TrkB-Induced Inhibition of R-SMAD/SMAD4 Activation is Essential for TGF-β-Mediated Tumor Suppressor Activity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
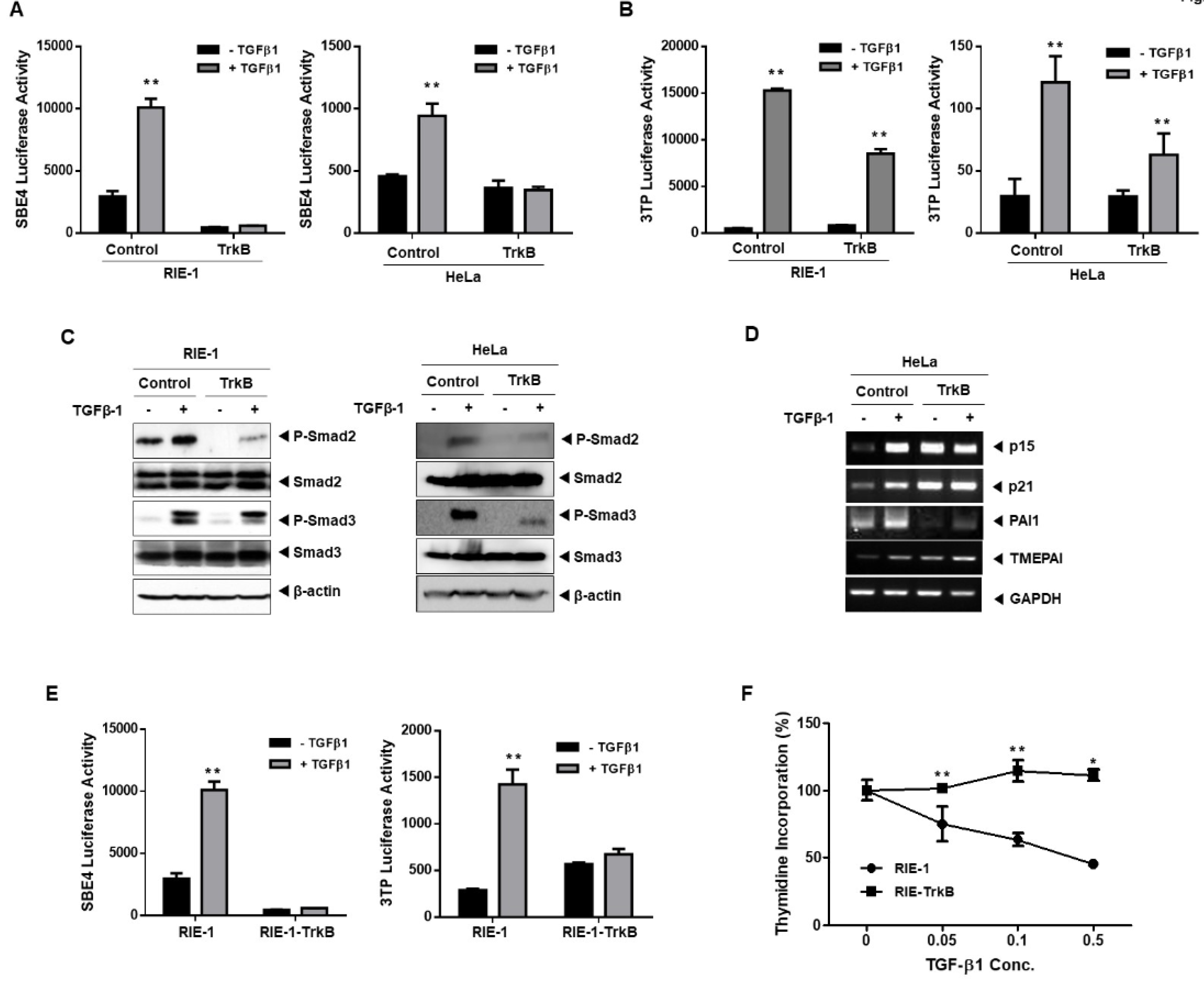
2.1. TrkB Regulates TGF-β-Mediated Tumor Suppressor Activity
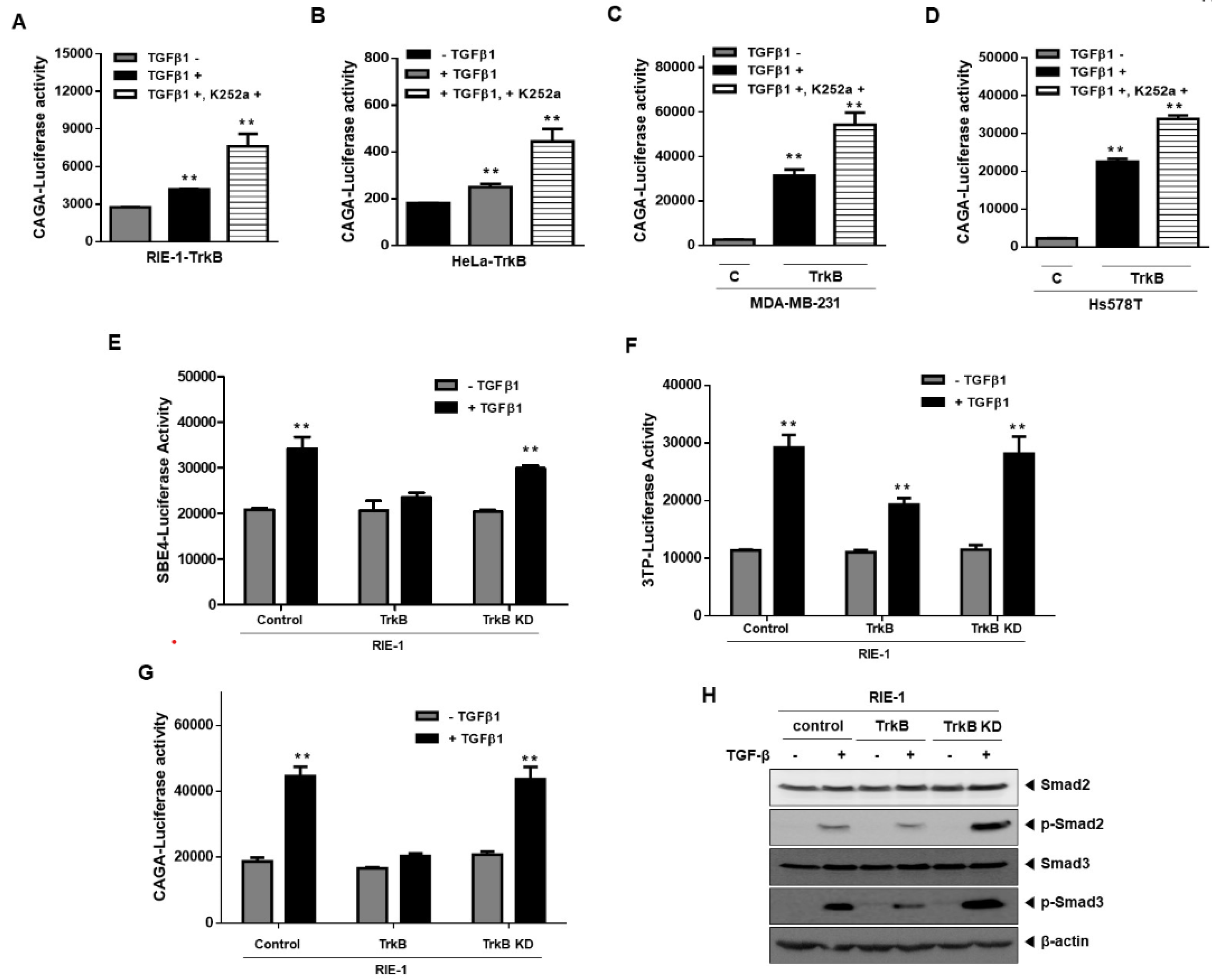
2.2. Significance of TrkB Tyrosine Kinase Activation in Inhibiting TGF-β Signaling
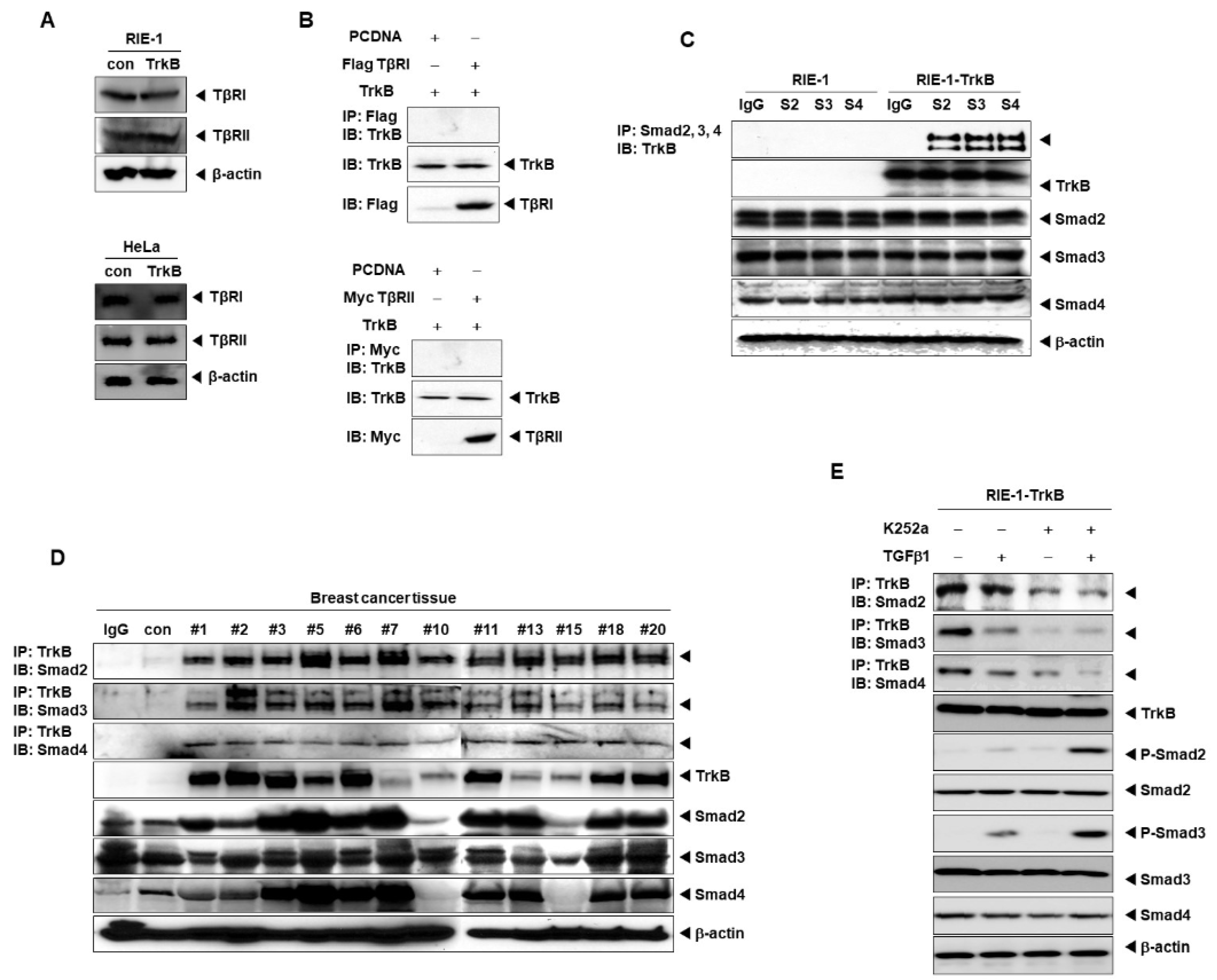
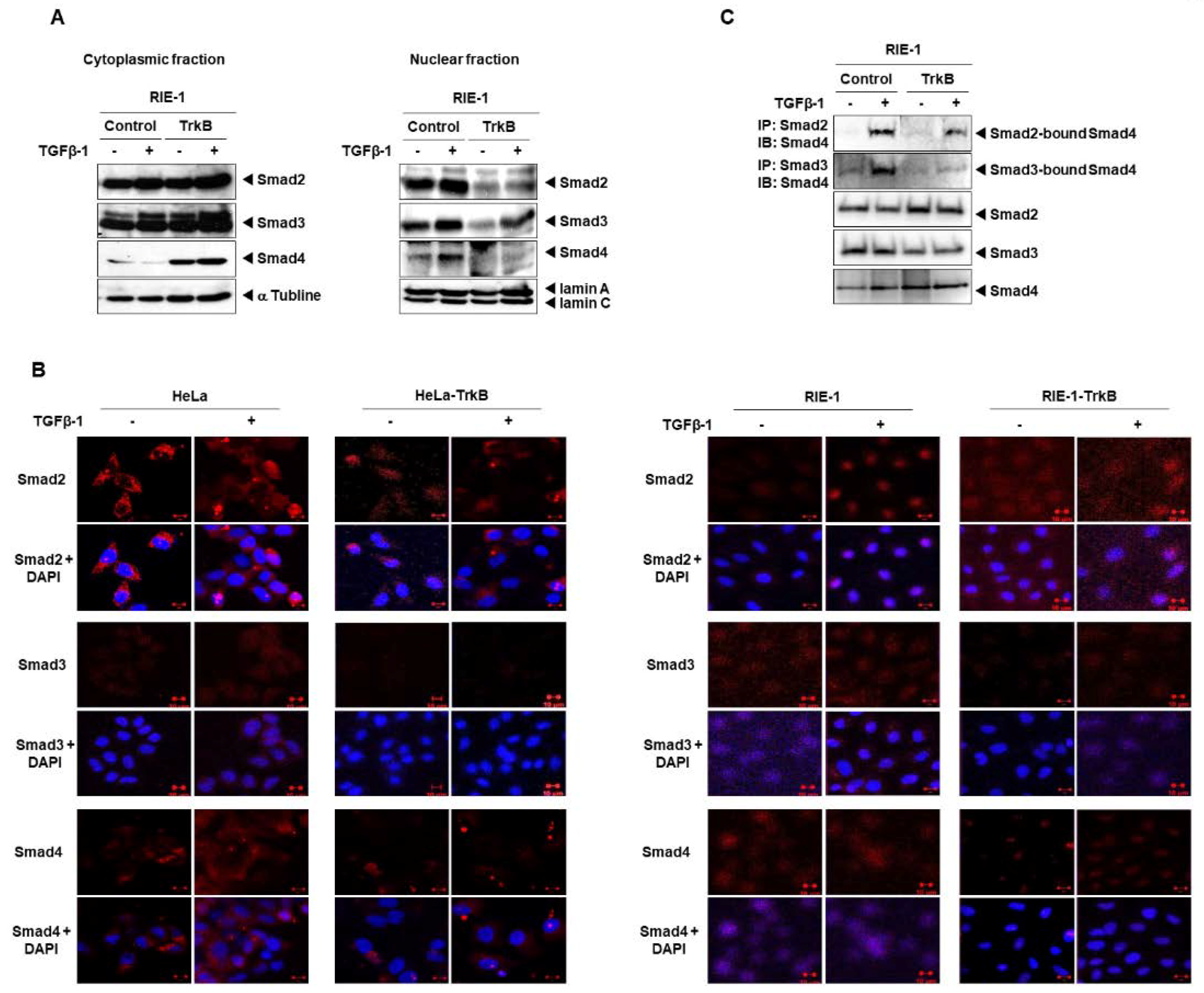
2.3. Direct Interaction between TrkB and SMADs Inhibits the TGF-β1 Signaling Pathway
2.4. TrkB Knockdown Restored TGF-β Signaling
3. Discussion
4. Material and Methods
4.1. Cell Lines, Culture Conditions and Chemical Inhibitors
4.2. Plasmids and Viral Production
4.3. Human Breast Tumor Samples
4.4. Antibodies, Western Blotting, Immunoprecipitation, and Immunofluorescence
4.5. Luciferase Reporter Assay
4.6. Thymidine Incorporation Assay
4.7. RNA Preparation and RT-PCR Analysis
4.8. Migration Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Heldin, C.H.; Vanlandewijck, M.; Moustakas, A. Regulation of EMT by TGF beta in cancer. FEBS Lett. 2012, 586, 1959–1970. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Derynck, R.; Muthusamy, B.P.; Saeteurn, K.Y. Signaling pathway cooperation in TGF-beta-induced epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 2014, 31, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Puehringer, D.; Orel, N.; Luningschror, P.; Subramanian, N.; Herrmann, T.; Chao, M.V.; Sendtner, M. EGF transactivation of Trk receptors regulates the migration of newborn cortical neurons. Nat. Neurosci. 2013, 16, 407–463. [Google Scholar] [CrossRef] [PubMed]
