Current Approaches for Combination Therapy of Cancer: The Role of Immunogenic Cell Death


 ,
,  ,
,  and
and 
Abstract
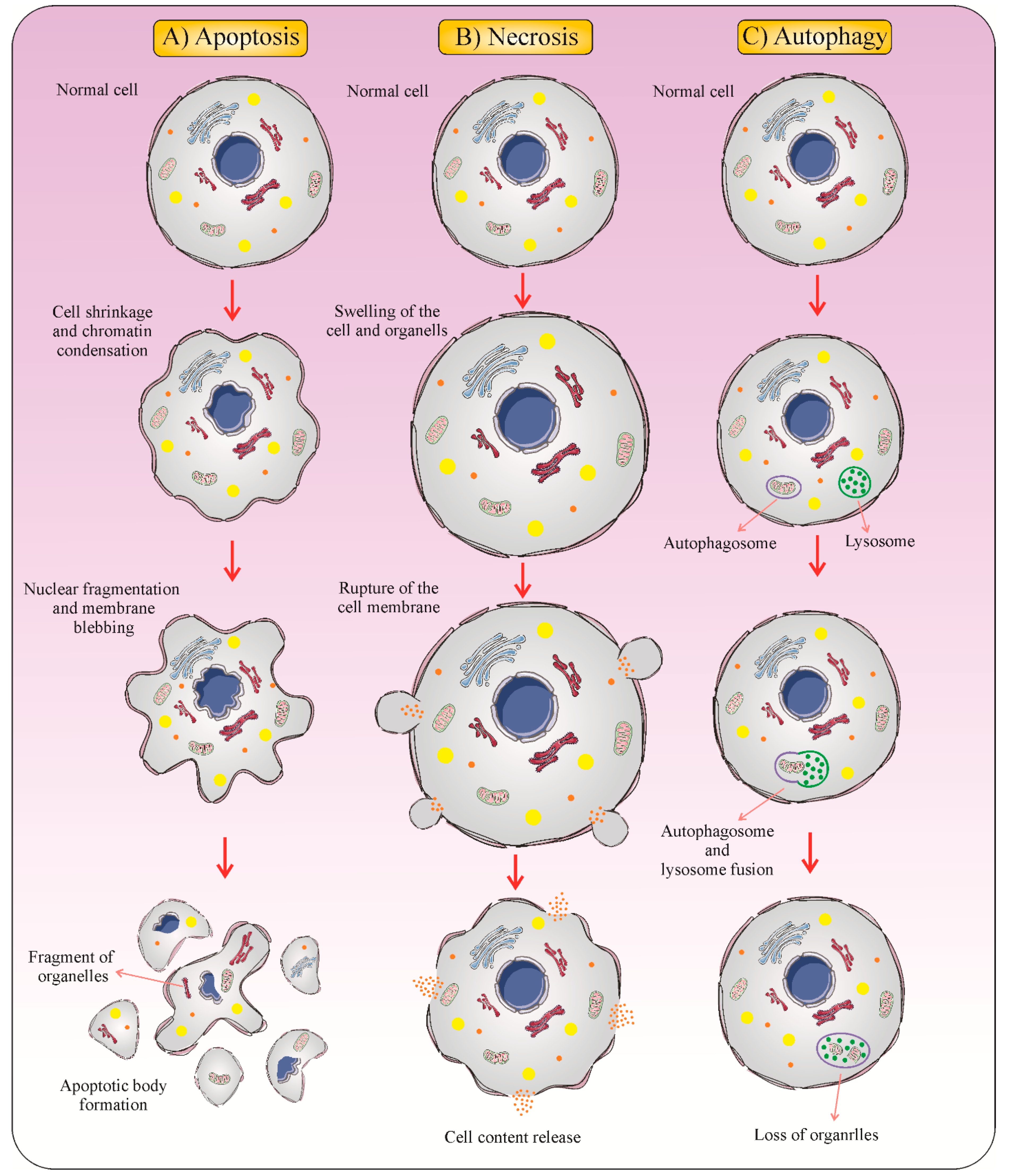
1. Introduction
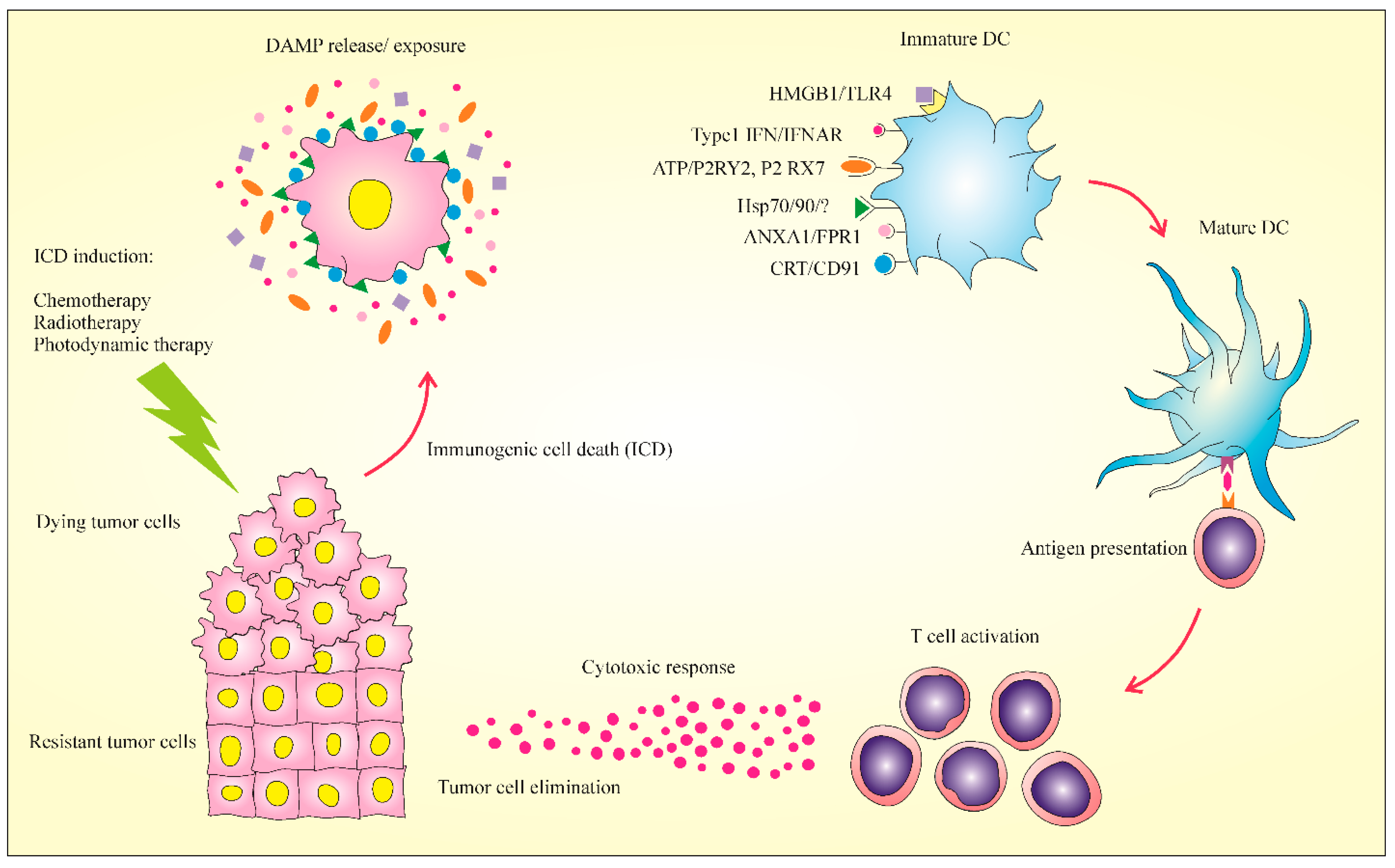
2. Immunogenic Cell Death: New Meaning in Cancer Therapy
3. Major Hallmarks of ICD
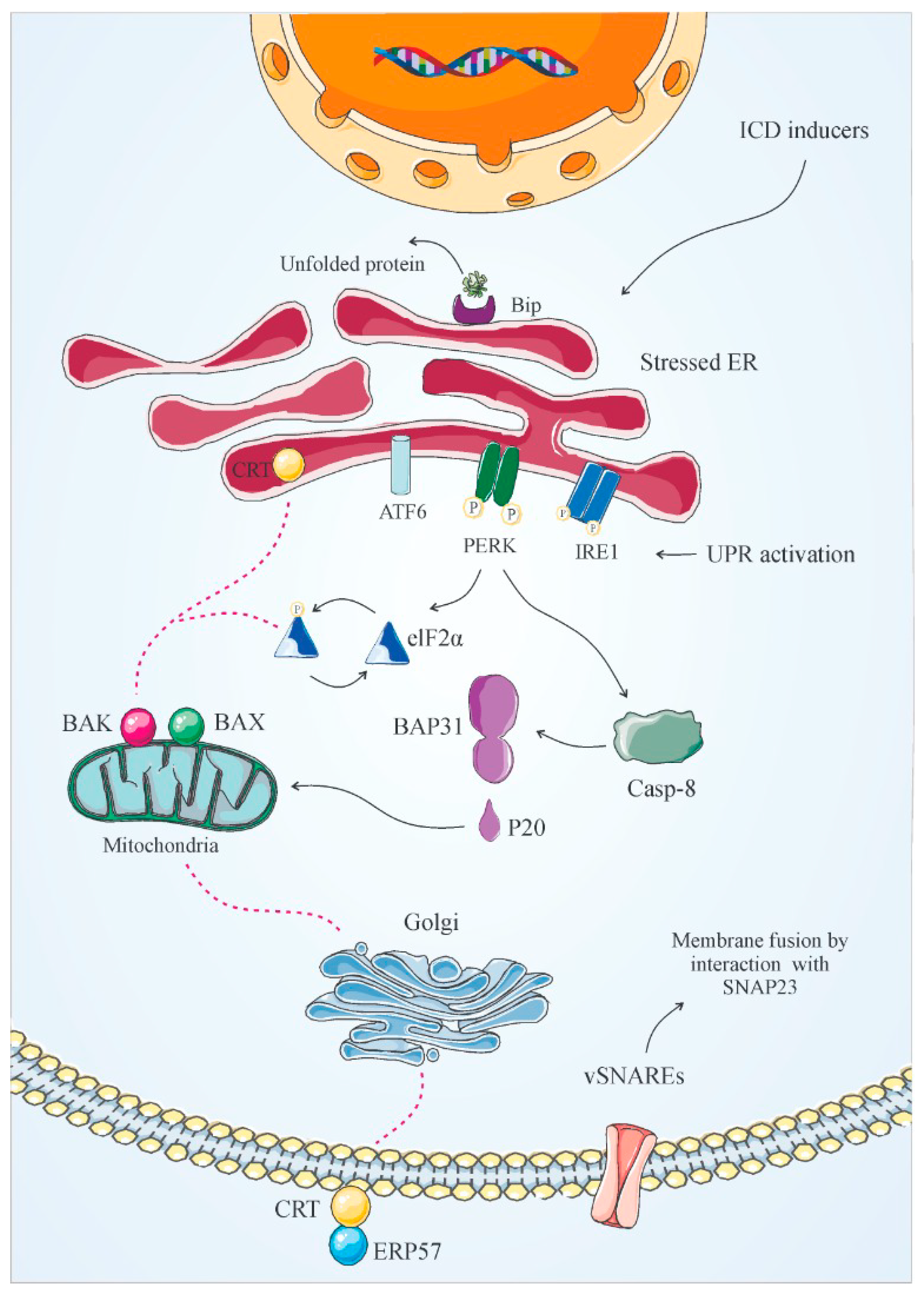
3.1. Calreticulin (CRT)
3.2. Adenosine Triphosphate (ATP)
3.3. High Mobility Group Box 1 Release (HMGB1)
3.4. Heat-Shock Proteins (HSPs)
3.5. Type I IFN
3.6. Annexin A1 (ANXA1)
4. ICD Inducers
4.1. Type I ICD Inducers
4.1.1. Anthracyclines
4.1.2. Cardiac Glycosides
4.1.3. Capsaicin (CPS)
4.1.4. Clostridium Difficile Toxin
4.1.5. High Hydrostatic Pressure (HHP)
4.1.6. Ultraviolet Light (UV)
4.2. Type II ICD Inducers
4.2.1. Photodynamic Therapy (PDT)
4.2.2. Pt-NHC
4.2.3. Coxsackievirus B3
5. Subversion of ICD
6. ICD and Combination Therapy in Cancer
6.1. Therapies Combining Chemotherapy-Induced ICD
6.2. Therapies Combining Radiotherapy-Induced ICD
6.3. Therapies Combining Photodynamic Therapy-Induced ICD
7. Conclusions
Funding
Conflicts of Interest
References
- Sambi, M.; Bagheri, L.; Szewczuk, M.R. Current challenges in cancer immunotherapy: Multimodal approaches to improve efficacy and patient response rates. J. Oncol. 2019, 2019, 4508794. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Nowis, D.; Golab, J.