Frequent FOXA1-Activating Mutations in Extramammary Paget’s Disease

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
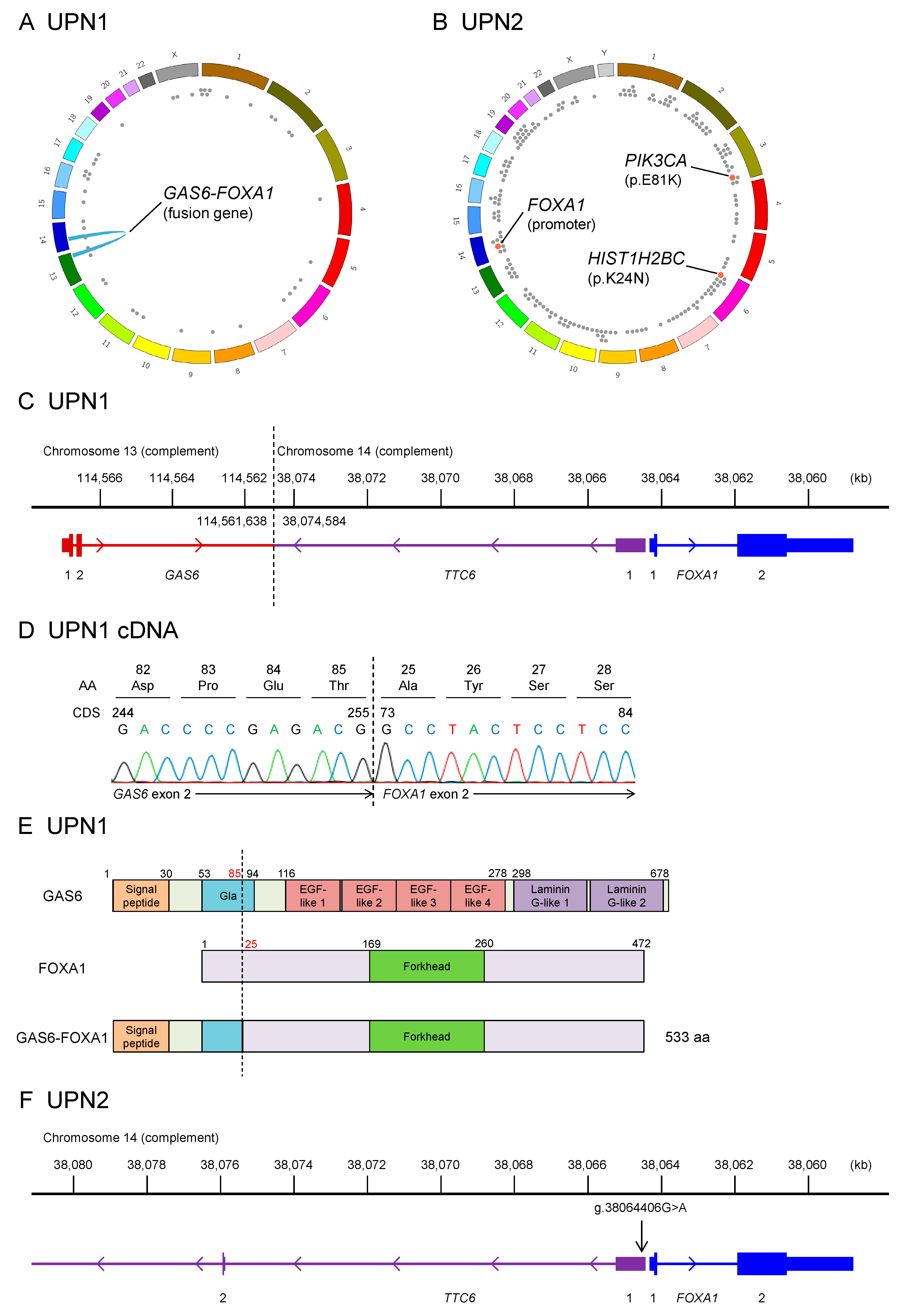
2.1. Whole-Genome Sequencing of Two Patients with EMPD
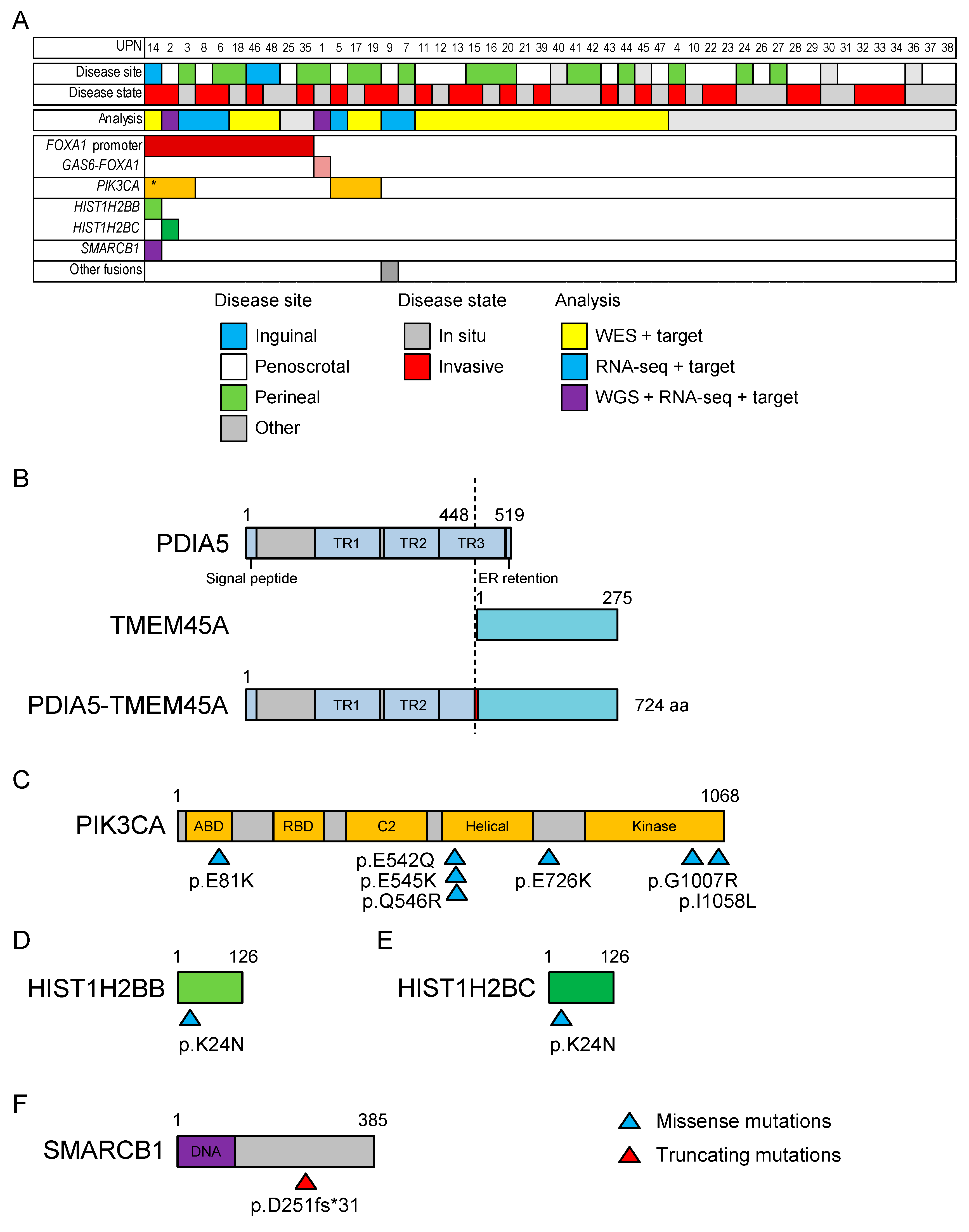
2.2. Whole-Exome Sequencing of 21 Patients with EMPD
2.3. RNA Sequencing of Six Patients with EMPD
2.4. Targeted Sequencing in 48 Patients with EMPD or MPD
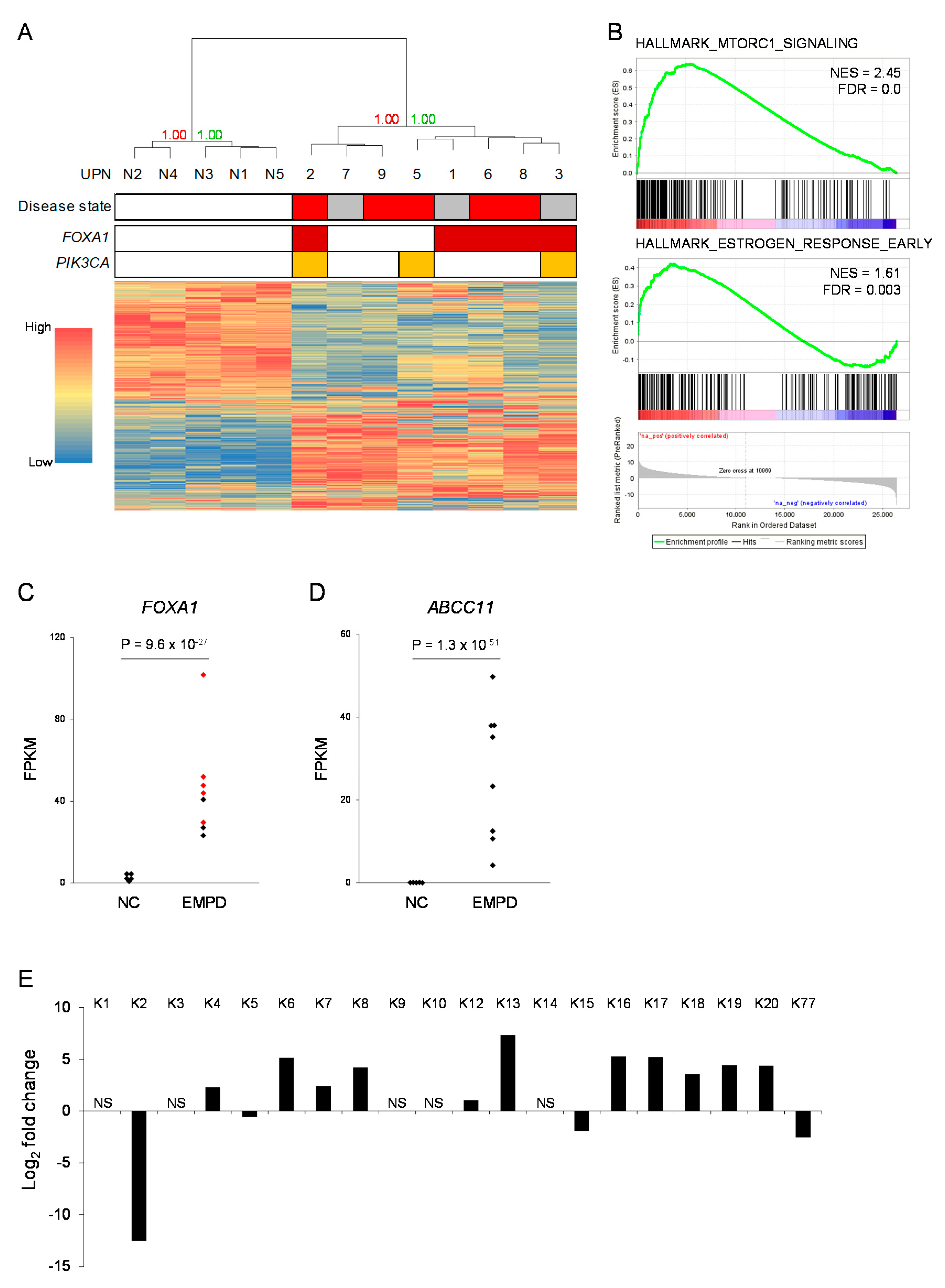
2.5. Global Gene Expression Profiling of EMPD
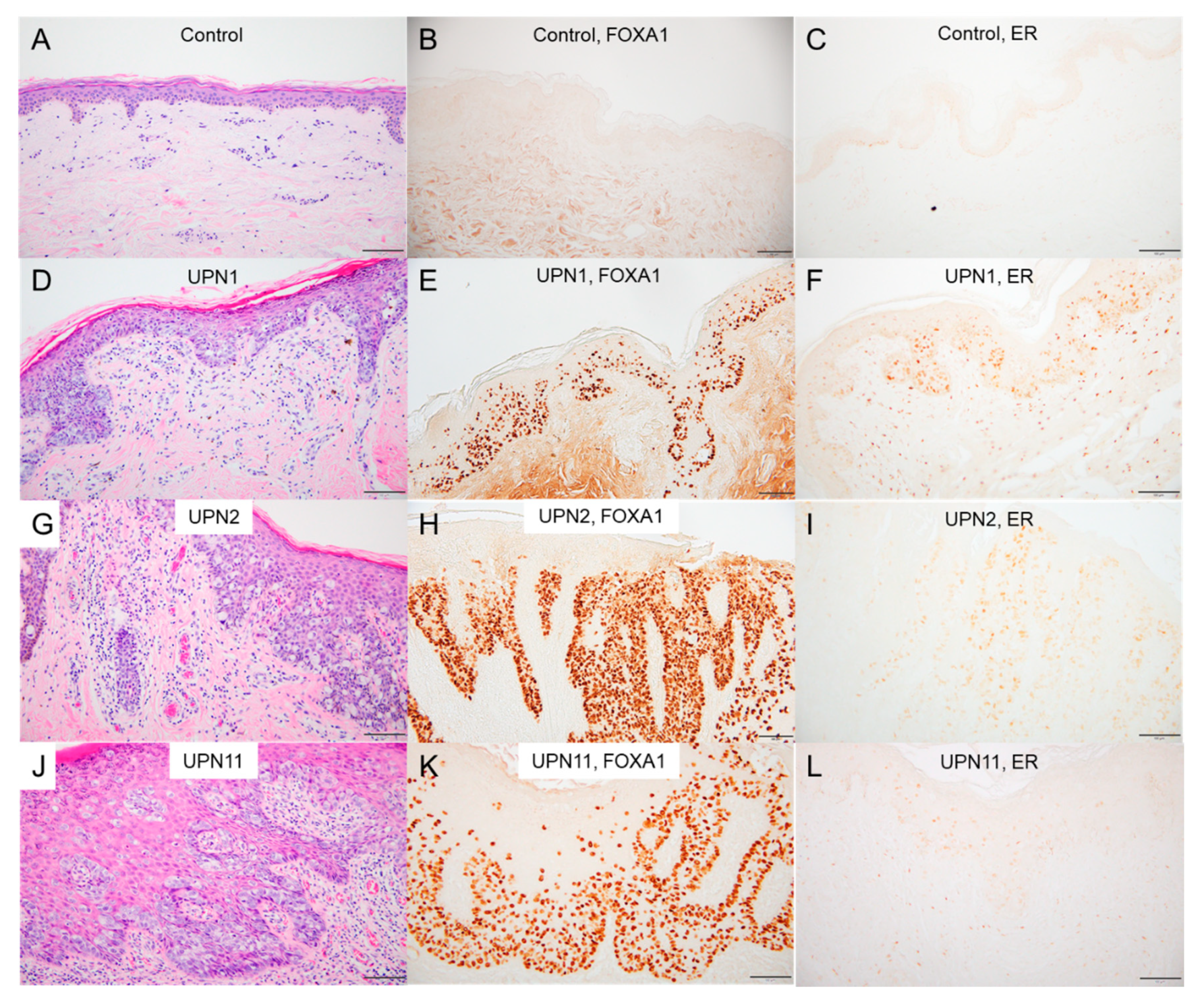
2.6. Immunostaining in EMPD/MPD Samples
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Nucleic Acid Preparation
4.3. Whole-Genome Sequencing
4.4. Whole-Exome Sequencing
4.5. Targeted Gene Sequencing
4.6. RNA Sequencing
4.7. Immunohistochemistry
4.8. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kanitakis, J. Mammary and extramammary Paget’s disease. J. Eur. Acad. Dermatol. Venereol. 2007, 21, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Crocker, H.R. Paget’s disease, affecting the scrotum and penis. Trans. Pathol. Soc. Lond. 1889, 40, 187–191. [Google Scholar]