- Luther, J.A.; Birren, S.J. Neurotrophins and target interactions in the development and regulation of sympathetic neuron electrical and synaptic properties. Auton. Neurosci. Basic Clin. 2009, 151, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.J.; Herman, M.M.; Kleinman, J.E.; Weickert, C.S. BDNF and trkB mRNA expression in the hippocampus and temporal cortex during the human lifespan. Gene Expr. Patterns 2006, 6, 941–951. [Google Scholar] [CrossRef]
- Yeo, G.S.H.; Hung, C.C.C.; Rochford, J.; Keogh, J.; Gray, J.; Sivaramakrishnan, S.; O’Rahilly, S.; Farooqi, I.S. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat. Neurosci. 2004, 7, 1187–1189. [Google Scholar] [CrossRef]
- Gray, J.; Yeo, G.S.H.; Cox, J.J.; Morton, J.; Adlam, A.L.R.; Keogh, J.M.; Yanovski, J.A.; El Gharbawy, A.; Han, J.C.; Tung, Y.C.L.; et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes 2006, 55, 3366–3371. [Google Scholar] [CrossRef]
- Liao, G.Y.; Kinney, C.E.; An, J.J.; Xu, B.J. TrkB-expressing neurons in the dorsomedial hypothalamus are necessary and sufficient to suppress homeostatic feeding. Proc. Natl. Acad. Sci. USA 2019, 116, 3256–3261. [Google Scholar] [CrossRef]
- Wang, Z.H.; Xiang, J.; Liu, X.; Yu, S.P.; Manfredsson, F.P.; Sandoval, I.M.; Wu, S.X.; Wang, J.Z.; Ye, K.Q. Deficiency in BDNF/TrkB neurotrophic activity stimulates delta-secretase by upregulating C/EBP beta in Alzheimer’s Disease. Cell Rep. 2019, 28, 655. [Google Scholar] [CrossRef]