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Immunogenic cell death, DAMPs and anticancer therapeutics: An emerging amalgamation. Biochim. Biophys. Acta Rev. Cancer 2010, 1805, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Tesniere, A.; Panaretakis, T.; Kepp, O.; Apetoh, L.; Ghiringhelli, F.; Zitvogel, L.; Kroemer, G. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 2008, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Montico, B.; Nigro, A.; Casolaro, V.; Dal Col, J. Immunogenic Apoptosis as a Novel Tool for Anticancer Vaccine Development. Int. J. Mol. Sci. 2018, 19, 594. [Google Scholar] [CrossRef]
- Clarke, C.; Smyth, M.J. Calreticulin exposure increases cancer immunogenicity. Nat. Biotechnol. 2007, 25, 192. [Google Scholar] [CrossRef]
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 2009, 9, 353. [Google Scholar] [CrossRef]
- Krysko, D.V.; Agostinis, P.; Krysko, O.; Garg, A.D.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011, 32, 157–164. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860. [Google Scholar] [CrossRef]
- Krysko, O.; Aaes, T.L.; Bachert, C.; Vandenabeele, P.; Krysko, D. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013, 4, e631. [Google Scholar] [CrossRef]
- Panzarini, E.; Inguscio, V.; Dini, L. Immunogenic cell death: Can it be exploited in PhotoDynamic Therapy for cancer? Biomed Res. Int. 2013, 2013, 482160. [Google Scholar] [CrossRef]
