the pathogenesis of OAML apparently have been identified, as we detected only one novel gene mutated in more than 10% of the cases (JAK3). Importantly, we also identified a substantial number of recurrently mutated genes with frequencies of mutated cases between 2–10%. This means that the landscape of genetic lesions in OAML is quite complex and diverse, and that besides a number of frequently mutated oncogenes and tumor suppressor genes, a large variety of additional genes are mutated. For most of these genes mutated at low frequency, it remains unclear as to whether they are driver genes in this lymphoma, or whether their contribution to the malignant transformation process is minor.
We confirmed previously published data on mutations occurring in OAML, observed no hits in a search for bacterial and viral triggering factors in OAML, and identified involvement of the JAK/STAT signaling pathway in OAML by detecting frequent mutations in JAK3, linked to a disadvantage in PFS in these patients. Evaluation of the WGS data for copy number aberrations confirmed frequent loss of TNFAIP3, and revealed recurrent gains of the NOTCH target HES4, and of members of the CEBP transcription factor family.
Supplementary Materials
The following are available online at https://www.mdpi.com/2072-6694/12/4/986/s1, Figure S1: Gene copy number alteration profiles of six OAML, Table S1: Overview of clinical data and the sequencing analyses performed, Table S2: Mutations identified in the 38 genes analyzed by targeted deep sequencing, Table S3: Significant enrichment of gains and losses in six OAML studied by WGS, Table S4: Significant recurrent enrichment of gains and losses in six OAML studied by WGS.
Author Contributions
Conceptualization, P.J., J.D., and R.K.; Methodology and Investigation, P.J. and L.K.-H.; Investigation and data curation, B.B., M.K., L.K.-H., C.L., M.S., and I.R.; Resources and essential material, U.D., J.D., R.P., M.S., and A.E.; Writing of the manuscript, P.J. and R.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Werner Jackstädt-Stiftung and by the Deutsche Forschungsgemeinschaft (FOR 1961, Control-T consortium). P.J. was supported by the Interne Forschungsförderung Essen (IFORES) program of the Medical Faculty of the University Duisburg-Essen. B.B. is employed through a grant from the Deutsche Krebshilfe (70112628).
Acknowledgments
We thank Sabine Senkel and Michael Möllmann for expert technical assistance. We thank René Scholtysik for data processing. We thank the Imaging Center Essen (IMCES) for support in cell sorting. We acknowledge support by the Open Access Publication Fund of the University of Duisburg-Essen.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Freeman, C.; Berg, J.W.; Cutler, S.J. Occurrence and prognosis of extranodal lymphomas. Cancer 1972, 29, 252–260. [Google Scholar] [CrossRef]
- Coupland, S.E.; Krause, L.; Delecluse, H.J.; Anagnostopoulos, I.; Foss, H.D.; Hummel, M.; Bornfeld, N.; Lee, W.R.; Stein, H. Lymphoproliferative lesions of the ocular adnexa. Analysis of 112 cases. Ophthalmology 1998, 105, 1430–1441. [Google Scholar] [CrossRef]
- Ferry, J.A.; Fung, C.Y.; Zukerberg, L.; Lucarelli, M.J.; Hasserjian, R.P.; Preffer, F.I.; Harris, N.L. Lymphoma of the ocular adnexa: A study of 353 cases. Am. J. Surg. Pathol. 2007, 31, 170–184. [Google Scholar] [CrossRef]
