[18F]FET-βAG-TOCA: The Design, Evaluation and Clinical Translation of a Fluorinated Octreotide


Abstract
1. Introduction
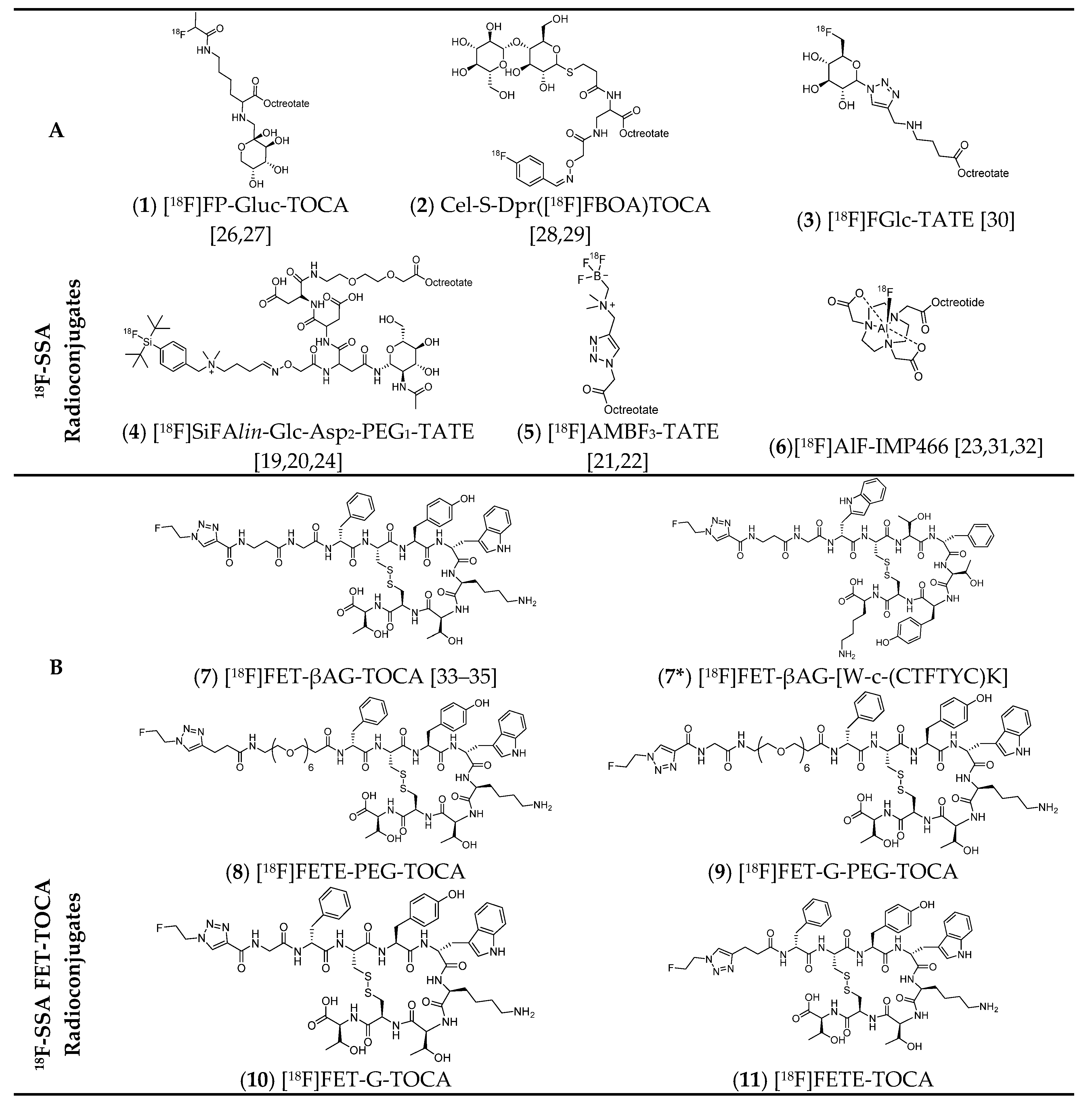
2. Fluorine-18 Radiolabelled Somatostatin Analogues
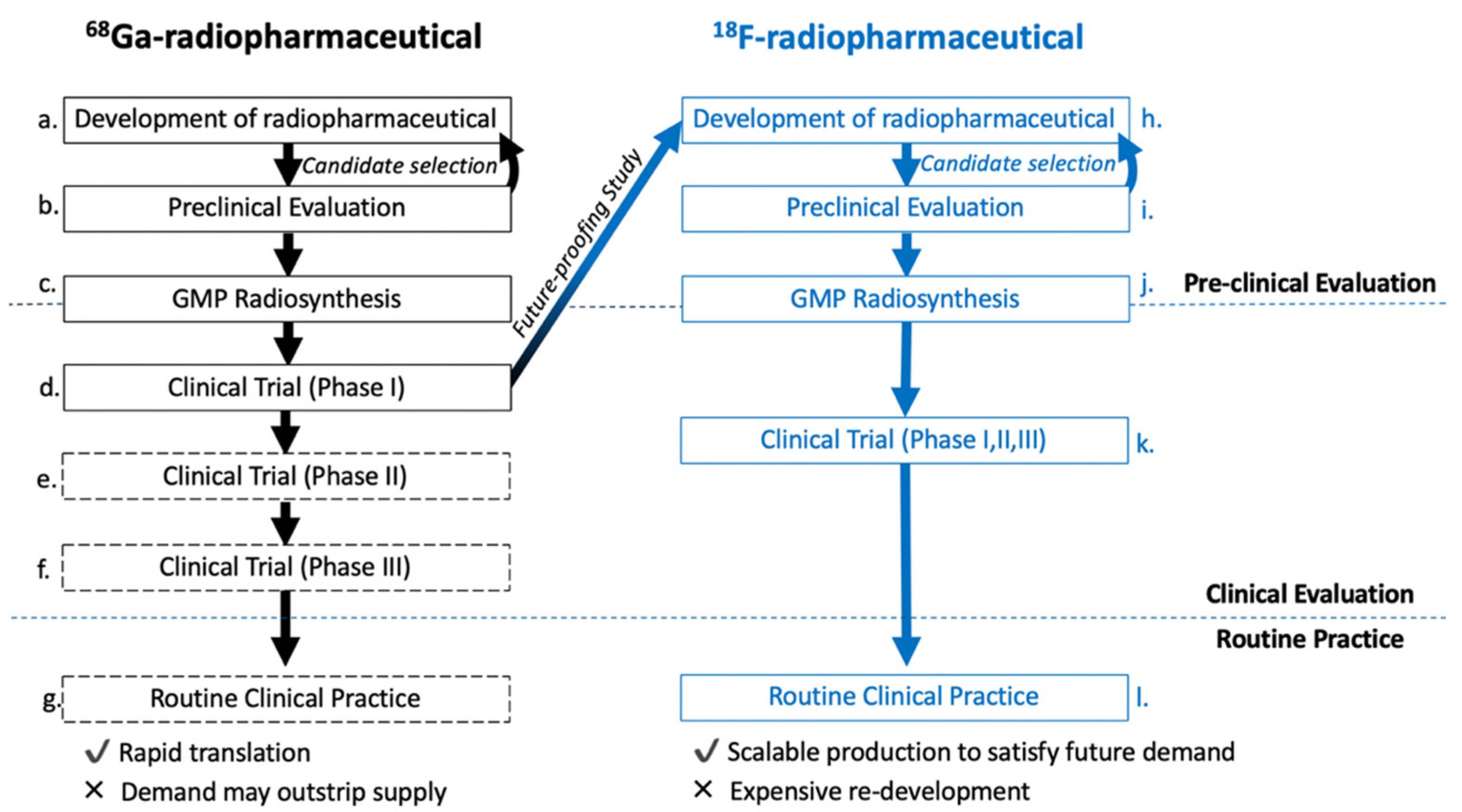
3. Strategic Aim
4. Development of the [18F]FET-TOCA Library