- Urabe, A.; Matsukuma, A.; Shimizu, N.; Nishimura, M.; Wada, H.; Hori, Y. Extramammary Paget’s disease: Comparative histopathologic studies of intraductal carcinoma of the breast and apocrine adenocarcinoma. J. Cutan. Pathol. 1990, 17, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Hatta, N.; Yamada, M.; Hirano, T.; Fujimoto, A.; Morita, R. Extramammary Paget’s disease: Treatment, prognostic factors and outcome in 76 patients. Br. J. Dermatol. 2008, 158, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Regauer, S. Extramammary Paget’s disease—A proliferation of adnexal origin? Histopathology 2006, 48, 723–729. [Google Scholar] [CrossRef]
- Toker, C. Clear cells of the nipple epidermis. Cancer 1970, 25, 601–610. [Google Scholar] [CrossRef]
- Kang, Z.; Xu, F.; Zhang, Q.A.; Wu, Z.; Zhang, X.; Xu, J.; Luo, Y.; Guan, M. Oncogenic mutations in extramammary Paget’s disease and their clinical relevance. Int. J. Cancer 2013, 132, 824–831. [Google Scholar] [CrossRef]
- Zhang, G.; Zhou, S.; Zhong, W.; Hong, L.; Wang, Y.; Lu, S.; Pan, J.; Huang, Y.; Su, M.; Crawford, R.; et al. Whole-Exome Sequencing Reveals Frequent Mutations in Chromatin Remodeling Genes in Mammary and Extramammary Paget’s Diseases. J. Investig. Dermatol. 2019, 139, 789–795. [Google Scholar] [CrossRef]
- Kiniwa, Y.; Yasuda, J.; Saito, S.; Saito, R.; Motoike, I.N.; Danjoh, I.; Kinoshita, K.; Fuse, N.; Yamamoto, M.; Okuyama, R. Identification of genetic alterations in extramammary Paget disease using whole exome analysis. J. Dermatol. Sci. 2019. [Google Scholar] [CrossRef]