- Knowles, D.M.; Jakobiec, F.A.; McNally, L.; Burke, J.S. Lymphoid hyperplasia and malignant lymphoma occurring in the ocular adnexa (orbit, conjunctiva, and eyelids): A prospective multiparametric analysis of 108 cases during 1977 to 1987. Hum. Pathol. 1990, 21, 959–973. [Google Scholar] [CrossRef]
- Hara, Y.; Nakamura, N.; Kuze, T.; Hashimoto, Y.; Sasaki, Y.; Shirakawa, A.; Furuta, M.; Yago, K.; Kato, K.; Abe, M. Immunoglobulin heavy chain gene analysis of ocular adnexal extranodal marginal zone B-cell lymphoma. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2450–2457. [Google Scholar]
- McKelvie, P.A. Ocular adnexal lymphomas: A review. Adv. Anat. Pathol. 2010, 17, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Chanudet, E.; Zhou, Y.; Bacon, C.M.; Wotherspoon, A.C.; Muller-Hermelink, H.K.; Adam, P.; Dong, H.Y.; de Jong, D.; Li, Y.; Wei, R.; et al. Chlamydia psittaci is variably associated with ocular adnexal MALT lymphoma in different geographical regions. J. Pathol. 2006, 209, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Ferreri, A.J.; Guidoboni, M.; Ponzoni, M.; De Conciliis, C.; Dell’Oro, S.; Fleischhauer, K.; Caggiari, L.; Lettini, A.A.; Dal Cin, E.; Ieri, R.; et al. Evidence for an association between Chlamydia psittaci and ocular adnexal lymphomas. J. Natl. Cancer Inst. 2004, 96, 586–594. [Google Scholar] [CrossRef]
- Du, M.Q. MALT lymphoma: Recent advances in aetiology and molecular genetics. J. Clin. Exp. Hematop. 2007, 47, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Du, M.Q. MALT lymphoma: Many roads lead to nuclear factor-kappab activation. Histopathology 2011, 58, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Streubel, B.; Vinatzer, U.; Lamprecht, A.; Raderer, M.; Chott, A. T(3;14)(p14.1;q32) involving IGH and FOXP1 is a novel recurrent chromosomal aberration in MALT lymphoma. Leukemia 2005, 19, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Ikpatt, O.F.; Dubovy, S.R.; Lossos, C.; Natkunam, Y.; Chapman-Fredricks, J.R.; Fan, Y.S.; Lossos, I.S. Molecular and genomic aberrations in Chlamydophila psittaci negative ocular adnexal marginal zone lymphomas. Am. J. Hematol. 2013, 88, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Du, M.Q. MALT lymphoma: A paradigm of NF-kappaB dysregulation. Semin. Cancer Biol. 2016, 39, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Wang, M.; Moody, S.; Xue, X.; Huang, Y.; Bi, Y.; Du, M.Q. Distinct involvement of NF-kappaB regulators by somatic mutation in ocular adnexal malt lymphoma. Br. J. Haematol. 2013, 160, 851–854. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wei, R.L.; Pi, Y.L.; Guo, Q. Significance of Bcl10 gene mutations in the clinical diagnosis of MALT-type ocular adnexal lymphoma in the Chinese population. Genet. Mol. Res. 2013, 12, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Klein-Hitpass, L.; Grabellus, F.; Arnold, G.; Klapper, W.; Pfortner, R.; Duhrsen, U.; Eckstein, A.; Durig, J.; Kuppers, R. Recurrent mutations in NF-kappaB pathway components, KMT2D, and NOTCH1/2 in ocular adnexal MALT-type marginal zone lymphomas. Oncotarget 2016, 7, 62627–62639. [Google Scholar] [CrossRef]
