Development of Prostate Cancer Organoid Culture Models in Basic Medicine and Translational Research

 ,
,  ,
, 
Abstract
1. Introduction
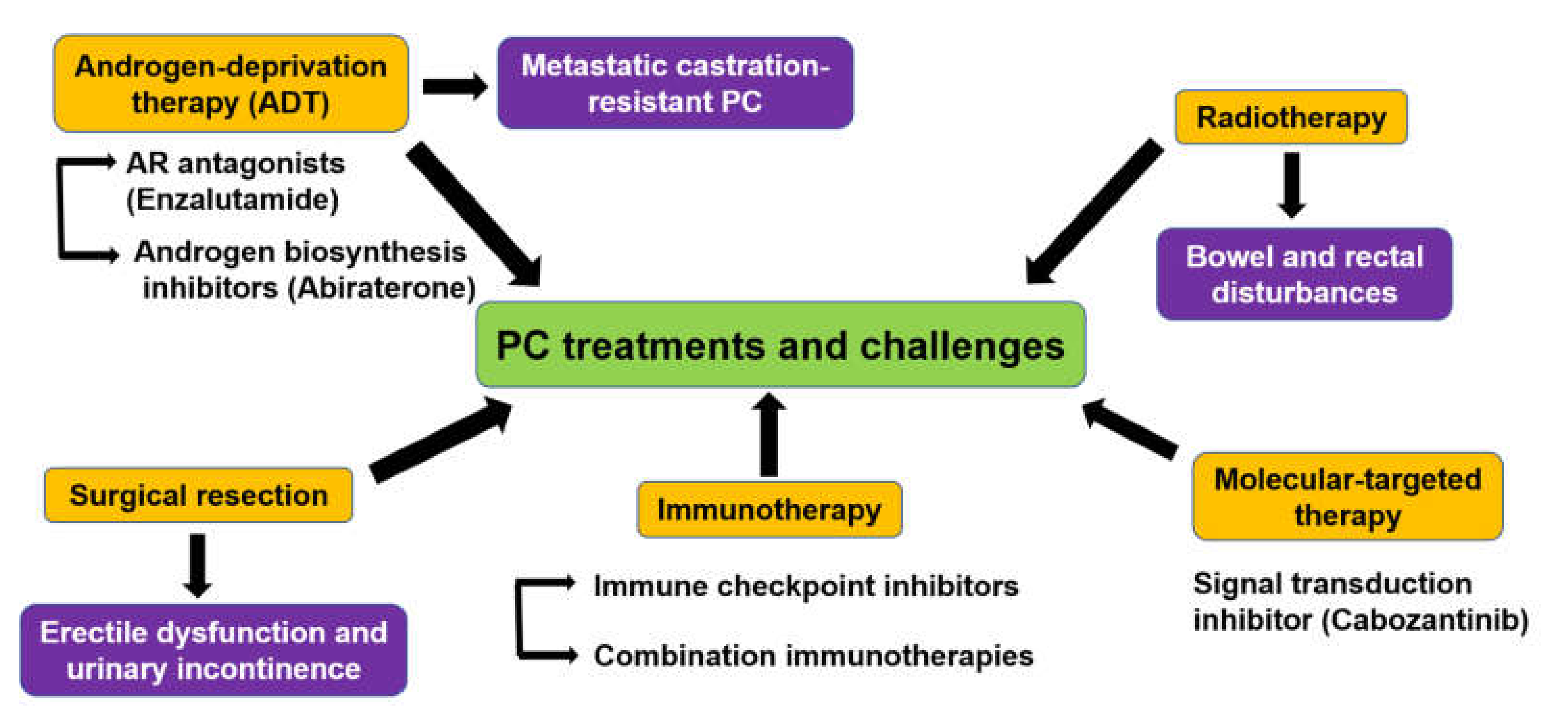
2. Treatment Challenges Against PC
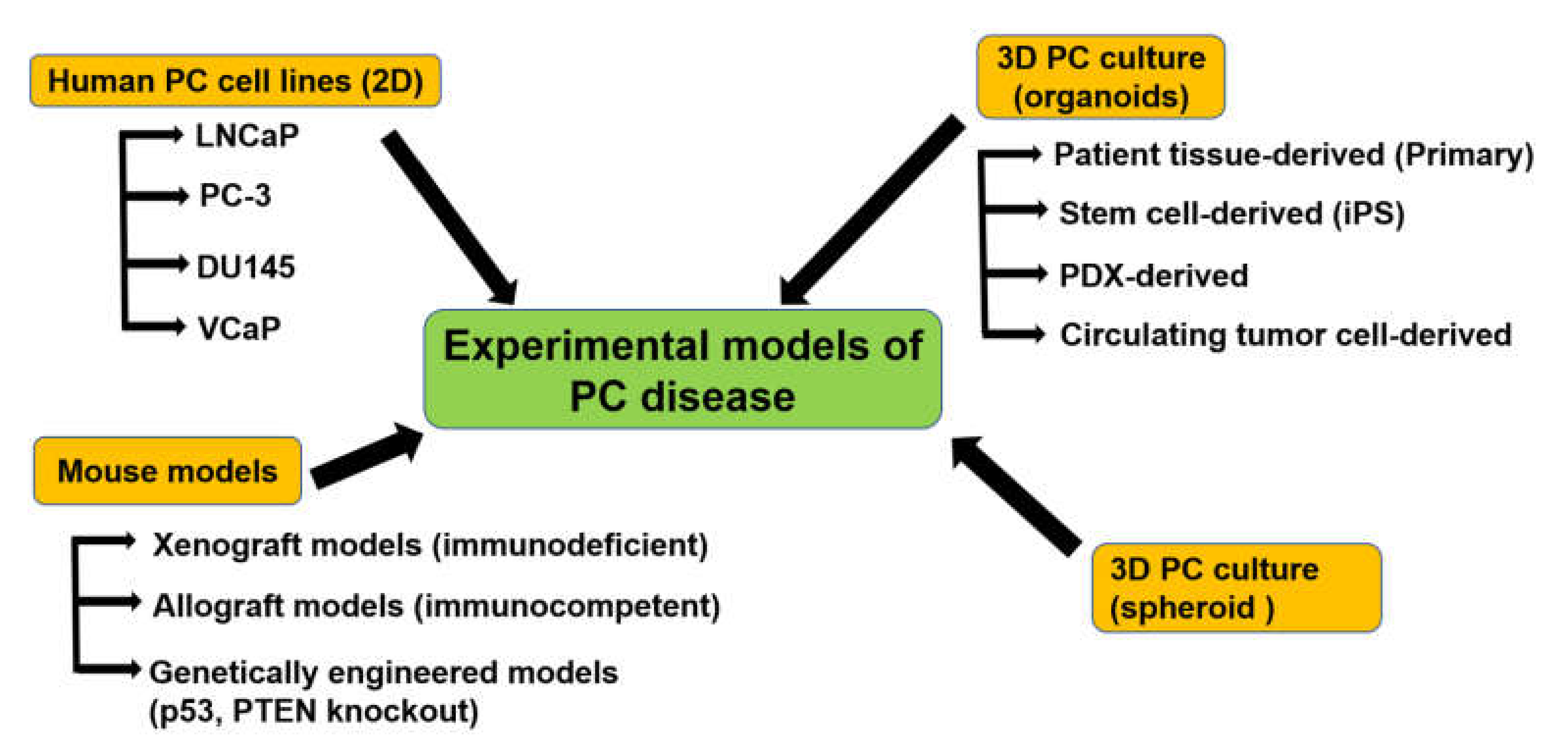
3. Experimental Models to Study PC
3.1. PC Cell Lines

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Origin | Type | PSA | AR | ER-α, β | Reference |
|---|---|---|---|---|---|---|
| LNCaP | Lymph node metastasis | Adenocarcinoma | + | + | + ER-β | [41,42] |
| PC-3 | Bone metastasis | Adenocarcinoma | - | - | + ER-α, ER-β | [43,44] |
| DU145 | Brain metastasis | Adenocarcinoma | - | - | + ER-β | [45,46] |
| VCap | Xenograft from metastasis | Adenocarcinoma | + | + | + ER-β | [47,48] |
| NCI-H660 | Metastatic extrapulmonary small cell carcinoma arising from the PC | Adenocarcinoma | - | - | - | [49] |
| PC346 | Primary tumor xenograft | Adenocarcinoma | + | ± | - | [50] |
| 22Rv1 (CWR22Rv1) | CRPC derivative of CWR22 xenograft | Adenocarcinoma | + | + | + ER-α, ER-β | [51] |
| CWR22Pc | CWR22 xenograft | Adenocarcinoma | + | + | + ER-β | [63,64] |
| MDA PCa 2a & MDA PCa 2b | Bone metastasis | Adenocarcinoma | + | + | + ER-β | [53,54] |
| KUCaP | Xenograft from liver metastasis | Adenocarcinoma | + | + | - | [52] |
| Subline | Original Cell Line | Resistance | Established Method | Reference |
|---|---|---|---|---|
| LNCaP-abl | LNCaP | Castration | Culture in ADM | [57] |
| LNCaP-LTAD | LNCaP | Castration | Culture in ADM | [58] |
| LNCaP-BicR | LNCaP | Anti-androgens | Culture with flutamide | [65] |
| PC346Flu1/2 | PC346 | Anti-androgens | Culture in ADM with flutamide | [66] |
| PC-3R | PC-3 | Chemotherapy | Docetaxel | [67] |
| DU145-TxR | DU145 | Chemotherapy | Culture with paclitaxel | [68] |
| DU145R | DU145 | Chemotherapy | Culture with docetaxel | [67] |
| PC-3CR | PC-3 | Chemotherapy | Culture with cabazitaxel | [69] |
| PC-3 D12 | PC-3 | Chemotherapy | Culture with docetaxel | [70] |
3.2. Xenograft Mouse Models
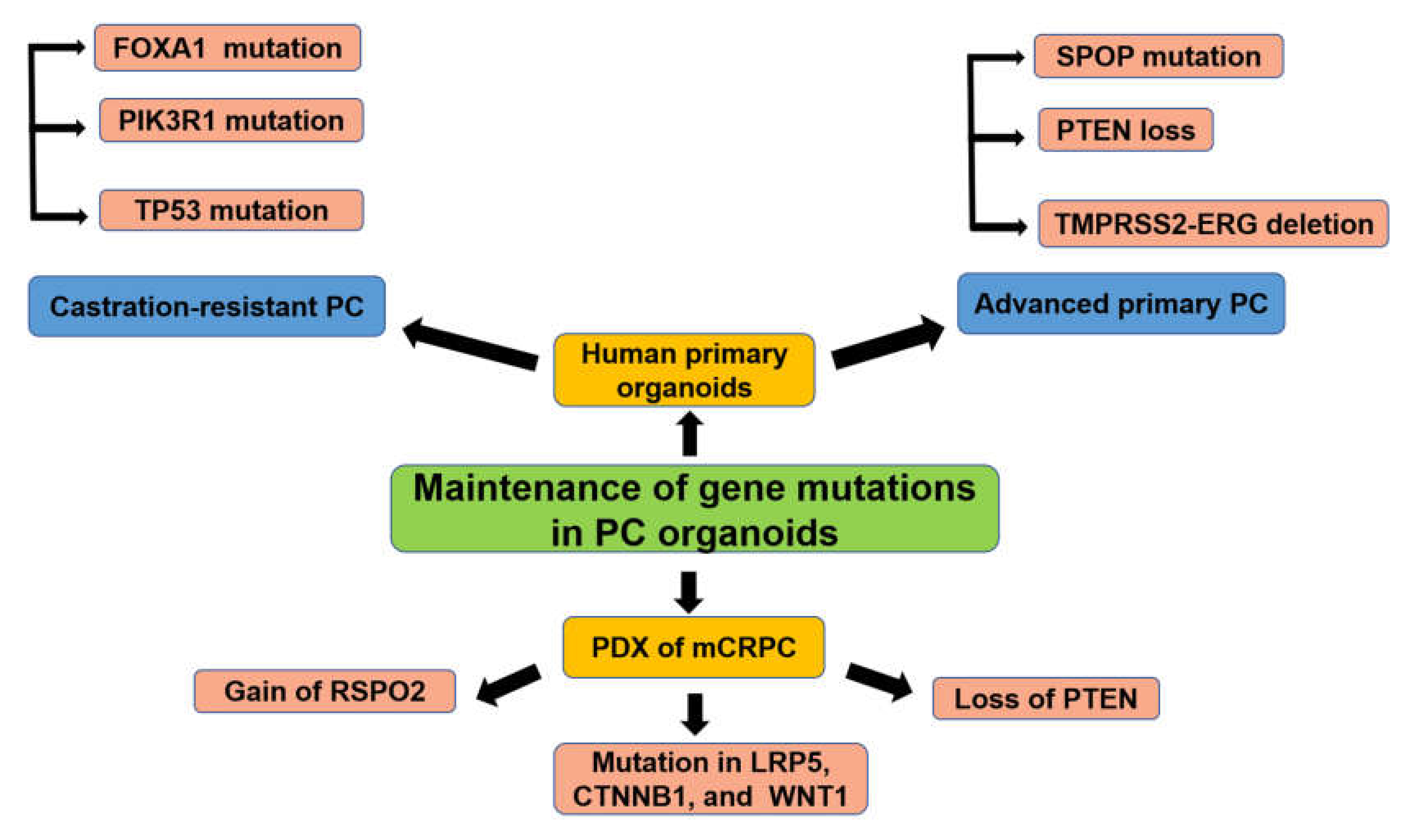
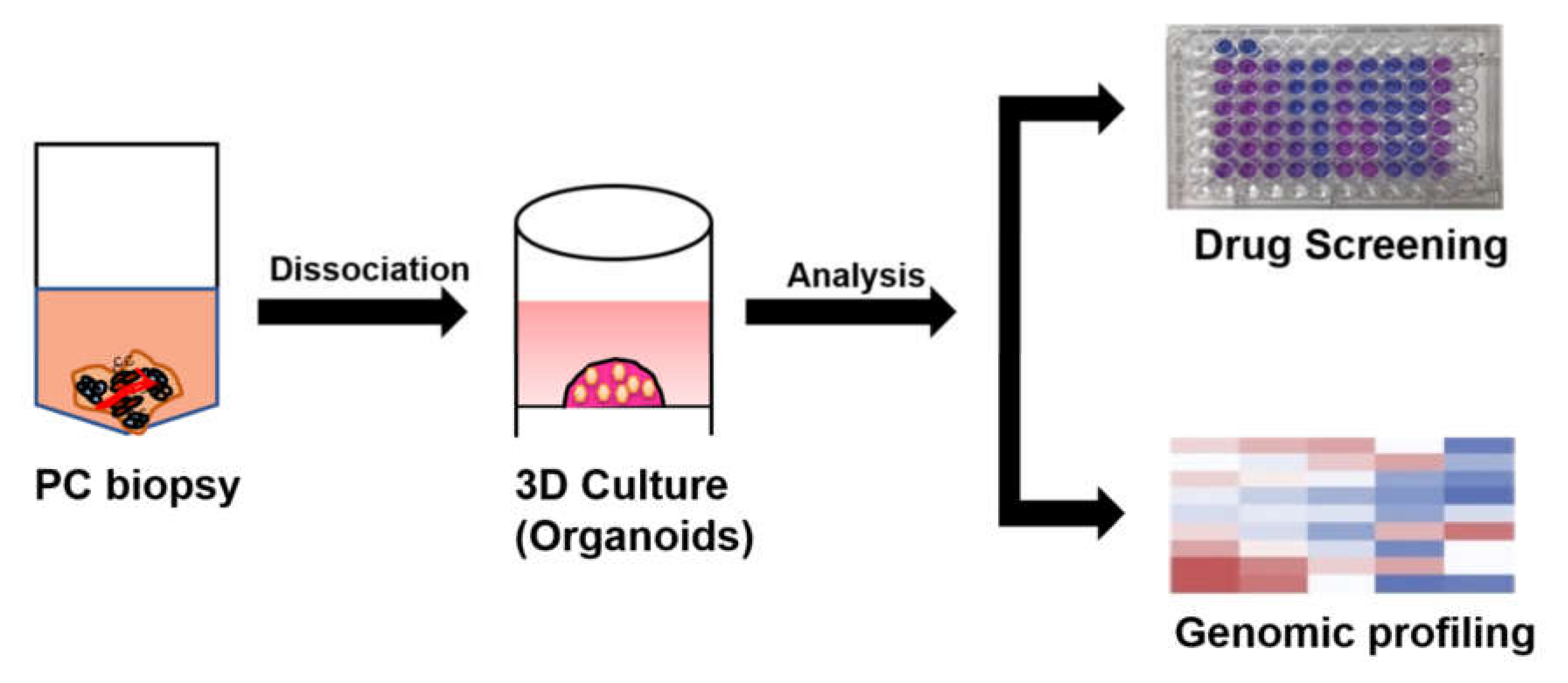
3.3. Organoid Culture of Patient-Derived PC Cells
3.3.1. PC Organoids for Screening of Gene Mutations
3.3.2. PC Organoids for Predicting Anti-Cancer Drugs Sensitivity or Resistance
3.3.3. PC Organoids as An Effective Model to Study Mechanisms of Resistance to ADT
3.3.4. PC Organoids to Study Cell of Origin of PC
4. Conclusions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S. Tobacco smoke carcinogens and lung cancer. J. Natl. Cancer Inst. 1999, 91, 1194–1210. [Google Scholar] [CrossRef] [PubMed]
- Bullock, T.L.; Andriole, G.L., Jr. Emerging drug therapies for benign prostatic hyperplasia. Expert Opin. Emerg. Drugs 2006, 11, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, S.V.; Roobol, M.J. Improving the evaluation and diagnosis of clinically significant prostate cancer in 2017. Curr. Opin. Urol. 2017, 27, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Ozguroglu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone acetate plus prednisone in patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (LATITUDE): Final overall survival analysis of a randomised, double-blind, phase 3 trial. Lancet Oncol. 2019, 20, 686–700. [Google Scholar] [CrossRef]
- Maitland, N.J. Stem cells in the normal and malignant prostate. In Prostate Cancer; Tindall, D.J., Ed.; Springer: New York, NY, USA, 2013; pp. 3–41. [Google Scholar] [CrossRef]
- Wang, Y.; Hayward, S.; Cao, M.; Thayer, K.; Cunha, G. Cell differentiation lineage in the prostate. Differ. Res. Biol. Divers. 2001, 68, 270–279. [Google Scholar] [CrossRef]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Di Santi, A.; Cernera, G.; Rossi, V.; Abbondanza, C.; Moncharmont, B.; Sinisi, A.A.; et al. Prostate cancer stem cells: The role of androgen and estrogen receptors. Oncotarget 2016, 7, 193–208. [Google Scholar] [CrossRef]
- Frame, F.M.; Maitland, N.J. Cancer stem cells, models of study and implications of therapy resistance mechanisms. In Human Cell Transformation: Role of Stem Cells and the Microenvironment; Rhim, J.S., Kremer, R., Eds.; Springer: New York, NY, USA, 2012; pp. 105–118. [Google Scholar]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Bilancio, A.; Perillo, B.; Sinisi, A.A.; Migliaccio, A.; Castoria, G. Estrogen receptors in epithelial-mesenchymal transition of prostate cancer. Cancers 2019, 1418. [Google Scholar] [CrossRef]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Castoria, G. Estrogens and their receptors in prostate cancer: Therapeutic implications. Front. Oncol. 2018, 8, 2. [Google Scholar] [CrossRef]