- Xiang, J.; Wang, Z.H.; Ahn, E.H.; Liu, X.; Yu, S.P.; Manfredsson, F.P.; Sandoval, I.M.; Ju, G.; Wu, S.X.; Ye, K.Q. Delta-secretase-cleaved Tau antagonizes TrkB neurotrophic signalings, mediating Alzheimer’s disease pathologies. Proc. Natl. Acad. Sci. USA 2019, 116, 9094–9102. [Google Scholar] [CrossRef] [PubMed]
- Couly, S.; Paucard, A.; Bonneaud, N.; Maurice, T.; Benigno, L.; Jourdan, C.; Cohen-Solal, C.; Vignes, M.; Maschat, F. Improvement of BDNF signalling by P42 peptide in Huntington’s disease. Hum. Mol. Genet. 2018, 27, 3012–3028. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Zhang, C.L.; Vincent, J.; Zala, D.; Benstaali, C.; Sainlos, M.; Grillo-Bosch, D.; Daburon, S.; Coussen, F.; Cho, Y.; et al. Modulation of AMPA receptor surface diffusion restores hippocampal plasticity and memory in Huntington’s disease models. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Zhang, Z.T.; Liu, X.; Manfredsson, F.P.; Benskey, M.J.; Cao, X.B.; Xu, J.; Sun, Y.E.; Ye, K.Q. TrkB neurotrophic activities are blocked by alpha-synuclein, triggering dopaminergic cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 10773–10778. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.X.; Huang, C.L.; Yun, W.W. Peripheral BDNF/TrkB protein expression is decreased in Parkinson’s disease but not in Essential tremor. J. Clin. Neurosci. 2019, 63, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Lee, W.S.; Jeong, J.; Kim, S.J.; Jin, W. Induction of metastatic potential by TrkB via activation of IL6/JAK2/STAT3 and PI3K/AKT signaling in breast cancer. Oncotarget 2015, 6, 40158–40171. [Google Scholar] [CrossRef]