- Moody, S.; Thompson, J.S.; Chuang, S.S.; Liu, H.; Raderer, M.; Vassiliou, G.; Wlodarska, I.; Wu, F.; Cogliatti, S.; Robson, A.; et al. Novel GPR34 and CCR6 mutation and distinct genetic profiles in MALT lymphomas of different sites. Haematologica 2018, 103, 1329–1336. [Google Scholar] [CrossRef]
- Cascione, L.; Rinaldi, A.; Bruscaggin, A.; Tarantelli, C.; Arribas, A.J.; Kwee, I.; Pecciarini, L.; Mensah, A.A.; Spina, V.; Chung, E.Y.L.; et al. Novel insights into the genetics and epigenetics of MALT lymphoma unveiled by next generation sequencing analyses. Haematologica 2019, 104, e558–e561. [Google Scholar] [CrossRef]
- Jung, H.; Yoo, H.Y.; Lee, S.H.; Shin, S.; Kim, S.C.; Lee, S.; Joung, J.G.; Nam, J.Y.; Ryu, D.; Yun, J.W.; et al. The mutational landscape of ocular marginal zone lymphoma identifies frequent alterations in TNFAIP3 followed by mutations in TBL1XR1 and CREBBP. Oncotarget 2017, 8, 17038–17049. [Google Scholar] [CrossRef]
- Yoon, H.G.; Chan, D.W.; Huang, Z.Q.; Li, J.; Fondell, J.D.; Qin, J.; Wong, J. Purification and functional characterization of the human N-CoR complex: The roles of HDAC3, TBL1 and TBLR1. EMBO J. 2003, 22, 1336–1346. [Google Scholar] [CrossRef]
- De Decker, M.; Lavaert, M.; Roels, J.; Tilleman, L.; Vandekerckhove, B.; Leclercq, G.; Van Nieuwerburgh, F.; Van Vlierberghe, P.; Taghon, T. HES1 and HES4 have non-redundant roles downstream of Notch during early human T cell development. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed]
- Heckman, C.A.; Wheeler, M.A.; Boxer, L.M. Regulation of Bcl-2 expression by C/EBP in t(14;18) lymphoma cells. Oncogene 2003, 22, 7891–7899. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Janz, M.; Galson, D.L.; Gries, M.; Li, S.; Johrens, K.; Anagnostopoulos, I.; Dorken, B.; Mapara, M.Y.; Borghesi, L.; et al. C/EBPbeta regulates transcription factors critical for proliferation and survival of multiple myeloma cells. Blood 2009, 114, 3890–3898. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vegesna, V.; Takeuchi, S.; Hofmann, W.K.; Ikezoe, T.; Tavor, S.; Krug, U.; Fermin, A.C.; Heaney, A.; Miller, C.W.; Koeffler, H.P. C/EBP-beta, C/EBP-delta, PU.1, AML1 genes: Mutational analysis in 381 samples of hematopoietic and solid malignancies. Leuk. Res. 2002, 26, 451–457. [Google Scholar] [CrossRef]
- Bi, Y.; Zeng, N.; Chanudet, E.; Huang, Y.; Hamoudi, R.A.; Liu, H.; Dong, G.; Watkins, A.J.; Ley, S.C.; Zou, L.; et al. A20 inactivation in ocular adnexal MALT lymphoma. Haematologica 2012, 97, 926–930. [Google Scholar] [CrossRef]
- Chanudet, E.; Ye, H.; Ferry, J.; Bacon, C.M.; Adam, P.; Muller-Hermelink, H.K.; Radford, J.; Pileri, S.A.; Ichimura, K.; Collins, V.P.; et al. A20 deletion is associated with copy number gain at the TNFA/B/C locus and occurs preferentially in translocation-negative MALT lymphoma of the ocular adnexa and salivary glands. J. Pathol. 2009, 217, 420–430. [Google Scholar] [CrossRef]
- Honma, K.; Tsuzuki, S.; Nakagawa, M.; Karnan, S.; Aizawa, Y.; Kim, W.S.; Kim, Y.D.; Ko, Y.H.; Seto, M. TNFAIP3 is the target gene of chromosome band 6q23.3-q24.1 loss in ocular adnexal marginal zone B cell lymphoma. Genes Chromosomes Cancer 2008, 47, 1–7. [Google Scholar] [CrossRef]