- Fukuda, K.; Funakoshi, T. Metastatic Extramammary Paget’s Disease: Pathogenesis and Novel Therapeutic Approach. Front. Oncol. 2018, 8, 38. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Rheinbay, E.; Parasuraman, P.; Grimsby, J.; Tiao, G.; Engreitz, J.M.; Kim, J.; Lawrence, M.S.; Taylor-Weiner, A.; Rodriguez-Cuevas, S.; Rosenberg, M.; et al. Recurrent and functional regulatory mutations in breast cancer. Nature 2017, 547, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Loconte, D.C.; Grossi, V.; Bozzao, C.; Forte, G.; Bagnulo, R.; Stella, A.; Lastella, P.; Cutrone, M.; Benedicenti, F.; Susca, F.C.; et al. Molecular and Functional Characterization of Three Different Postzygotic Mutations in PIK3CA-Related Overgrowth Spectrum (PROS) Patients: Effects on PI3K/AKT/mTOR Signaling and Sensitivity to PIK3 Inhibitors. PLoS ONE 2015, 10, e0123092. [Google Scholar] [CrossRef] [PubMed]
- Charlop-Powers, Z.; Zeng, L.; Zhang, Q.; Zhou, M.M. Structural insights into selective histone H3 recognition by the human Polybromo bromodomain 2. Cell Res. 2010, 20, 529–538. [Google Scholar] [CrossRef]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Martin, A.; Saathoff, M.; Kuhn, F.; Max, H.; Terstegen, L.; Natsch, A. A functional ABCC11 allele is essential in the biochemical formation of human axillary odor. J. Investig. Dermatol. 2010, 130, 529–540. [Google Scholar] [CrossRef]