- Claessens, F.; Denayer, S.; Van Tilborgh, N.; Kerkhofs, S.; Helsen, C.; Haelens, A. Diverse roles of androgen receptor (AR) domains in AR-mediated signaling. Nucl. Recept. Signal. 2008, 6, e008. [Google Scholar] [CrossRef]
- Cato, A.C.; Nestl, A.; Mink, S. Rapid actions of steroid receptors in cellular signaling pathways. Sci. STKE 2002, 2002, re9. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, P.; Di Donato, M.; Giraldi, T.; Migliaccio, A.; Castoria, G.; Auricchio, F. Targeting rapid action of sex-steroid receptors in breast and prostate cancers. Front. Biosci. 2012, 4, 453–461. [Google Scholar] [CrossRef]
- Castoria, G.; Lombardi, M.; Barone, M.V.; Bilancio, A.; Di Domenico, M.; Bottero, D.; Vitale, F.; Migliaccio, A.; Auricchio, F. Androgen-Stimulated DNA synthesis and cytoskeletal changes in fibroblasts by a nontranscriptional receptor action. J. Cell Biol. 2003, 161, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Castoria, G.; D’Amato, L.; Ciociola, A.; Giovannelli, P.; Giraldi, T.; Sepe, L.; Paolella, G.; Barone, M.V.; Migliaccio, A.; Auricchio, F. Androgen-induced cell migration: Role of androgen receptor/filamin A association. PLoS ONE 2011, 6, e17218. [Google Scholar] [CrossRef]
- Cunha, G.R.; Wang, Y.Z.; Hayward, S.W.; Risbridger, G.P. Estrogenic effects on prostatic differentiation and carcinogenesis. Reprod. Fertil. Dev. 2001, 13, 285–296. [Google Scholar] [CrossRef]
- Hu, W.Y.; Shi, G.B.; Lam, H.M.; Hu, D.P.; Ho, S.M.; Madueke, I.C.; Kajdacsy-Balla, A.; Prins, G.S. Estrogen-Initiated transformation of prostate epithelium derived from normal human prostate stem-progenitor cells. Endocrinology 2011, 152, 2150–2163. [Google Scholar] [CrossRef]
- Migliaccio, A.; Castoria, G.; Di Domenico, M.; de Falco, A.; Bilancio, A.; Lombardi, M.; Barone, M.V.; Ametrano, D.; Zannini, M.S.; Abbondanza, C.; et al. Steroid-Induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000, 19, 5406–5417. [Google Scholar] [CrossRef]
- Grisanzio, C.; Signoretti, S. p63 in prostate biology and pathology. J. Cell. Biochem. 2008, 103, 1354–1368. [Google Scholar] [CrossRef]
- Berry, P.A.; Maitland, N.J.; Collins, A.T. Androgen receptor signalling in prostate: Effects of stromal factors on normal and cancer stem cells. Mol. Cell. Endocrinol. 2008, 288, 30–37. [Google Scholar] [CrossRef]
- Elbadawy, M.; Usui, T.; Mori, T.; Tsunedomi, R.; Hazama, S.; Nabeta, R.; Uchide, T.; Fukushima, R.; Yoshida, T.; Shibutani, M.; et al. Establishment of a novel experimental model for muscle-invasive bladder cancer using a dog bladder cancer organoid culture. Cancer Sci. 2019, 110, 2806–2821. [Google Scholar] [CrossRef]
- Elbadawy, M.; Yamanaka, M.; Goto, Y.; Hayashi, K.; Tsunedomi, R.; Hazama, S.; Nagano, H.; Yoshida, T.; Shibutani, M.; Ichikawa, R.; et al. Efficacy of primary liver organoid culture from different stages of non-alcoholic steatohepatitis (NASH) mouse model. Biomaterials 2020, 237, 119823. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gao, D.; Chen, Y. The potential of organoids in urological cancer research. Nat. Rev. Urol. 2017, 14, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Sanda, M.G.; Cadeddu, J.A.; Kirkby, E.; Chen, R.C.; Crispino, T.; Fontanarosa, J.; Freedland, S.J.; Greene, K.; Klotz, L.H.; Makarov, D.V.; et al. Clinically localized prostate cancer: AUA/ASTRO/SUO guideline. Part II: Recommended approaches and details of specific care options. J. Urol. 2018, 199, 990–997. [Google Scholar] [CrossRef]
- Paller, C.J.; Antonarakis, E.S.; Eisenberger, M.A.; Carducci, M.A. Management of patients with biochemical recurrence after local therapy for prostate cancer. Hematol. Oncol. Clin. N. Am. 2013, 27, 1205–1219. [Google Scholar] [CrossRef] [PubMed]
- Alemozaffar, M.; Regan, M.M.; Cooperberg, M.R.; Wei, J.T.; Michalski, J.M.; Sandler, H.M.; Hembroff, L.; Sadetsky, N.; Saigal, C.S.; Litwin, M.S.; et al. Prediction of erectile function following treatment for prostate cancer. JAMA 2011, 306, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Mullins, B.T.; Basak, R.; Broughman, J.R.; Chen, R.C. Patient-Reported sexual quality of life after different types of radical prostatectomy and radiotherapy: Analysis of a population-based prospective cohort. Cancer 2019, 125, 3657–3665. [Google Scholar] [CrossRef]
- Resnick, M.J.; Koyama, T.; Fan, K.H.; Albertsen, P.C.; Goodman, M.; Hamilton, A.S.; Hoffman, R.M.; Potosky, A.L.; Stanford, J.L.; Stroup, A.M.; et al. Long-Term functional outcomes after treatment for localized prostate cancer. N. Engl. J. Med. 2013, 368, 436–445. [Google Scholar] [CrossRef]
- Venturini, N.J.; Drake, C.G. Immunotherapy for prostate cancer. Cold Spring Harb. Perspect. Med. 2019, 9. [Google Scholar] [CrossRef]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Ozguroglu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef]