- Choung, H.; Kim, Y.A.; Kim, N.; Lee, M.J.; Khwarg, S.I. Clinicopathologic Study of Chromosomal Aberrations in Ocular Adnexal Lymphomas of Korean Patients. Korean J. Ophthalmol. 2015, 29, 285–293. [Google Scholar] [CrossRef][Green Version]
- Ferreri, A.J.; Dolcetti, R.; Du, M.Q.; Doglioni, C.; Resti, A.G.; Politi, L.S.; De Conciliis, C.; Radford, J.; Bertoni, F.; Zucca, E.; et al. Ocular adnexal MALT lymphoma: An intriguing model for antigen-driven lymphomagenesis and microbial-targeted therapy. Ann. Oncol. 2008, 19, 835–846. [Google Scholar] [CrossRef]
- Kim, W.S.; Honma, K.; Karnan, S.; Tagawa, H.; Kim, Y.D.; Oh, Y.L.; Seto, M.; Ko, Y.H. Genome-wide array-based comparative genomic hybridization of ocular marginal zone B cell lymphoma: Comparison with pulmonary and nodal marginal zone B cell lymphoma. Genes Chromosomes Cancer 2007, 46, 776–783. [Google Scholar] [CrossRef]
- Liu, F.; He, Y.; Cao, Q.; Liu, N.; Zhang, W. TBL1XR1 Is Highly Expressed in Gastric Cancer and Predicts Poor Prognosis. Dis. Markers 2016, 2016, 2436518. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Liu, C.; Yu, Y.; Geng, J.; Meng, Q.; Xu, S.; Zhou, F.; Chen, Y.; Jin, S.; Shen, J.; et al. TBL1XR1 is involved in c-Met-mediated tumorigenesis of human nonsmall cell lung cancer. Cancer Gene Ther. 2019. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011, 471, 189–195. [Google Scholar] [CrossRef]
- Degryse, S.; de Bock, C.E.; Cox, L.; Demeyer, S.; Gielen, O.; Mentens, N.; Jacobs, K.; Geerdens, E.; Gianfelici, V.; Hulselmans, G.; et al. JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model. Blood 2014, 124, 3092–3100. [Google Scholar] [CrossRef] [PubMed]
- Elliott, N.E.; Cleveland, S.M.; Grann, V.; Janik, J.; Waldmann, T.A.; Dave, U.P. FERM domain mutations induce gain of function in JAK3 in adult T-cell leukemia/lymphoma. Blood 2011, 118, 3911–3921. [Google Scholar] [CrossRef] [PubMed]
- Lupardus, P.J.; Ultsch, M.; Wallweber, H.; Bir Kohli, P.; Johnson, A.R.; Eigenbrot, C. Structure of the pseudokinase-kinase domains from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 8025–8030. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Kojima, M.; Shioya, M.; Tamaki, Y.; Saitoh, J.; Sakurai, H.; Kitamoto, Y.; Suzuki, Y.; Niibe, H.; Nakano, T. Treatment results of radiotherapy for malignant lymphoma of the orbit and histopathologic review according to the WHO classification. Int. J. Radiat. Oncol. Biol. Phys. 2003, 57, 172–176. [Google Scholar] [CrossRef]
- Wenzel, C.; Fiebiger, W.; Dieckmann, K.; Formanek, M.; Chott, A.; Raderer, M. Extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue of the head and neck area: High rate of disease recurrence following local therapy. Cancer 2003, 97, 2236–2241. [Google Scholar] [CrossRef]
- Derenzini, E.; Younes, A. Targeting the JAK-STAT pathway in lymphoma: A focus on pacritinib. Expert Opin. Investig. Drugs 2013, 22, 775–785. [Google Scholar] [CrossRef]
- Liu, J.; Li, H.; Shen, S.; Sun, L.; Yuan, Y.; Xing, C. Alternative splicing events implicated in carcinogenesis and prognosis of colorectal cancer. J. Cancer 2018, 9, 1754–1764. [Google Scholar] [CrossRef]