- Krysko, D. Immunogenic cell death and emission of damps: Calreticulin and ATP. J. Nanomed. Biother. Discov. 2012, 2. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.; Protti, M.P. Tumor-derived factors affecting immune cells. Cytokine Growth Factor Rev. 2017, 36, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.F.; Cotter, T.G. Historical perspectives. Essays Biochem. 2003, 39, 1–10. [Google Scholar]
- Galluzzi, L.; Vitale, I.; Abrams, J.; Alnemri, E.; Baehrecke, E.; Blagosklonny, M.; Dawson, T.; Dawson, V.; El-Deiry, W.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107. [Google Scholar] [CrossRef]
- Brenner, C.; Kroemer, G. Mitochondria—the death Signal integrators. Science 2000, 289, 1150–1151. [Google Scholar] [CrossRef]
- Melino, G. The Sirens’ song. Nature 2001, 412, 23. [Google Scholar] [CrossRef]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef]
- Garrido, C.; Kroemer, G. Life’s smile, death’s grin: Vital functions of apoptosis-executing proteins. Curr. Opin. Cell Biol. 2004, 16, 639–646. [Google Scholar] [CrossRef]
- De la Rosa, E.J.; de Pablo, F. Cell death in early neural development: Beyond the neurotrophic theory. Trends Neurosci. 2000, 23, 454–458. [Google Scholar] [CrossRef]
- Savill, J.; Dransfield, I.; Gregory, C.; Haslett, C. A blast from the past: Clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2002, 2, 965. [Google Scholar] [CrossRef] [PubMed]
- Napirei, M.; Mannherz, H.G. Molecules involved in recognition and clearance of apoptotic/necrotic cells and cell debris. In Phagocytosis of Dying Cells: From Molecular Mechanisms to Human Diseases; Springer: Berlin/Heidelberg, Germany, 2009; pp. 103–145. [Google Scholar]