- James, N.D.; de Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.S.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. Abiraterone for prostate cancer not previously treated with hormone therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Morris, M.J.; Stadler, W.M.; Higano, C.; Basch, E.; Fizazi, K.; Antonarakis, E.S.; Beer, T.M.; Carducci, M.A.; Chi, K.N.; et al. Trial design and objectives for castration-resistant prostate cancer: Updated recommendations from the prostate cancer clinical trials working group 3. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 1402–1418. [Google Scholar] [CrossRef]
- Lau, K.M.; To, K.F. Importance of estrogenic signaling and its mediated receptors in prostate cancer. Int. J. Mol. Sci. 2016, 1434. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, A.N.; Usman, A.; Morgans, A.; VanderWeele, D.J.; Sosman, J.; Wu, J.D. Past, current, and future of immunotherapies for prostate cancer. Front. Oncol. 2019, 9, 884. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Drake, C.G.; Lipson, E.J.; Brahmer, J.R. Breathing new life into immunotherapy: Review of melanoma, lung and kidney cancer. Nat. Rev. Clin. Oncol. 2014, 11, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef]
- Solit, D.B.; Garraway, L.A.; Pratilas, C.A.; Sawai, A.; Getz, G.; Basso, A.; Ye, Q.; Lobo, J.M.; She, Y.; Osman, I.; et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006, 439, 358–362. [Google Scholar] [CrossRef]
- Horoszewicz, J.S.; Leong, S.S.; Chu, T.M.; Wajsman, Z.L.; Friedman, M.; Papsidero, L.; Kim, U.; Chai, L.S.; Kakati, S.; Arya, S.K.; et al. The LNCaP cell line—A new model for studies on human prostatic carcinoma. Prog. Clin. Biol. Res. 1980, 37, 115–132. [Google Scholar]
- Horoszewicz, J.S.; Leong, S.S.; Kawinski, E.; Karr, J.P.; Rosenthal, H.; Chu, T.M.; Mirand, E.A.; Murphy, G.P. LNCaP model of human prostatic carcinoma. Cancer Res. 1983, 43, 1809–1818. [Google Scholar]
- Kaighn, M.E.; Narayan, K.S.; Ohnuki, Y.; Lechner, J.F.; Jones, L.W. Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Investig. Urol. 1979, 17, 16–23. [Google Scholar]
- Wang, M.; Stearns, M.E. Isolation and characterization of PC-3 human prostatic tumor sublines which preferentially metastasize to select organs in S.C.I.D. mice. Differ. Res. Biol. Divers. 1991, 48, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Mickey, D.D.; Stone, K.R.; Wunderli, H.; Mickey, G.H.; Vollmer, R.T.; Paulson, D.F. Heterotransplantation of a human prostatic adenocarcinoma cell line in nude mice. Cancer Res. 1977, 37, 4049–4058. [Google Scholar] [PubMed]
- Stone, K.R.; Mickey, D.D.; Wunderli, H.; Mickey, G.H.; Paulson, D.F. Isolation of a human prostate carcinoma cell line (DU 145). Int. J. Cancer 1978, 21, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Rubin, M.A.; Putzi, M.; Mucci, N.; Smith, D.C.; Wojno, K.; Korenchuk, S.; Pienta, K.J. Rapid (“warm”) autopsy study for procurement of metastatic prostate cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 1038–1045. [Google Scholar]
- Korenchuk, S.; Lehr, J.E.; MCLean, L.; Lee, Y.G.; Whitney, S.; Vessella, R.; Lin, D.L.; Pienta, K.J. VCaP, a cell-based model system of human prostate cancer. Vivo 2001, 15, 163–168. [Google Scholar]
- Mertz, K.D.; Setlur, S.R.; Dhanasekaran, S.M.; Demichelis, F.; Perner, S.; Tomlins, S.; Tchinda, J.; Laxman, B.; Vessella, R.L.; Beroukhim, R.; et al. Molecular characterization of TMPRSS2-ERG gene fusion in the NCI-H660 prostate cancer cell line: A new perspective for an old model. Neoplasia 2007, 9, 200–206. [Google Scholar] [CrossRef]
- Marques, R.B.; van Weerden, W.M.; Erkens-Schulze, S.; de Ridder, C.M.; Bangma, C.H.; Trapman, J.; Jenster, G. The human PC346 xenograft and cell line panel: A model system for prostate cancer progression. Eur. Urol. 2006, 49, 245–257. [Google Scholar] [CrossRef]
- Sramkoski, R.M.; Pretlow, T.G., II; Giaconia, J.M.; Pretlow, T.P.; Schwartz, S.; Sy, M.S.; Marengo, S.R.; Rhim, J.S.; Zhang, D.; Jacobberger, J.W. A new human prostate carcinoma cell line, 22Rv1. In Vitro Cell. Dev. Biol. Anim. 1999, 35, 403–409. [Google Scholar] [CrossRef]
- Yoshida, T.; Kinoshita, H.; Segawa, T.; Nakamura, E.; Inoue, T.; Shimizu, Y.; Kamoto, T.; Ogawa, O. Antiandrogen bicalutamide promotes tumor growth in a novel androgen-dependent prostate cancer xenograft model derived from a bicalutamide-treated patient. Cancer Res. 2005, 65, 9611–9616. [Google Scholar] [CrossRef]