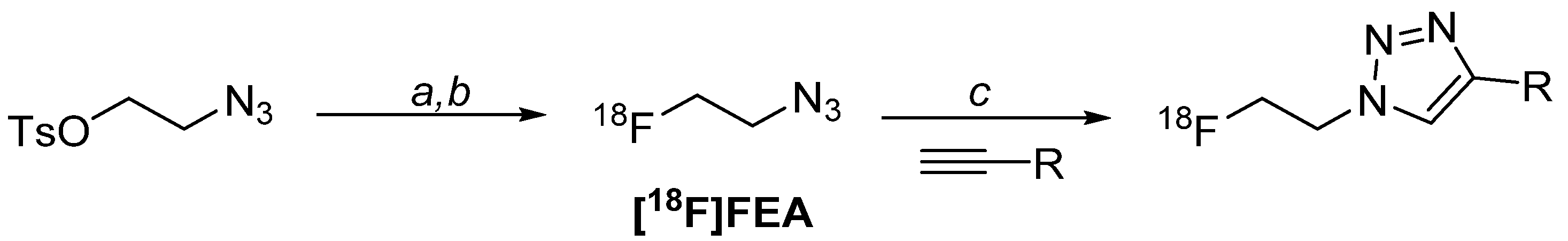
4.1. Radiolabelling Chemistry
4.2. Peptide Sequence
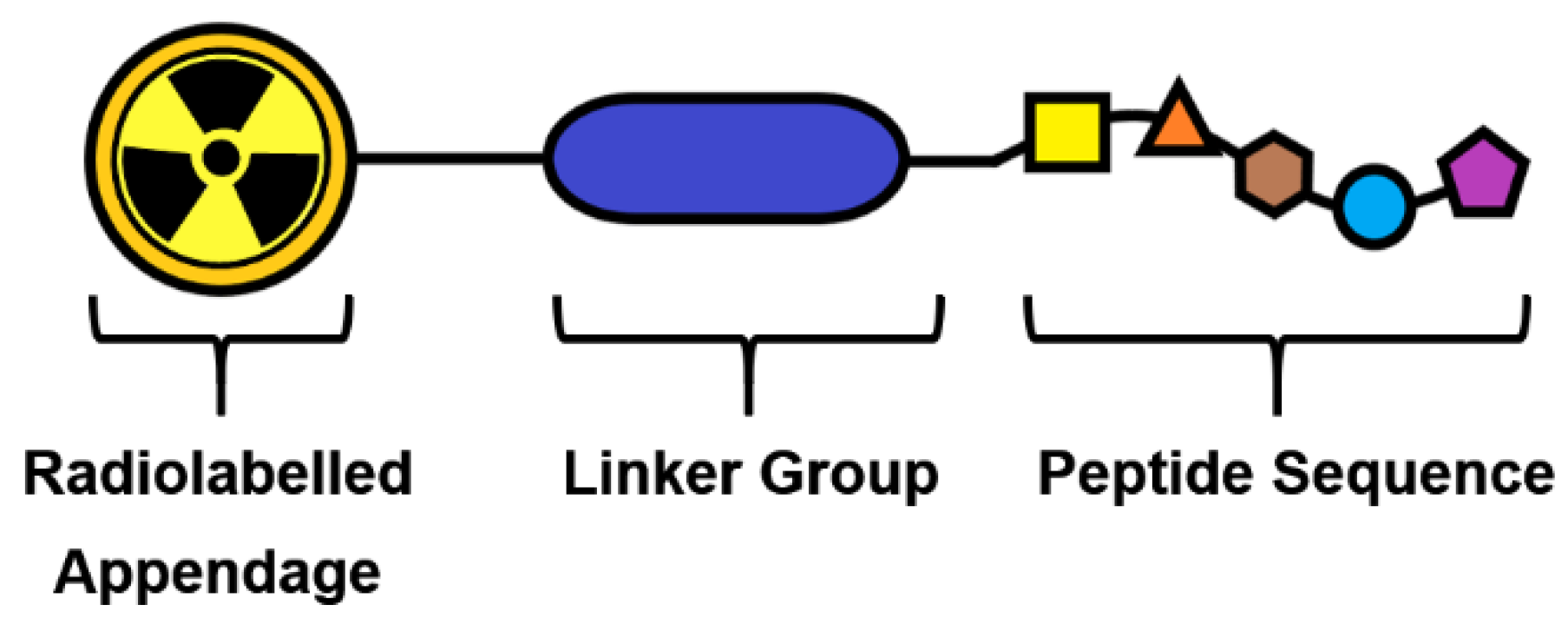
4.3. Linker Group
4.4. The Library
5. Radiochemistry for Pre-Clinical Studies
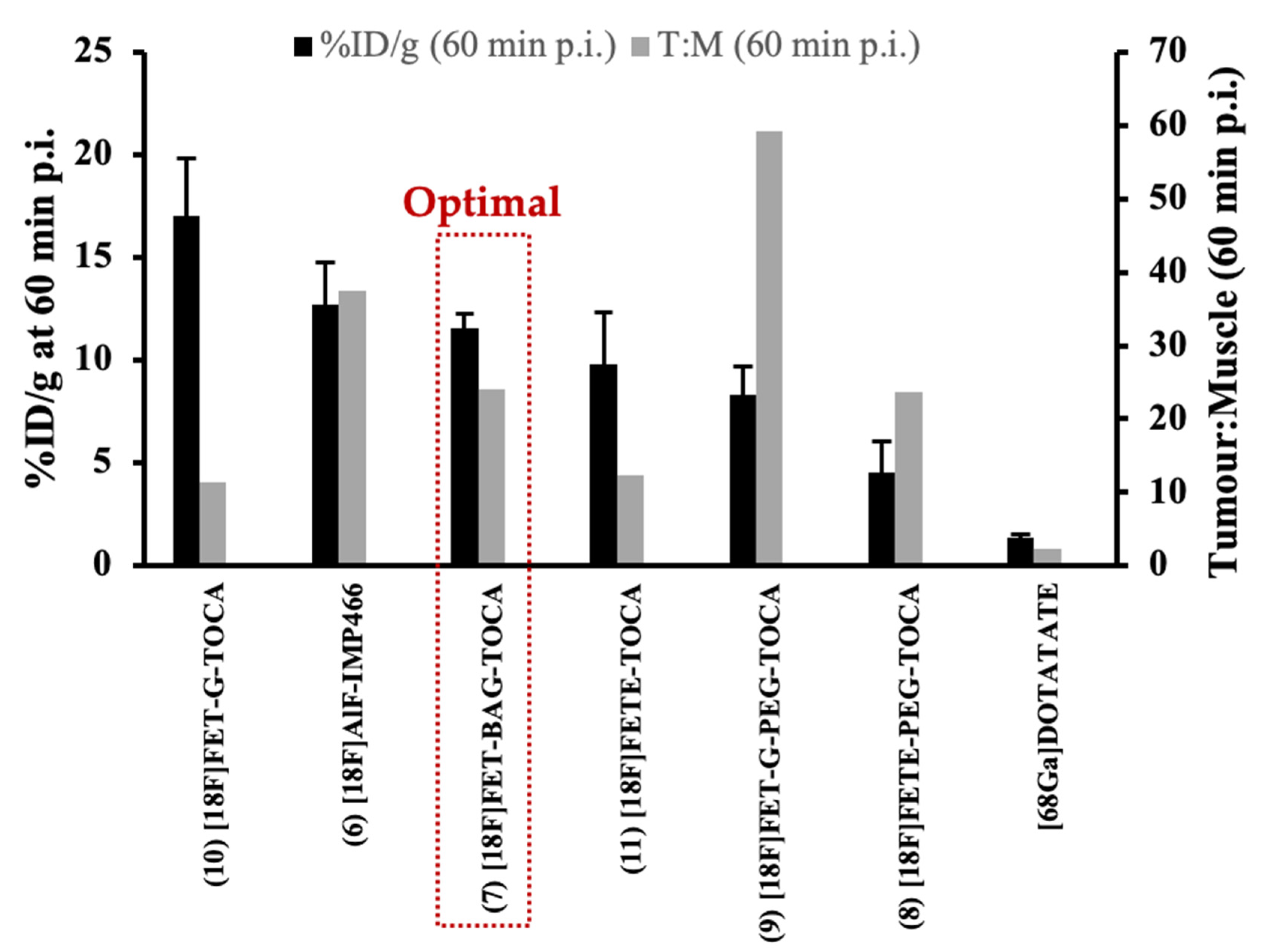
6. Biological Evaluation and Candidate Selection
7. Radiochemistry for Clinical Studies