- Yang, Y.; Klionsky, D.J. Autophagy and disease: Unanswered questions. Cell Death Differ. 2020, 27, 858–871. [Google Scholar] [CrossRef] [PubMed]
- Vanlangenakker, N.; Berghe, T.V.; Krysko, D.V.; Festjens, N.; Vandenabeele, P. Molecular mechanisms and pathophysiology of necrotic cell death. Curr. Mol. Med. 2008, 8, 207–220. [Google Scholar] [CrossRef]
- Golstein, P.; Kroemer, G. Cell death by necrosis: Towards a molecular definition. Trends Biochem. Sci. 2007, 32, 37–43. [Google Scholar] [CrossRef]
- Vakkila, J.; Lotze, M.T. Inflammation and necrosis promote tumour growth. Nat. Rev. Immunol. 2004, 4, 641. [Google Scholar] [CrossRef]
- Hemmat, N.; Bannazadeh Baghi, H. Association of human papillomavirus infection and inflammation in cervical cancer. Pathog. Dis. 2019, 77, ftz048. [Google Scholar] [CrossRef]
- Fadok, V.A.; Bratton, D.L.; Guthrie, L.; Henson, P.M. Differential effects of apoptotic versus lysed cells on macrophage production of cytokines: Role of proteases. J. Immunol. 2001, 166, 6847–6854. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Razavi, S.M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune evasion strategies of glioblastoma. Front. Surg. 2016, 3, 11. [Google Scholar] [CrossRef]