- Cui, C.Y.; Schlessinger, D. Eccrine sweat gland development and sweat secretion. Exp. Dermatol. 2015, 24, 644–650. [Google Scholar] [CrossRef]
- Chu, P.G.; Weiss, L.M. Keratin expression in human tissues and neoplasms. Histopathology 2002, 40, 403–439. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, D.; Li, Q.; Liang, J.; Sun, L.; Yi, X.; Chen, Z.; Yan, R.; Xie, G.; Li, W.; et al. Nucleation of DNA repair factors by FOXA1 links DNA demethylation to transcriptional pioneering. Nat. Genet. 2016, 48, 1003–1013. [Google Scholar] [CrossRef]
- Mai, R.; Zhou, S.; Zhou, S.; Zhong, W.; Hong, L.; Wang, Y.; Lu, S.; Pan, J.; Huang, Y.; Su, M.; et al. Transcriptome analyses reveal FOXA1 dysregulation in mammary and extramammary Paget’s disease. Hum. Pathol. 2018, 77, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Fujino, K.; Monteiro, L.J.; Gomes, A.R.; Drost, R.; Davidson-Smith, H.; Takeda, S.; Khoo, U.S.; Jonkers, J.; Sproul, D.; et al. FOXA1 repression is associated with loss of BRCA1 and increased promoter methylation and chromatin silencing in breast cancer. Oncogene 2015, 34, 5012–5024. [Google Scholar] [CrossRef] [PubMed]