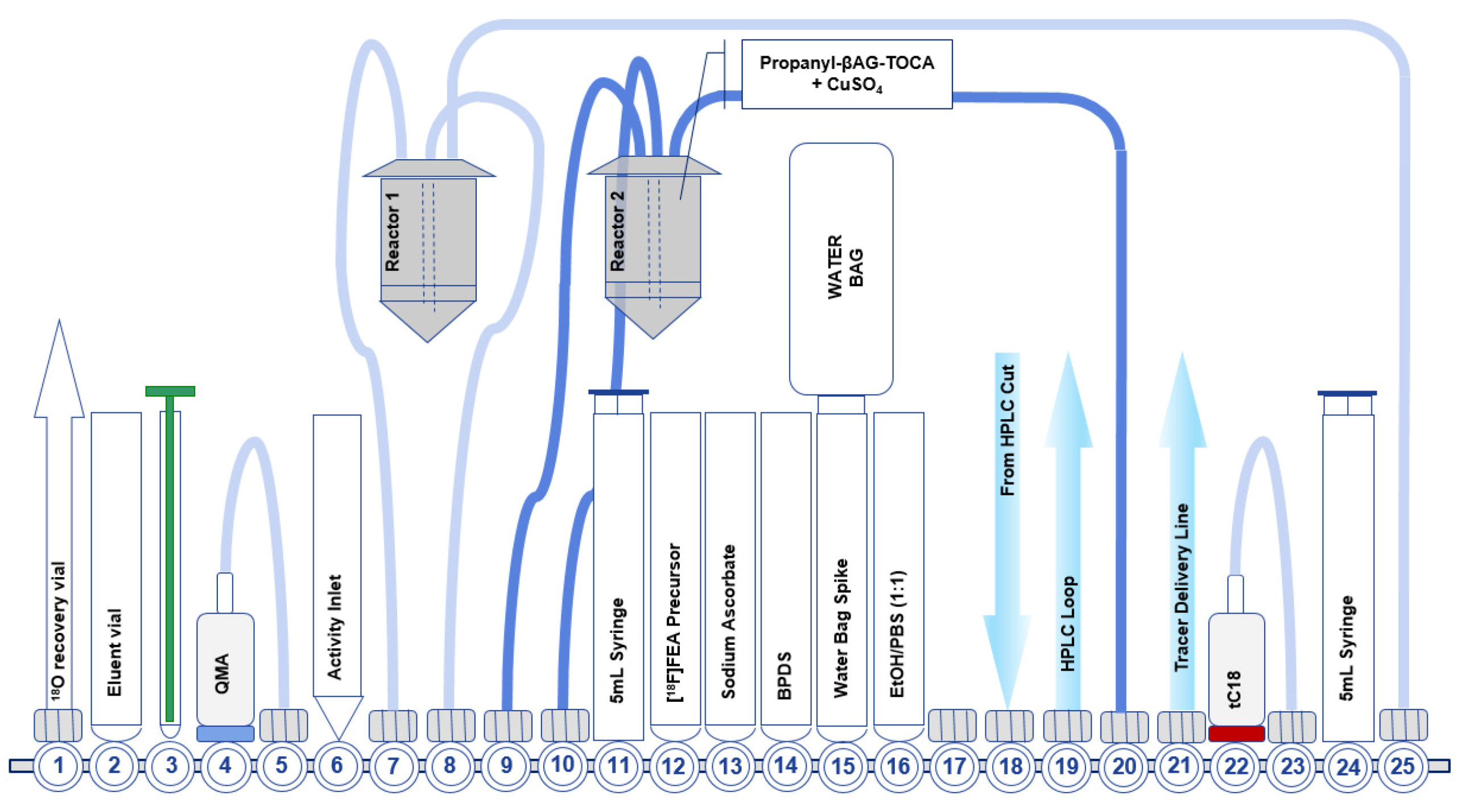
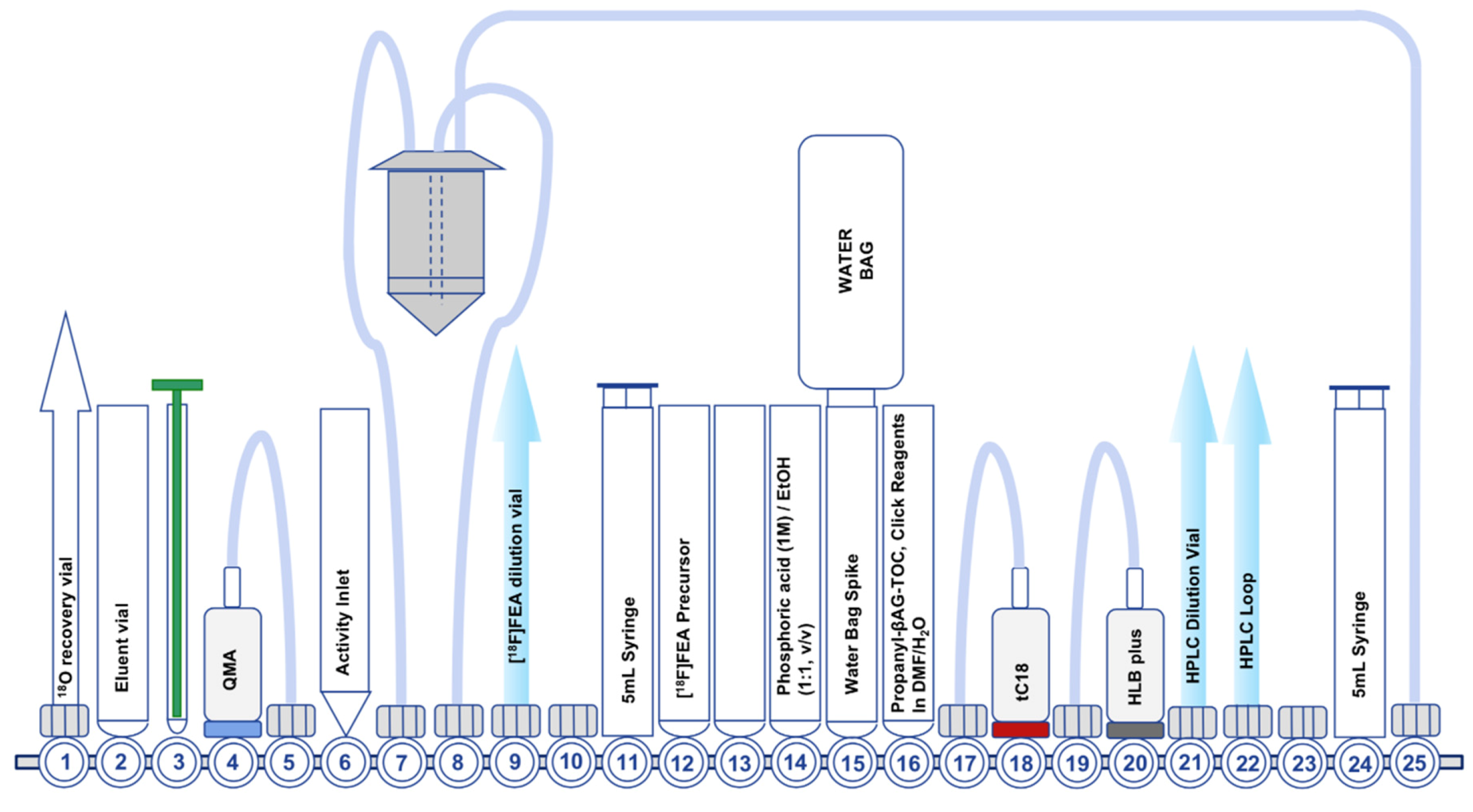
7.1. Development of the First-Generation Radiosynthesis of [18F]FET-βAG-TOCA
7.2. Development of the Second-Generation Radiosynthesis of [18F]FET-βAG-TOCA
8. Additional Pre-Clinical Evaluation to Support First-in-Human Use of [18F]FET-βAG-TOCA
9. Clinical Evaluation
10. Comparison of 18F-Radiolabelled Octreotide Analogues
11. Lessons for the Clinical Translation of 18F-Radiolabelled Peptides
- Defining clear aims of what a successful 18F-peptide will achieve, in terms of radiolabelling chemistry, biological properties and clinical translation.