- Navone, N.M.; Olive, M.; Ozen, M.; Davis, R.; Troncoso, P.; Tu, S.M.; Johnston, D.; Pollack, A.; Pathak, S.; von Eschenbach, A.C.; et al. Establishment of two human prostate cancer cell lines derived from a single bone metastasis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1997, 3, 2493–2500. [Google Scholar]
- Zhao, X.Y.; Boyle, B.; Krishnan, A.V.; Navone, N.M.; Peehl, D.M.; Feldman, D. Two mutations identified in the androgen receptor of the new human prostate cancer cell line MDA PCa 2a. J. Urol. 1999, 162, 2192–2199. [Google Scholar] [CrossRef]
- Leister, P.; Felten, A.; Chasan, A.I.; Scheidtmann, K.H. ZIP kinase plays a crucial role in androgen receptor-mediated transcription. Oncogene 2008, 27, 3292–3300. [Google Scholar] [CrossRef] [PubMed]
- Elbadawy, M.; Usui, T.; Yamawaki, H.; Sasaki, K. novel functions of death-associated protein kinases through mitogen-activated protein kinase-related signals. Int. J. Mol. Sci. 2018, 19, 3031. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z.; Hoffmann, J.; Erdel, M.; Eder, I.E.; Hobisch, A.; Hittmair, A.; Bartsch, G.; Utermann, G.; Schneider, M.R.; Parczyk, K.; et al. Switch from antagonist to agonist of the androgen receptor bicalutamide is associated with prostate tumour progression in a new model system. Br. J. Cancer 1999, 81, 242–251. [Google Scholar] [CrossRef]
- Takayama, K.; Horie-Inoue, K.; Suzuki, T.; Urano, T.; Ikeda, K.; Fujimura, T.; Takahashi, S.; Homma, Y.; Ouchi, Y.; Inoue, S. TACC2 is an androgen-responsive cell cycle regulator promoting androgen-mediated and castration-resistant growth of prostate cancer. Mol. Endocrinol. 2012, 26, 748–761. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef]
- Vela, I.; Chen, Y. Prostate cancer organoids: A potential new tool for testing drug sensitivity. Expert Rev. Anticancer 2015, 15, 261–263. [Google Scholar] [CrossRef]
- Pretlow, T.G.; Wolman, S.R.; Micale, M.A.; Pelley, R.J.; Kursh, E.D.; Resnick, M.I.; Bodner, D.R.; Jacobberger, J.W.; Delmoro, C.M.; Giaconia, J.M.; et al. Xenografts of primary human prostatic carcinoma. J. Natl. Cancer Inst. 1993, 85, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Dagvadorj, A.; Tan, S.H.; Liao, Z.; Cavalli, L.R.; Haddad, B.R.; Nevalainen, M.T. Androgen-Regulated and highly tumorigenic human prostate cancer cell line established from a transplantable primary CWR22 tumor. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 6062–6072. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Misawa, A.; Suzuki, T.; Takagi, K.; Hayashizaki, Y.; Fujimura, T.; Homma, Y.; Takahashi, S.; Urano, T.; Inoue, S. TET2 repression by androgen hormone regulates global hydroxymethylation status and prostate cancer progression. Nat. Commun. 2015, 6, 8219. [Google Scholar] [CrossRef] [PubMed]
- Marques, R.B.; Dits, N.F.; Erkens-Schulze, S.; van Ijcken, W.F.; van Weerden, W.M.; Jenster, G. Modulation of androgen receptor signaling in hormonal therapy-resistant prostate cancer cell lines. PLoS ONE 2011, 6, e23144. [Google Scholar] [CrossRef] [PubMed]
- Marin-Aguilera, M.; Codony-Servat, J.; Kalko, S.G.; Fernandez, P.L.; Bermudo, R.; Buxo, E.; Ribal, M.J.; Gascon, P.; Mellado, B. Identification of docetaxel resistance genes in castration-resistant prostate cancer. Mol. Cancer Ther. 2012, 11, 329–339. [Google Scholar] [CrossRef]
- Takeda, M.; Mizokami, A.; Mamiya, K.; Li, Y.Q.; Zhang, J.; Keller, E.T.; Namiki, M. The establishment of two paclitaxel-resistant prostate cancer cell lines and the mechanisms of paclitaxel resistance with two cell lines. Prostate 2007, 67, 955–967. [Google Scholar] [CrossRef]
- Hongo, H.; Kosaka, T.; Oya, M. Analysis of cabazitaxel-resistant mechanism in human castration-resistant prostate cancer. Cancer Sci. 2018, 109, 2937–2945. [Google Scholar] [CrossRef]
- O’Neill, A.J.; Prencipe, M.; Dowling, C.; Fan, Y.; Mulrane, L.; Gallagher, W.M.; O’Connor, D.; O’Connor, R.; Devery, A.; Corcoran, C.; et al. Characterisation and manipulation of docetaxel resistant prostate cancer cell lines. Mol. Cancer 2011, 10, 126. [Google Scholar] [CrossRef]
- Shappell, S.B.; Thomas, G.V.; Roberts, R.L.; Herbert, R.; Ittmann, M.M.; Rubin, M.A.; Humphrey, P.A.; Sundberg, J.P.; Rozengurt, N.; Barrios, R.; et al. Prostate pathology of genetically engineered mice: Definitions and classification. The consensus report from the Bar Harbor meeting of the mouse models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res. 2004, 64, 2270–2305. [Google Scholar] [CrossRef]
- Rea, D.; Del Vecchio, V.; Palma, G.; Barbieri, A.; Falco, M.; Luciano, A.; De Biase, D.; Perdonà, S.; Facchini, G.; Arra, C. Mouse models in prostate cancer translational research: From xenograft to PDX. Biomed. Res. Int. 2016, 2016, 9750795. [Google Scholar] [CrossRef]