- Kim, M.S.; Lee, W.S.; Jin, W. TrkB promotes breast cancer metastasis via suppression of Runx3 and Keap1 expression. Mol. Cells 2016, 39, 258–265. [Google Scholar] [CrossRef]
- Smit, M.A.; Geiger, T.R.; Song, J.Y.; Gitelman, I.; Peeper, D.S. A twist-snail axis critical for TrkB-induced epithelial-mesenchymal transition-like transformation, anoikis resistance, and metastasis. Mol. Cell. Biol. 2009, 29, 3722–3737. [Google Scholar] [CrossRef]
- Jin, W.; Kim, B.C.; Tognon, C.; Lee, H.J.; Patel, S.; Lannon, C.L.; Maris, J.M.; Triche, T.J.; Sorensen, P.H.B.; Kim, S.J. The ETV6-NTRK3 chimeric tyrosine kinase suppresses TGF-beta signaling by inactivating the TGF-beta type II receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 16239–16244. [Google Scholar] [CrossRef]
- Jin, W.; Yun, C.; Kwak, M.K.; Kim, T.A.; Kim, S.J. TrkC binds to the type II TGF-beta receptor to suppress TGF-beta signaling. Oncogene 2007, 26, 7684–7691. [Google Scholar] [CrossRef][Green Version]
- Feng, X.H.; Liang, Y.Y.; Liang, M.; Zhai, W.G.; Lin, X. Direct Interaction of c-Myc with Smad2 and Smad3 to Inhibit TGF-beta-Mediated Induction of the CDK Inhibitor p15(Ink4B). Mol. Cell 2016, 63, 1089. [Google Scholar] [CrossRef]