- Implement a focused library with appropriate candidates and a generic radiolabelling method that can be applied to all, which ultimately simplifies their biological evaluation.
- Develop chemistry that supports clinical translation, but does not stop innovation when phase I and II trials have been completed; look to the future, asking whether the current method is adequate for taking this compound forwards into routine clinical practice? Are there better radiolabelling methods available which may alleviate challenges or improve production yield, that were not available for the first GMP compliant radiosynthesis?
12. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Yordanova, A.; Eppard, E.; Kürpig, S.; Bundschuh, R.A.; Schönberger, S.; Gonzalez-Carmona, M.; Feldmann, G.; Ahmadzadehfar, H.; Essler, M. Theranostics in nuclear medicine practice. Onco Targets Ther. 2017, 10, 4821–4828. [Google Scholar] [CrossRef] [PubMed]
- Hennrich, U.; Kopka, K. Lutathera®: The first FDA-and EMA-approved radiopharmaceutical for peptide receptor radionuclide therapy. Pharmaceuticals 2019, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. NETTER-1: Phase 3 Trial of 177 Lu-Dotatate for Midgut Neuroendocrine Tumors (pancreatic approved as well). N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.H. An introduction to the clinical practice of theranostics in oncology. Br. J. Radiol. 2018, 91, 20180440. [Google Scholar] [CrossRef] [PubMed]
- Alves, F.; Bertrand, S.; Degrado, T.; Gagnon, K.; Guérin, B.; Hoehr, C.; Pandey, M.; Tremblay, S.; Jalilian, A. Gallium-68 Cyclotron Production; International Atomic Energy Agency: Vienna, Austria, 2019. [Google Scholar]
- Mueller, D.; Breeman, W.A.P.; Klette, I.; Gottschaldt, M.; Odparlik, A.; Baehre, M.; Tworowska, I.; Schultz, M.K. Radiolabeling of DOTA-like conjugated peptides with generator-produced 68 Ga and using NaCl-based cationic elution method. Nat. Protoc. 2016, 11, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Velikyan, I. 68Ga-based radiopharmaceuticals: Production and application relationship. Molecules 2015, 20, 12912–12943. [Google Scholar] [CrossRef]
- Banerjee, S.R.; Pomper, M.G. Clinical applications of Gallium-68. Appl. Radiat. Isot. 2013, 76, 2–13. [Google Scholar] [CrossRef]
- Lin, M.; Waligorski, G.J.; Lepera, C.G. Production of curie quantities of 68Ga with a medical cyclotron via the 68Zn(p,n)68Ga reaction. Appl. Radiat. Isot. 2018, 133, 1–3. [Google Scholar] [CrossRef]
- Delpassand, E.S.; Ranganathan, D.; Wagh, N.; Shafie, A.; Gaber, A.; Abbasi, A.; Kjaer, A.; Tworowska, I.; Nunez, R. (64)Cu-DOTATATE PET/CT for imaging patients with known or suspected somatostatin receptor-positive neuroendocrine tumors: Results of the first US prospective, reader-blinded clinical trial. J. Nucl. Med. 2020, jnumed-119. [Google Scholar] [CrossRef]
- Johnbeck, C.B.; Knigge, U.; Loft, A.; Berthelsen, A.K.; Mortensen, J.; Oturai, P.; Langer, S.W.; Elema, D.R.; Kjaer, A. Head-to-Head Comparison of (64)Cu-DOTATATE and (68)Ga-DOTATOC PET/CT: A prospective study of 59 patients with neuroendocrine tumors. J. Nucl. Med. 2017, 58, 451–457. [Google Scholar] [CrossRef]
- Pfeifer, A.; Knigge, U.; Mortensen, J.; Oturai, P.; Berthelsen, A.K.; Loft, A.; Binderup, T.; Rasmussen, P.; Elema, D.; Klausen, T.L.; et al. Clinical PET of neuroendocrine tumors using 64Cu-DOTATATE: First-in-humans study. J. Nucl. Med. 2012, 53, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Kjaer, A.; Binderup, T.; Johnbeck, C.; Carlsen, E.; Langer, S.; Federspiel, B.; Knigge, U. 64Cu-DOTATATE somatostatin receptor imaging in neuroendocrine tumors: Experience from 500 patients at Copenhagen ENETS Center of Excellence. J. Nucl. Med 2019, 60, 504. [Google Scholar]
- Anderson, C.J.; Ferdani, R. Copper-64 radiopharmaceuticals for PET imaging of cancer: Advances in preclinical and clinical research. Cancer Biother. Radiopharm. 2009, 24, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Qaim, S.M. The present and future of medical radionuclide production. Radiochim. Acta 2012, 100, 635–651. [Google Scholar] [CrossRef]
- Waldmann, C.M.; Stuparu, A.D.; van Dam, R.M.; Slavik, R. The search for an alternative to [68Ga]Ga-DOTA-TATE in neuroendocrine tumor theranostics: Current state of 18F-labeled somatostatin analog development. Theranostics 2019, 9, 1336–1347. [Google Scholar] [CrossRef] [PubMed]
- Hope, T.A.; Bergsland, E.K.; Bozkurt, M.F.; Graham, M.; Heaney, A.P.; Herrmann, K.; Howe, J.R.; Kulke, M.H.; Kunz, P.L.; Mailman, J.; et al. Appropriate use criteria for somatostatin receptor PET imaging in neuroendocrine tumors. J. Nucl. Med. 2018, 59, 66–74. [Google Scholar] [CrossRef]