- Pettaway, C.A.; Pathak, S.; Greene, G.; Ramirez, E.; Wilson, M.R.; Killion, J.J.; Fidler, I.J. Selection of highly metastatic variants of different human prostatic carcinomas using orthotopic implantation in nude mice. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1996, 2, 1627–1636. [Google Scholar]
- Veldscholte, J.; Ris-Stalpers, C.; Kuiper, G.G.; Jenster, G.; Berrevoets, C.; Claassen, E.; van Rooij, H.C.; Trapman, J.; Brinkmann, A.O.; Mulder, E. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem. Biophys. Res. Commun. 1990, 173, 534–540. [Google Scholar] [CrossRef]
- Van Weerden, W.M.; de Ridder, C.M.; Verdaasdonk, C.L.; Romijn, J.C.; van der Kwast, T.H.; Schroder, F.H.; van Steenbrugge, G.J. Development of seven new human prostate tumor xenograft models and their histopathological characterization. Am. J. Pathol. 1996, 149, 1055–1062. [Google Scholar] [PubMed]
- Craft, N.; Shostak, Y.; Carey, M.; Sawyers, C.L. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat. Med. 1999, 5, 280–285. [Google Scholar] [CrossRef]
- Nemeth, J.A.; Harb, J.F.; Barroso, U., Jr.; He, Z.; Grignon, D.J.; Cher, M.L. Severe combined immunodeficient-hu model of human prostate cancer metastasis to human bone. Cancer Res. 1999, 59, 1987–1993. [Google Scholar]
- D’Antonio, J.M.; Vander Griend, D.J.; Antony, L.; Ndikuyeze, G.; Dalrymple, S.L.; Koochekpour, S.; Isaacs, J.T. Loss of androgen receptor-dependent growth suppression by prostate cancer cells can occur independently from acquiring oncogenic addiction to androgen receptor signaling. PLoS ONE 2010, 5, e11475. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, X.J.; Kundu, S.D.; Pins, M.; Javonovic, B.; Meyer, R.; Kim, S.J.; Greenberg, N.M.; Kuzel, T.; Meagher, R.; et al. Blockade of transforming growth factor-{beta} signaling in tumor-reactive CD8(+) T cells activates the antitumor immune response cycle. Mol. Cancer Ther. 2006, 5, 1733–1743. [Google Scholar] [CrossRef]
- Kopetz, S.; Lemos, R.; Powis, G. The promise of patient-derived xenografts: The best laid plans of mice and men. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 5160–5162. [Google Scholar] [CrossRef]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef]
- Wang, Y.; Revelo, M.P.; Sudilovsky, D.; Cao, M.; Chen, W.G.; Goetz, L.; Xue, H.; Sadar, M.; Shappell, S.B.; Cunha, G.R.; et al. Development and characterization of efficient xenograft models for benign and malignant human prostate tissue. Prostate 2005, 64, 149–159. [Google Scholar] [CrossRef]
- Russell, P.J.; Russell, P.; Rudduck, C.; Tse, B.W.; Williams, E.D.; Raghavan, D. Establishing prostate cancer patient derived xenografts: Lessons learned from older studies. Prostate 2015, 75, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.M.; Vessella, R.L.; Morrissey, C.; Brown, L.G.; Coleman, I.M.; Higano, C.S.; Mostaghel, E.A.; Zhang, X.; True, L.D.; Lam, H.M.; et al. LuCaP prostate cancer patient-derived xenografts reflect the molecular heterogeneity of advanced disease an—D serve as models for evaluating cancer therapeutics. Prostate 2017, 77, 654–671. [Google Scholar] [CrossRef] [PubMed]
- Navone, N.M.; van Weerden, W.M.; Vessella, R.L.; Williams, E.D.; Wang, Y.; Isaacs, J.T.; Nguyen, H.M.; Culig, Z.; van der Pluijm, G.; Rentsch, C.A.; et al. Movember GAP1 PDX project: An international collection of serially transplantable prostate cancer patient-derived xenograft (PDX) models. Prostate 2018, 78, 1262–1282. [Google Scholar] [CrossRef] [PubMed]
- Toivanen, R.; Berman, D.M.; Wang, H.; Pedersen, J.; Frydenberg, M.; Meeker, A.K.; Ellem, S.J.; Risbridger, G.P.; Taylor, R.A. Brief report: A bioassay to identify primary human prostate cancer repopulating cells. Stem Cells 2011, 29, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Vaeteewoottacharn, K.; Kariya, R. Establishment of a patient-derived tumor xenograft model and application for precision cancer medicine. Chem. Pharm. Bull. 2018, 66, 225–230. [Google Scholar] [CrossRef]
- Gittes, R.F. The nude mouse—Its use as tumor-bearing model of the prostate. Prog. Clin. Biol. Res. 1980, 37, 31–37. [Google Scholar]
- Namekawa, T.; Ikeda, K.; Horie-Inoue, K.; Inoue, S. Application of prostate cancer models for preclinical study: Advantages and limitations of cell lines, patient-derived xenografts, and three-dimensional culture of patient-derived cells. Cells 2019, 8, 74. [Google Scholar] [CrossRef]
- Van Weerden, W.M.; Bangma, C.; de Wit, R. Human xenograft models as useful tools to assess the potential of novel therapeutics in prostate cancer. Br. J. Cancer 2009, 100, 13–18. [Google Scholar] [CrossRef]
- Corey, E.; Quinn, J.E.; Buhler, K.R.; Nelson, P.S.; Macoska, J.A.; True, L.D.; Vessella, R.L. LuCaP 35: A new model of prostate cancer progression to androgen independence. Prostate 2003, 55, 239–246. [Google Scholar] [CrossRef]