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.J.; Brown, G.D.; Gojis, O.; Ellis, I.O.; Green, A.R.; et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; Holmes, K.A.; Carroll, J.S. FOXA1 mutations in hormone-dependent cancers. Front. Oncol. 2013, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Ishii, K.; Mirosevich, J.; Kuwajima, S.; Oppenheimer, S.R.; Roberts, R.L.; Jiang, M.; Yu, X.; Shappell, S.B.; Caprioli, R.M.; et al. Forkhead box A1 regulates prostate ductal morphogenesis and promotes epithelial cell maturation. Development 2005, 132, 3431–3443. [Google Scholar] [CrossRef]
- Zhang, H.; Meng, F.; Liu, G.; Zhang, B.; Zhu, J.; Wu, F.; Ethier, S.P.; Miller, F.; Wu, G. Forkhead transcription factor foxq1 promotes epithelial-mesenchymal transition and breast cancer metastasis. Cancer Res. 2011, 71, 1292–1301. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The Pathway Interaction Database. Nucleic Acids Res. 2009, 37, D674–D679. [Google Scholar] [CrossRef]
- Takeichi, T.; Sugiura, K.; Nomura, T.; Sakamoto, T.; Ogawa, Y.; Oiso, N.; Futei, Y.; Fujisaki, A.; Koizumi, A.; Aoyama, Y.; et al. Pityriasis Rubra Pilaris Type V as an Autoinflammatory Disease by CARD14 Mutations. JAMA Dermatol. 2017, 153, 66–70. [Google Scholar] [CrossRef]