- Hostetler, E.; Edwards, W.; Anderson, C.; Welch, M. Poster session r-oncologic imaging & therapy-R-14-synthesis of 4-(18F) fluorobenzoyl octreotide and biodistribution in tumour-bearing Lewis rats. J. Label. Compd. Radiopharm. 1999, 42, S720. [Google Scholar]
- Litau, S.; Niedermoser, S.; Vogler, N.; Roscher, M.; Schirrmacher, R.; Fricker, G.; Wängler, B.; Wängler, C. Next generation of SiFAlin-based TATE derivatives for PET imaging of SSTR-positive tumors: Influence of molecular design on in vitro SSTR binding and in vivo pharmacokinetics. Bioconjug. Chem. 2015, 26, 2350–2359. [Google Scholar] [CrossRef]
- Niedermoser, S.; Chin, J.; Wangler, C.; Kostikov, A.; Bernard-Gauthier, V.; Vogler, N.; Soucy, J.P.; McEwan, A.J.; Schirrmacher, R.; Wangler, B. In vivo evaluation of 18F-SiFAlin-Modified TATE: A potential challenge for 68Ga-DOTATATE, the clinical gold standard for somatostatin receptor imaging with PET. J. Nucl. Med. 2015, 56, 1100–1105. [Google Scholar] [CrossRef]
- Liu, Z.; Pourghiasian, M.; Radtke, M.A.; Lau, J.; Pan, J.; Dias, G.M.; Yapp, D.; Lin, K.-S.; Bénard, F.; Perrin, D.M. An Organotrifluoroborate for Broadly Applicable One-Step 18 F-Labeling. Angew. Chem. 2014, 53, 11876–11880. [Google Scholar] [CrossRef]
- Liu, Z.; Pourghiasian, M.; Bénard, F.; Pan, J.; Lin, K.S.; Perrin, D.M. Preclinical evaluation of a high-affinity 18F-trifluoroborate octreotate derivative for somatostatin receptor imaging. J. Nucl. Med. 2014, 55, 1499–1505. [Google Scholar] [CrossRef]
- Laverman, P.; McBride, W.J.; Sharkey, R.M.; Eek, A.; Joosten, L.; Oyen, W.J.G.; Goldenberg, D.M.; Boerman, O.C. A novel facile method of labeling octreotide with 18F-fluorine. J. Nucl. Med. 2010, 51, 454–461. [Google Scholar] [CrossRef]
- Goffin, K. Al18F-NOTA-octreotide and 18F-SiFAlin-TATE: Two ‘new kids on the block’ in somatostatin receptor imaging. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2225–2227. [Google Scholar] [CrossRef]
- Fani, M.; Nicolas, G.P.; Wild, D. Somatostatin receptor antagonists for imaging and therapy. J. Nucl. Med. 2017, 58, 61S–66S. [Google Scholar] [CrossRef]
- Wester, H.J.; Schottelius, M.; Scheidhauer, K.; Meisetschläger, G.; Herz, M.; Rau, F.C.; Reubi, J.C.; Schwaiger, M. PET imaging of somatostatin receptors: Design, synthesis and preclinical evaluation of a novel 18F-labelled, carbohydrated analogue of octreotide. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 117–122. [Google Scholar] [PubMed]
- Meisetschläger, G.; Poethko, T.; Stah, A.; Wolf, I.; Scheidhauer, K.; Schottelius, M.; Herz, M.; Wester, H.J.; Schwaiger, M. Gluc-Lys([18F]FP)-TOCA PET in patients with SSTR-positive tumors: Biodistribution and diagnostic evaluation compared with [ 111In]DTPA-octreotide. J. Nucl. Med. 2006, 47, 566–573. [Google Scholar] [PubMed]
- Schottelius, M.; Poethko, T.; Herz, M.; Reubi, J.C.; Kessler, H.; Schwaiger, M.; Wester, H.J. First 18F-labeled tracer suitable for routine clinical imaging of sst receptor-expressing tumors using positron emission tomography. Clin. Cancer Res. 2004, 10, 3593–3606. [Google Scholar] [CrossRef] [PubMed]
- Poethko, T.; Schottelius, M.; Thumshirn, G.; Hersel, U.; Herz, M.; Henriksen, G.; Kessler, H.; Schwaiger, M.; Wester, H.J. Two-step methodology for high-yield routine radiohalogenation of peptides:18F-labeled RGD and octreotide analogs. J. Nucl. Med. 2004, 45, 892–902. [Google Scholar]
- Maschauer, S.; Heilmann, M.; Wängler, C.; Schirrmacher, R.; Prante, O. Radiosynthesis and preclinical evaluation of 18F-fluoroglycosylated octreotate for somatostatin receptor imaging. Bioconjug. Chem. 2016, 27, 2707–2714. [Google Scholar] [CrossRef]
- Tshibangu, T.; Cawthorne, C.; Serdons, K.; Pauwels, E.; Gsell, W.; Bormans, G.; Deroose, C.M.; Cleeren, F. Automated GMP compliant production of [18F]AlF-NOTA-octreotide. EJNMMI Radiopharm. Chem. 2020, 5, 4. [Google Scholar] [CrossRef]
- Pauwels, E.; Cleeren, F.; Tshibangu, T.; Koole, M.; Serdons, K.; Dekervel, J.; Van Cutsem, E.; Verslype, C.; Van Laere, K.; Bormans, G.; et al. Al18F-NOTA-octreotide: First comparison with 68Ga-DOTATATE in a neuroendocrine tumour patient. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2398–2399. [Google Scholar] [CrossRef] [PubMed]
- Iddon, L.; Leyton, J.; Indrevoll, B.; Glaser, M.; Robins, E.G.; George, A.J.T.; Cuthbertson, A.; Luthra, S.K.; Aboagye, E.O. Synthesis and in vitro evaluation of [18F]fluoroethyl triazole labelled [Tyr3]octreotate analogues using click chemistry. Bioorg.Med. Chem. Lett. 2011, 21, 3122–3127. [Google Scholar] [CrossRef] [PubMed]