- Tuxhorn, J.A.; McAlhany, S.J.; Dang, T.D.; Ayala, G.E.; Rowley, D.R. Stromal cells promote angiogenesis and growth of human prostate tumors in a differential reactive stroma (DRS) xenograft model. Cancer Res. 2002, 62, 3298–3307. [Google Scholar]
- Gao, D.; Chen, Y. Organoid development in cancer genome discovery. Curr. Opin. Genet. Dev. 2015, 30, 42–48. [Google Scholar] [CrossRef]
- Usui, T.; Sakurai, M.; Nishikawa, S.; Umata, K.; Nemoto, Y.; Haraguchi, T.; Itamoto, K.; Mizuno, T.; Noguchi, S.; Mori, T.; et al. Establishment of a dog primary prostate cancer organoid using the urine cancer stem cells. Cancer Sci. 2017, 108, 2383–2392. [Google Scholar] [CrossRef]
- Elbadawy, M.; Usui, T.; Yamawaki, H.; Sasaki, K. Emerging roles of C-Myc in cancer stem cell-related signaling and resistance to cancer chemotherapy: A potential therapeutic target against colorectal cancer. Int. J. Mol. Sci. 2019, 20, 2340. [Google Scholar] [CrossRef] [PubMed]
- Abugomaa, A.; Elbadawy, M.; Yamawaki, H.; Usui, T.; Sasaki, K. Emerging roles of cancer stem cells in bladder cancer progression, tumorigenesis, and resistance to chemotherapy: A potential therapeutic target for bladder cancer. Cells 2020, 9, 235. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Elbadawy, M.; Sasaki, K. Session 6: Advances in anticancer therapy. J. Vet. Pharmacol. Ther. 2018, 41, 28–30. [Google Scholar] [CrossRef][Green Version]
- Bartfeld, S.; Clevers, H. Stem cell-derived organoids and their application for medical research and patient treatment. J. Mol. Med. 2017, 95, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T. Organoids for drug discovery and personalized medicine. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 447–462. [Google Scholar] [CrossRef]
- Usui, T.; Sakurai, M.; Umata, K.; Elbadawy, M.; Ohama, T.; Yamawaki, H.; Hazama, S.; Takenouchi, H.; Nakajima, M.; Tsunedomi, R.; et al. Hedgehog signals mediate anti-cancer drug resistance in three-dimensional primary colorectal cancer organoid culture. Int. J. Mol. Sci. 2018, 19, 1098. [Google Scholar] [CrossRef]
- Elbadawy, M.; Usui, T.; Yamawaki, H.; Sasaki, K. Development of an experimental model for analyzing drug resistance in colorectal cancer. Cancers 2018, 10, 164. [Google Scholar] [CrossRef]
- Clevers, H. Modeling development and disease with organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef]
- Abugomaa, A.; Elbadawy, M. Patient-Derived organoid analysis of drug resistance in precision medicine: Is there a value? Expert Rev. Precis. Med. Drug Dev. 2020, 5, 1–5. [Google Scholar] [CrossRef]
- Gattazzo, F.; Urciuolo, A.; Bonaldo, P. Extracellular matrix: A dynamic microenvironment for stem cell niche. Biochim. Biophys. Acta 2014, 1840, 2506–2519. [Google Scholar] [CrossRef] [PubMed]
- Shamir, E.R.; Ewald, A.J. Three-Dimensional organotypic culture: Experimental models of mammalian biology and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 647–664. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Sachs, N.; Clevers, H. Organoid cultures for the analysis of cancer phenotypes. Curr. Opin. Genet. Dev. 2014, 24, 68–73. [Google Scholar] [CrossRef]
- Karthaus, W.R.; Iaquinta, P.J.; Drost, J.; Gracanin, A.; van Boxtel, R.; Wongvipat, J.; Dowling, C.M.; Gao, D.; Begthel, H.; Sachs, N.; et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 2014, 159, 163–175. [Google Scholar] [CrossRef]
- Ma, J.; Chang, K.; Peng, J.; Shi, Q.; Gan, H.; Gao, K.; Feng, K.; Xu, F.; Zhang, H.; Dai, B.; et al. SPOP promotes ATF2 ubiquitination and degradation to suppress prostate cancer progression. J. Exp. Clin. Cancer Res. CR 2018, 37, 145. [Google Scholar] [CrossRef]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef]
- Beshiri, M.L.; Tice, C.M.; Tran, C.; Nguyen, H.M.; Sowalsky, A.G.; Agarwal, S.; Jansson, K.H.; Yang, Q.; McGowen, K.M.; Yin, J.; et al. A PDX/organoid biobank of advanced prostate cancers captures genomic and phenotypic heterogeneity for disease modeling and therapeutic screening. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4332–4345. [Google Scholar] [CrossRef] [PubMed]
- Shoag, J.; Liu, D.; Blattner, M.; Sboner, A.; Park, K.; Deonarine, L.; Robinson, B.D.; Mosquera, J.M.; Chen, Y.; Rubin, M.A.; et al. SPOP mutation drives prostate neoplasia without stabilizing oncogenic transcription factor ERG. J. Clin. Invest. 2018, 128, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Blee, A.M.; He, Y.; Yang, Y.; Ye, Z.; Yan, Y.; Pan, Y.; Ma, T.; Dugdale, J.; Kuehn, E.; Kohli, M.; et al. TMPRSS2-ERG controls luminal epithelial lineage and antiandrogen sensitivity in PTEN and TP53-mutated prostate cancer. Clin. Cancer Res.Off. J. Am. Assoc. Cancer Res. 2018, 24, 4551–4565. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Kaochar, S.; Li, M.; Rajapakshe, K.; Fiskus, W.; Dong, J.; Foley, C.; Dong, B.; Zhang, L.; Kwon, O.J.; et al. SPOP regulates prostate epithelial cell proliferation and promotes ubiquitination and turnover of c-MYC oncoprotein. Oncogene 2017, 36, 4767–4777. [Google Scholar] [CrossRef] [PubMed]