- Lee, D.K.; Kim, B.C.; Brady, J.N.; Jeang, K.T.; Kim, S.J. Human T-cell lymphotropic virus type 1 tax inhibits transforming growth factor-beta signaling by blocking the association of smad proteins with smad-binding element. J. Biol. Chem. 2002, 277, 33766–33775. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.Y.; Heller, M.; Meng, Z.; Yu, L.R.; Tang, Y.; Zhou, M.; Zhang, Y.E. Transforming Growth Factor-beta (TGF-beta) directly activates the JAK1-STAT3 axis to induce hepatic fibrosis in coordination with the SMAD pathway. J. Biol. Chem. 2017, 292, 4302–4312. [Google Scholar] [CrossRef]
- Seoane, J.; Gomis, R.R. TGF-beta family signaling in tumor suppression and cancer progression. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Sandhu, C.; Garbe, J.; Bhattacharya, N.; Daksis, J.; Pan, C.H.; Yaswen, P.; Koh, J.; Slingerland, J.M.; Stampfer, M.R. Transforming growth factor beta stabilizes p15(INK4B) protein, increases p15(INK4B)-cdk4 complexes, and inhibits cyclin D1 cdk4 association in human mammary epithelial cells. Mol. Cell. Biol. 1997, 17, 2458–2467. [Google Scholar] [CrossRef] [PubMed]
- Cipriano, S.C.; Chen, Y.Q. Insensitivity to growth inhibition by TGF-beta 1 correlates with a lack of inhibition of the CDK2 activity in prostate carcinoma cells. Oncogene 1998, 17, 1549–1556. [Google Scholar] [CrossRef]
- Hill, C.S. Transcriptional control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Macias, M.J.; Martin-Malpartida, P.; Massague, J. Structural determinants of Smad function in TGF-beta signaling. Trends Biochem. Sci. 2015, 40, 296–308. [Google Scholar] [CrossRef]
- Xu, L.; Kang, Y.B.; Col, S.; Massague, J. Smad2 nucleocytoplasmic shuttling by nucleoporins CAN/Nup214 and Nup153 feeds TGF beta signaling complexes in the cytoplasm and nucleus. Mol. Cell 2002, 10, 271–282. [Google Scholar] [CrossRef]