- Dubash, S.R.; Nicholas, K.; Mapelli, P.; Twyman, F.; Carroll, L.; Kozlowski, K.; Al-Nahhas, A.; Saleem, A.; Huiban, M.; Janisch, R.; et al. Clinical translation of a click-labeled 18F-octreotate radioligand for imaging neuroendocrine tumors. J. Nucl. Med. 2016, 57, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Leyton, J.; Iddon, L.; Perumal, M.; Indrevoll, B.; Glaser, M.; Robins, E.; George, A.J.T.; Cuthbertson, A.; Luthra, S.K.; Aboagye, E.O. Targeting somatostatin receptors: Preclinical evaluation of novel 18F-fluoroethyltriazole-Tyr3-octreotate analogs for PET. J. Nucl. Med. 2011, 52, 1441–1448. [Google Scholar] [CrossRef][Green Version]
- Allott, L.; Da Pieve, C.; Turton, D.R.; Smith, G. A general [18 F]AlF radiochemistry procedure on two automated synthesis platforms. React. Chem. Eng. 2017, 2, 68–74. [Google Scholar] [CrossRef]
- Glaser, M.; Årstad, E. “Click labeling” with 2-[18F]fluoroethylazide for positron emission tomography. Bioconjug. Chem. 2007, 18, 989–993. [Google Scholar] [CrossRef]
- Zhou, D.; Chu, W.; Dence, C.S.; Mach, R.H.; Welch, M.J. Highly efficient click labeling using 2-[18F]fluoroethyl azide and synthesis of an 18FN-hydroxysuccinimide ester as conjugation agent. Nucl. Med. Biol. 2012, 39, 1175–1181. [Google Scholar] [CrossRef]
- Glaser, M.; Goggi, J.; Smith, G.; Morrison, M.; Luthra, S.K.; Robins, E.; Aboagye, E.O. Improved radiosynthesis of the apoptosis marker18F-ICMT11 including biological evaluation. Bioorg. Med. Chem. Lett. 2011, 21, 6945–6949. [Google Scholar] [CrossRef]
- Fortt, R.; Smith, G.; Awais, R.O.; Luthra, S.K.; Aboagye, E.O. Automated GMP synthesis of [18F]ICMT-11 for in vivo imaging of caspase-3 activity. Nucl. Med. Biol. 2012, 39, 1000–1005. [Google Scholar] [CrossRef]
- Bonandi, E.; Christodoulou, M.S.; Fumagalli, G.; Perdicchia, D.; Rastelli, G.; Passarella, D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today 2017, 22, 1572–1581. [Google Scholar] [CrossRef]
- Wen, P.L.; Lewis, J.S.; Kim, J.; Bugaj, J.E.; Johnson, M.A.; Erion, J.L.; Anderson, C.J. DOTA-D-Tyrl-octreotate: A somatostatin analogue for labeling with metal and halogen radionuclides for cancer imaging and therapy. Bioconjug. Chem. 2002, 13, 721–728. [Google Scholar]
- Weiner, R.E.; Thakur, M.L. Radiolabeled peptides in diagnosis and therapy. Semin. Nucl. Med. 2001, 31, 296–311. [Google Scholar] [CrossRef] [PubMed]
- de Jong, M.; Valkema, R.; Kwekkeboom, D.J.; Krenning, E.P. Somatostatin Receptor Targeted-Radio-Ablation of Tumors. In Somatostatin; Springer: Boston, MA, USA, 2004; pp. 233–249. ISBN 978-1-4020-7799-9. [Google Scholar]
- Wieder, H.; Beer, A.J.; Poethko, T.; Meisetschlaeger, G.; Wester, H.J.; Rummeny, E.; Schwaiger, M.; Stahl, A.R. PET/CT with Gluc-Lys-([18F]FP)-TOCA: Correlation between uptake, size and arterial perfusion in somatostatin receptor positive lesions. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Allott, L.; Barnes, C.; Brickute, D.; Aboagye, E.O. An improved automated radiosynthesis of [18 F]FET-βAG-TOCA. React. Chem. Eng. 2019, 4, 569–574. [Google Scholar] [CrossRef]
- Yan, R.; Sander, K.; Galante, E.; Rajkumar, V.; Badar, A.; Robson, M.; El-Emir, E.; Lythgoe, M.F.; Pedley, R.B.; Årstad, E. A one-pot three-component radiochemical reaction for rapid assembly of 125I-labeled molecular probes. J. Am. Chem. Soc. 2013, 135, 703–709. [Google Scholar] [CrossRef]
- Glaser, M.; Rajkumar, V.; Diocou, S.; Gendron, T.; Yan, R.; Sin, P.K.B.; Sander, K.; Carroll, L.; Pedley, R.B.; Aboagye, E.O.; et al. One-pot radiosynthesis and biological evaluation of a caspase-3 selective 5-[123,125I]iodo-1,2,3-triazole derived Isatin SPECT tracer. Sci. Rep. 2019, 9, 19299. [Google Scholar] [CrossRef]
- Lewis, W.G.; Magallon, F.G.; Fokin, V.V.; Finn, M.G. Discovery and characterization of catalysts for azide-alkyne cycloaddition by fluorescence quenching. J. Am. Chem. Soc. 2004, 126, 9152–9153. [Google Scholar] [CrossRef]
- Gupta, S.S.; Kuzelka, J.; Singh, P.; Lewis, W.G.; Manchester, M.; Finn, M.G. Accelerated bioorthogonal conjugation: A practical method for the ligation of diverse functional molecules to a polyvalent virus scaffold. Bioconjug. Chem. 2005, 16, 1572–1579. [Google Scholar] [CrossRef]
- Haldón, E.; Nicasio, M.C.; Pérez, P.J. Copper-catalysed azide-alkyne cycloadditions (CuAAC): An update. Org. Biomol. Chem. 2015, 13, 9528–9550. [Google Scholar] [CrossRef]