- Allred, D.C.; Harvey, J.M.; Berardo, M.; Clark, G.M. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod. Pathol. 1998, 11, 155–168. [Google Scholar]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeichi, T.; Okuno, Y.; Matsumoto, T.; Tsunoda, N.; Suzuki, K.; Tanahashi, K.; Kono, M.; Kikumori, T.; Muro, Y.; Akiyama, M. Frequent FOXA1-Activating Mutations in Extramammary Paget’s Disease. Cancers 2020, 12, 820. https://doi.org/10.3390/cancers12040820
Takeichi T, Okuno Y, Matsumoto T, Tsunoda N, Suzuki K, Tanahashi K, Kono M, Kikumori T, Muro Y, Akiyama M. Frequent FOXA1-Activating Mutations in Extramammary Paget’s Disease. Cancers. 2020; 12(4):820. https://doi.org/10.3390/cancers12040820
Chicago/Turabian StyleTakeichi, Takuya, Yusuke Okuno, Takaaki Matsumoto, Nobuyuki Tsunoda, Kyogo Suzuki, Kana Tanahashi, Michihiro Kono, Toyone Kikumori, Yoshinao Muro, and Masashi Akiyama. 2020. "Frequent FOXA1-Activating Mutations in Extramammary Paget’s Disease" Cancers 12, no. 4: 820. https://doi.org/10.3390/cancers12040820
APA StyleTakeichi, T., Okuno, Y., Matsumoto, T., Tsunoda, N., Suzuki, K., Tanahashi, K., Kono, M., Kikumori, T., Muro, Y., & Akiyama, M. (2020). Frequent FOXA1-Activating Mutations in Extramammary Paget’s Disease. Cancers, 12(4), 820. https://doi.org/10.3390/cancers12040820