- Geiger, T.R.; Song, J.Y.; Rosado, A.; Peeper, D.S. Functional characterization of human cancer-derived TRKB mutations. PLoS ONE 2011, 6, e16871. [Google Scholar] [CrossRef]
- Kim, M.S.; Jeong, J.; Seo, J.; Kim, H.S.; Kim, S.J.; Jin, W. Dysregulated JAK2 expression by TrkC promotes metastasis potential, and EMT program of metastatic breast cancer. Sci Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Lee, H.S.; Kim, Y.J.; Lee, D.Y.; Kang, S.G.; Jin, W. MEST induces Twist-1-mediated EMT through STAT3 activation in breast cancers. Cell Death Differ. 2019. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.S.; Jin, W. TrkB-Induced Inhibition of R-SMAD/SMAD4 Activation is Essential for TGF-β-Mediated Tumor Suppressor Activity. Cancers 2020, 12, 1048. https://doi.org/10.3390/cancers12041048
Kim MS, Jin W. TrkB-Induced Inhibition of R-SMAD/SMAD4 Activation is Essential for TGF-β-Mediated Tumor Suppressor Activity. Cancers. 2020; 12(4):1048. https://doi.org/10.3390/cancers12041048
Chicago/Turabian StyleKim, Min Soo, and Wook Jin. 2020. "TrkB-Induced Inhibition of R-SMAD/SMAD4 Activation is Essential for TGF-β-Mediated Tumor Suppressor Activity" Cancers 12, no. 4: 1048. https://doi.org/10.3390/cancers12041048
APA StyleKim, M. S., & Jin, W. (2020). TrkB-Induced Inhibition of R-SMAD/SMAD4 Activation is Essential for TGF-β-Mediated Tumor Suppressor Activity. Cancers, 12(4), 1048. https://doi.org/10.3390/cancers12041048