- Blattner, M.; Liu, D.; Robinson, B.D.; Huang, D.; Poliakov, A.; Gao, D.; Nataraj, S.; Deonarine, L.D.; Augello, M.A.; Sailer, V.; et al. SPOP mutation drives prostate tumorigenesis in vivo through coordinate regulation of PI3K/mTOR and AR signaling. Cancer Cell 2017, 31, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Shen, M.M. Cell types of origin for prostate cancer. Curr. Opin. Cell Biol. 2015, 37, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lindberg, J.; Sui, G.; Luo, J.; Egevad, L.; Li, T.; Xie, C.; Wan, M.; Kim, S.T.; Wang, Z.; et al. Identification of novel CHD1-associated collaborative alterations of genomic structure and functional assessment of CHD1 in prostate cancer. Oncogene 2012, 31, 3939–3948. [Google Scholar] [CrossRef]
- Shenoy, T.R.; Boysen, G.; Wang, M.Y.; Xu, Q.Z.; Guo, W.; Koh, F.M.; Wang, C.; Zhang, L.Z.; Wang, Y.; Gil, V.; et al. CHD1 loss sensitizes prostate cancer to DNA damaging therapy by promoting error-prone double-strand break repair. Ann. Oncol. 2017, 28, 1495–1507. [Google Scholar] [CrossRef]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef]
- Scher, H.I.; Heller, G.; Molina, A.; Attard, G.; Danila, D.C.; Jia, X.; Peng, W.; Sandhu, S.K.; Olmos, D.; Riisnaes, R.; et al. Circulating tumor cell biomarker panel as an individual-level surrogate for survival in metastatic castration-resistant prostate cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1348–1355. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Gan, W.; Li, X.; Wang, S.; Zhang, W.; Huang, L.; Liu, S.; Zhong, Q.; Guo, J.; Zhang, J.; et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat. Med. 2017, 23, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Janouskova, H.; El Tekle, G.; Bellini, E.; Udeshi, N.D.; Rinaldi, A.; Ulbricht, A.; Bernasocchi, T.; Civenni, G.; Losa, M.; Svinkina, T.; et al. Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nat. Med. 2017, 23, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, D.; Zhao, Y.; Ren, S.; Gao, K.; Ye, Z.; Wang, S.; Pan, C.W.; Zhu, Y.; Yan, Y.; et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat. Med. 2017, 23, 1055–1062. [Google Scholar] [CrossRef]
- Yan, Y.; Ma, J.; Wang, D.; Lin, D.; Pang, X.; Wang, S.; Zhao, Y.; Shi, L.; Xue, H.; Pan, Y.; et al. The novel BET-CBP/p300 dual inhibitor NEO2734 is active in SPOP mutant and wild-type prostate cancer. EMBO Mol. Med. 2019, 11, e10659. [Google Scholar] [CrossRef]
- Chua, C.W.; Shibata, M.; Lei, M.; Toivanen, R.; Barlow, L.J.; Bergren, S.K.; Badani, K.K.; McKiernan, J.M.; Benson, M.C.; Hibshoosh, H.; et al. Single luminal epithelial progenitors can generate prostate organoids in culture. Nat. Cell Biol. 2014, 16, 951–961. [Google Scholar] [CrossRef]
- Floc’h, N.; Kinkade, C.W.; Kobayashi, T.; Aytes, A.; Lefebvre, C.; Mitrofanova, A.; Cardiff, R.D.; Califano, A.; Shen, M.M.; Abate-Shen, C. Dual targeting of the Akt/mTOR signaling pathway inhibits castration-resistant prostate cancer in a genetically engineered mouse model. Cancer Res. 2012, 72, 4483–4493. [Google Scholar] [CrossRef]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef]
- Pappas, K.J.; Choi, D.; Sawyers, C.L.; Karthaus, W.R. Prostate organoid cultures as tools to translate genotypes and mutational profiles to pharmacological responses. J. Vis. Exp. JoVE 2019. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef] [PubMed]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Puca, L.; Bareja, R.; Prandi, D.; Shaw, R.; Benelli, M.; Karthaus, W.R.; Hess, J.; Sigouros, M.; Donoghue, A.; Kossai, M.; et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat. Commun. 2018, 9, 2404. [Google Scholar] [CrossRef] [PubMed]
- Xin, L. Cells of origin for cancer: An updated view from prostate cancer. Oncogene 2013, 32, 3655–3663. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.A.; Shen, M.M. Revisiting the concept of cancer stem cells in prostate cancer. Oncogene 2011, 30, 1261–1271. [Google Scholar] [CrossRef]
- Vander Griend, D.J.; D’Antonio, J.; Gurel, B.; Antony, L.; Demarzo, A.M.; Isaacs, J.T. Cell-Autonomous intracellular androgen receptor signaling drives the growth of human prostate cancer initiating cells. Prostate 2010, 70, 90–99. [Google Scholar] [CrossRef]