- Pisaneschi, F.; Kelderhouse, L.E.; Hardy, A.; Engel, B.J.; Mukhopadhyay, U.; Gonzalez-Lepera, C.; Gray, J.P.; Ornelas, A.; Takahashi, T.T.; Roberts, R.W.; et al. Automated, resin-based method to enhance the specific activity of fluorine-18 clicked PET radiotracers. Bioconjug.Chem. 2017, 28, 583–589. [Google Scholar] [CrossRef]
- Pettinato, C.; Sarnelli, A.; Di Donna, M.; Civollani, S.; Nanni, C.; Montini, G.; Di Pierro, D.; Ferrari, M.; Marengo, M.; Bergamini, C. 68Ga-DOTANOC: Biodistribution and dosimetry in patients affected by neuroendocrine tumors. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, H.; Zöphel, K.; Freudenberg, R.; Oehme, L.; Andreeff, M.; Wunderlich, G.; Eisenhofer, G.; Kotzerke, J. Radiation exposure of patients during 68Ga-DOTATOC PET/CT examinations. NuklearMedizin 2009, 48, 201–207. [Google Scholar] [PubMed]
- Walker, R.C.; Smith, G.T.; Liu, E.; Moore, B.; Clanton, J.; Stabin, M. Measured human dosimetry of 68Ga-DOTATATE. J. Nucl. Med. 2013, 54, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Ilhan, H.; Lindner, S.; Todica, A.; Cyran, C.C.; Tiling, R.; Auernhammer, C.J.; Spitzweg, C.; Boeck, S.; Unterrainer, M.; Gildehaus, F.J.; et al. Biodistribution and first clinical results of 18F-SiFAlin-TATE PET: A novel 18F-labeled somatostatin analog for imaging of neuroendocrine tumors. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 870–880. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry. | Compounds | Synthesis Time (min) | Synthetic Steps | Automated synthesis? | GMP Compliant? | Evaluated in Humans? | Ref | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RCP (%) | RCY (%) | Am (GBq/μmol) | SSTR2 IC50 (nM) | Model | LogD | Tumour (%ID/g at 60 min p.i.) | Tumour:Bone (1 h) | Tumour:Muscle (1 h) | Tumour:Liver (1 h) | |||||||||
| Published 18F-SSTRadioconjugates | 1 | [18F]FP-Gluc-TOCA | 180 | 5 | ND | Y | Y | >98 | 20–30 | >37 | 2.8 ± 0.4 | AR42J | ND | 13.54 ± 1.47 | 20 | 56 | 19 | [26,27] |
| 2 | Cel-S-Dpr([18F]FBOA)TOCA | 50 | 2 | ND | N | N | >98 | 40–60 | ND | 1.8 ± 1.1 | AR42J | ND | 24.04 ± 2.50 | 28 | 200 | 27 | [28,29] | |
| 3 | [18F]FGlc-TATE | 70 | 3 | Y | N | N | >98 | 19–22 | 32–106 | 4.2 | AR42J | ND | 5.62 ± 1.53 | 9.7 | 12.2 | 6.2 | [30] | |
| 4 | [18F]SiFAlin-Glc-Asp 2 -PEG1-TATE ([18F]SiFAlin-TATE) | 25 | 1 | ND | Y | Y | >98 | ~50 | 44–63 | 14.4 ± 1.2 | AR42J | ND | 18.51 ± 4.89 | ND | 39.35 | 8.97 | [19,20,24] | |
| 5 | [18F]AMBF3-TATE | 25 | 1 | ND | N | N | >99 | >30 | >111 | 0.13 ± 0.03 | AR42J | ND | 10.11 ± 1.67 | 21.3 | 92 | 26.2 | [21,22] | |
| 6 | [18F]AlF-NOTA-octreotide ([18F]AlF-IMP466) | 45 | 2 | Y | Y | Y | >98 | ~50 | 36.1 | 3.6 ± 0.6 | AR42J | ND | 12.73 ± 2.05 | 86 (2h) | 37.44 | ND | [23,31,32,36] | |
| [18F]FET-TOCA analogues | 7 | [18F]FET-βAG-TOCA | 90 | 2 | Y | Y | Y | >98 | ~6.5 66dc | 224–562 | 1.6 ± 0.2 | AR42J | −2.26 | 11.58 ± 0.67 | ND | 24.12 | ND | [33,34,35] |
| 7 | [18F]FET-βAG-TOCA | HCT116 | 0.52 ± 0.39 | ND | 0.97 | ND | [33,34,35] | |||||||||||
| 7* | [18F]FET-βAG-[W-c-(CTFTYC)K] | 90 | 2 | Y | N | N | >98 | 64dc | 12.3 | > 10.0 mM | AR42J | −1.14 | 0.22 ± 0.12 | ND | 0.65 | ND | [33,35] | |
| 8 | [18F]FETE-PEG-TOCA | 90 | 2 | Y | N | N | >98 | 52dc | 5.9 | 13.2 ± 7.8 | AR42J | −2.77 | 4.51 ± 1.52 | ND | 23.74 | ND | [33,35] | |
| 9 | [18F]FET-G-PEG-TOCA | 90 | 2 | Y | N | N | >98 | 40dc | 4.8 | 10.8 ± 5.9 | AR42J | −2.68 | 8.29 ± 1.42 | ND | 59.21 | ND | [33,35] | |
| 10 | [18F]FET-G-TOCA | 90 | 2 | Y | N | N | >98 | 50dc | 5.9 | 4.0 ± 1.4 | AR42J | −1.83 | 17.05 ± 2.77 | ND | 11.44 | ND | [33,35] | |
| 11 | [18F]FETE-TOCA | 90 | 2 | Y | N | N | >98 | 51dc | 8.4 | 2.9 ± 1.3 | AR42J | −1.5 | 9.79 ± 2.57 | ND | 12.39 | ND | [33,35] | |
| [68Ga]Ga-DOTA-TATE | -- | 1 | -- | N | N | >98 | ND | ND | 14.7 ± 7.7 | AR42J | N/A | 1.37 ± 0.16 | ND | 2.28 | ND | -- | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allott, L.; Dubash, S.; Aboagye, E.O. [18F]FET-βAG-TOCA: The Design, Evaluation and Clinical Translation of a Fluorinated Octreotide. Cancers 2020, 12, 865. https://doi.org/10.3390/cancers12040865
Allott L, Dubash S, Aboagye EO. [18F]FET-βAG-TOCA: The Design, Evaluation and Clinical Translation of a Fluorinated Octreotide. Cancers. 2020; 12(4):865. https://doi.org/10.3390/cancers12040865
Chicago/Turabian StyleAllott, Louis, Suraiya Dubash, and Eric O. Aboagye. 2020. "[18F]FET-βAG-TOCA: The Design, Evaluation and Clinical Translation of a Fluorinated Octreotide" Cancers 12, no. 4: 865. https://doi.org/10.3390/cancers12040865
APA StyleAllott, L., Dubash, S., & Aboagye, E. O. (2020). [18F]FET-βAG-TOCA: The Design, Evaluation and Clinical Translation of a Fluorinated Octreotide. Cancers, 12(4), 865. https://doi.org/10.3390/cancers12040865