- Qiu, Z.; Lin, A.; Li, K.; Lin, W.; Wang, Q.; Wei, T.; Zhu, W.; Luo, P.; Zhang, J. A novel mutation panel for predicting etoposide resistance in small-cell lung cancer. Drug Des. Devel. Ther. 2019, 13, 2021–2041. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Ren, Y.; Pang, L.; Qi, Y.; Jia, W.; Tao, L.; Hu, Z.; Zhao, J.; Zhang, H.; Li, L.; et al. Papillary renal cell carcinoma: A clinicopathological and whole-genome exon sequencing study. Int. J. Clin. Exp. Pathol. 2015, 8, 8311–8335. [Google Scholar] [PubMed]
- Sung, C.O.; Kim, S.C.; Karnan, S.; Karube, K.; Shin, H.J.; Nam, D.H.; Suh, Y.L.; Kim, S.H.; Kim, J.Y.; Kim, S.J.; et al. Genomic profiling combined with gene expression profiling in primary central nervous system lymphoma. Blood 2011, 117, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Anderton, J.A.; Lindsey, J.C.; Lusher, M.E.; Gilbertson, R.J.; Bailey, S.; Ellison, D.W.; Clifford, S.C. Global analysis of the medulloblastoma epigenome identifies disease-subgroup-specific inactivation of COL1A2. Neuro Oncol. 2008, 10, 981–994. [Google Scholar] [CrossRef]
- Kalmar, A.; Peterfia, B.; Hollosi, P.; Galamb, O.; Spisak, S.; Wichmann, B.; Bodor, A.; Toth, K.; Patai, A.V.; Valcz, G.; et al. DNA hypermethylation and decreased mRNA expression of MAL, PRIMA1, PTGDR and SFRP1 in colorectal adenoma and cancer. BMC Cancer 2015, 15, 736. [Google Scholar] [CrossRef]
- Randall, K.L.; Lambe, T.; Johnson, A.L.; Treanor, B.; Kucharska, E.; Domaschenz, H.; Whittle, B.; Tze, L.E.; Enders, A.; Crockford, T.L.; et al. Dock8 mutations cripple B cell immunological synapses, germinal centers and long-lived antibody production. Nat. Immunol. 2009, 10, 1283–1291. [Google Scholar] [CrossRef]
- Schramm, A.; Koster, J.; Assenov, Y.; Althoff, K.; Peifer, M.; Mahlow, E.; Odersky, A.; Beisser, D.; Ernst, C.; Henssen, A.G.; et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 2015, 47, 872–877. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Rimmer, A.; Phan, H.; Mathieson, I.; Iqbal, Z.; Twigg, S.R.; Consortium, W.G.S.; Wilkie, A.O.; McVean, G.; Lunter, G. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014, 46, 912–918. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Kamburov, A.; Stelzl, U.; Lehrach, H.; Herwig, R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2013, 41, D793–D800. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Bhaduri, A.; Qu, K.; Lee, C.S.; Ungewickell, A.; Khavari, P.A. Rapid identification of non-human sequences in high-throughput sequencing datasets. Bioinformatics 2012, 28, 1174–1175. [Google Scholar] [CrossRef]
- Chen, K.; Wallis, J.W.; McLellan, M.D.; Larson, D.E.; Kalicki, J.M.; Pohl, C.S.; McGrath, S.D.; Wendl, M.C.; Zhang, Q.; Locke, D.P.; et al. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 2009, 6, 677–681. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Stollberg, S.; Kammerer, D.; Neubauer, E.; Schulz, S.; Simonitsch-Klupp, I.; Kiesewetter, B.; Raderer, M.; Lupp, A. Differential somatostatin and CXCR4 chemokine receptor expression in MALT-type lymphoma of gastric and extragastric origin. J. Cancer Res. Clin. Oncol. 2016, 142, 2239–2247. [Google Scholar] [CrossRef] [PubMed]
- Aksenova, V.; Turoverova, L.; Khotin, M.; Magnusson, K.E.; Tulchinsky, E.; Melino, G.; Pinaev, G.P.; Barlev, N.; Tentler, D. Actin-binding protein alpha-actinin 4 (ACTN4) is a transcriptional co-activator of RelA/p65 sub-unit of NF-kB. Oncotarget 2013, 4, 362–372. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).