- Humphrey, P.A. Histological variants of prostatic carcinoma and their significance. Histopathology 2012, 60, 59–74. [Google Scholar] [CrossRef]
- Goldstein, A.S.; Huang, J.; Guo, C.; Garraway, I.P.; Witte, O.N. Identification of a cell of origin for human prostate cancer. Science 2010, 329, 568–571. [Google Scholar] [CrossRef]
- Stoyanova, T.; Cooper, A.R.; Drake, J.M.; Liu, X.; Armstrong, A.J.; Pienta, K.J.; Zhang, H.; Kohn, D.B.; Huang, J.; Witte, O.N.; et al. Prostate cancer originating in basal cells progresses to adenocarcinoma propagated by luminal-like cells. Proc. Natl. Acad. Sci. USA 2013, 110, 20111–20116. [Google Scholar] [CrossRef]
- Drost, J.; Karthaus, W.R.; Gao, D.; Driehuis, E.; Sawyers, C.L.; Chen, Y.; Clevers, H. Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc. 2016, 11, 347–358. [Google Scholar] [CrossRef]
- Li, J.J.; Shen, M.M. Prostate stem cells and cancer stem cells. Cold Spring Harb. Perspect. Med. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Zong, Y.; Memarzadeh, S.; Xin, L.; Huang, J.; Witte, O.N. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 2610–2615. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kruithof-de Julio, M.; Economides, K.D.; Walker, D.; Yu, H.; Halili, M.V.; Hu, Y.P.; Price, S.M.; Abate-Shen, C.; Shen, M.M. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature 2009, 461, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Grogan, T.R.; Hieronymus, H.; Hashimoto, T.; Mottahedeh, J.; Cheng, D.; Zhang, L.; Huang, K.; Stoyanova, T.; Park, J.W.; et al. Low CD38 identifies progenitor-like inflammation-associated luminal cells that can initiate human prostate cancer and predict poor outcome. Cell Rep. 2016, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Lee, J.K.; Phillips, J.W.; Huang, P.; Cheng, D.; Huang, J.; Witte, O.N. Prostate epithelial cell of origin determines cancer differentiation state in an organoid transformation assay. Proc. Natl. Acad. Sci. USA 2016, 113, 4482–4487. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.G.; Wang, B.E.; Johnson, L.; Gao, W.Q. Generation of a prostate from a single adult stem cell. Nature 2008, 456, 804–808. [Google Scholar] [CrossRef]
- Jiao, J.; Hindoyan, A.; Wang, S.; Tran, L.M.; Goldstein, A.S.; Lawson, D.; Chen, D.; Li, Y.; Guo, C.; Zhang, B.; et al. Identification of CD166 as a surface marker for enriching prostate stem/progenitor and cancer initiating cells. PLoS ONE 2012, 7, e42564. [Google Scholar] [CrossRef]
- Wang, Z.A.; Mitrofanova, A.; Bergren, S.K.; Abate-Shen, C.; Cardiff, R.D.; Califano, A.; Shen, M.M. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat. Cell Biol. 2013, 15, 274–283. [Google Scholar] [CrossRef]
- Shibata, M.; Shen, M.M. Stem cells in genetically-engineered mouse models of prostate cancer. Endocr. Relat. Cancer 2015, 22, T199–T208. [Google Scholar] [CrossRef]
- Kwon, O.J.; Zhang, L.; Xin, L. Stem cell antigen-1 identifies a distinct androgen-independent murine prostatic luminal cell lineage with bipotent potential. Stem Cells 2016, 34, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Hynes, P.G.; Tillman, H.S.; Lake, R.; Abou-Kheir, W.G.; Fang, L.; Casey, O.M.; Ameri, A.H.; Martin, P.L.; Yin, J.J.; et al. Identification of different classes of luminal progenitor cells within prostate tumors. Cell Rep. 2015, 13, 2147–2158. [Google Scholar] [CrossRef]
- Leroy, B.E.; Northrup, N. Prostate cancer in dogs: Comparative and clinical aspects. Vet. J. 2009, 180, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, C.; Lean, F.Z.; Akter, S.H.; Romussi, S.; Grieco, V. A retrospective analysis of 111 canine prostatic samples: Histopathological findings and classification. Res. Vet. Sci. 2014, 97, 568–573. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elbadawy, M.; Abugomaa, A.; Yamawaki, H.; Usui, T.; Sasaki, K. Development of Prostate Cancer Organoid Culture Models in Basic Medicine and Translational Research. Cancers 2020, 12, 777. https://doi.org/10.3390/cancers12040777
Elbadawy M, Abugomaa A, Yamawaki H, Usui T, Sasaki K. Development of Prostate Cancer Organoid Culture Models in Basic Medicine and Translational Research. Cancers. 2020; 12(4):777. https://doi.org/10.3390/cancers12040777
Chicago/Turabian StyleElbadawy, Mohamed, Amira Abugomaa, Hideyuki Yamawaki, Tatsuya Usui, and Kazuaki Sasaki. 2020. "Development of Prostate Cancer Organoid Culture Models in Basic Medicine and Translational Research" Cancers 12, no. 4: 777. https://doi.org/10.3390/cancers12040777
APA StyleElbadawy, M., Abugomaa, A., Yamawaki, H., Usui, T., & Sasaki, K. (2020). Development of Prostate Cancer Organoid Culture Models in Basic Medicine and Translational Research. Cancers, 12(4), 777. https://doi.org/10.3390/cancers12040777