- Preston, C.C.; Goode, E.L.; Hartmann, L.C.; Kalli, K.R.; Knutson, K.L. Immunity and immune suppression in human ovarian cancer. Immunotherapy 2011, 3, 539–556. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Tonnus, W.; Meyer, C.; Paliege, A.; Belavgeni, A.; von Mässenhausen, A.; Bornstein, S.R.; Hugo, C.; Becker, J.U.; Linkermann, A. The pathological features of regulated necrosis. J. Pathol. 2019, 247, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Showalter, A.; Limaye, A.; Oyer, J.L.; Igarashi, R.; Kittipatarin, C.; Copik, A.J.; Khaled, A.R. Cytokines in immunogenic cell death: Applications for cancer immunotherapy. Cytokine 2017, 97, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Agostinis, P. immunogenic Cell death in Cancer: From Benchside research to Bedside reality. Front. Immunol. 2016, 7, 110. [Google Scholar] [CrossRef]
- Garg, A.D.; Galluzzi, L.; Apetoh, L.; Baert, T.; Birge, R.B.; Pedro, B.S.; Manuel, J.; Breckpot, K.; Brough, D.; Chaurio, R.; et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front. Immunol. 2015, 6, 588. [Google Scholar] [CrossRef]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. Therapeutic cancer vaccines: Past, present, and future. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2013; Volume 119, pp. 421–475. [Google Scholar]
- Montico, B.; Lapenta, C.; Ravo, M.; Martorelli, D.; Muraro, E.; Zeng, B.; Comaro, E.; Spada, M.; Donati, S.; Santini, S.; et al. Exploiting a new strategy to induce immunogenic cell death to improve dendritic cell-based vaccines for lymphoma immunotherapy. OncoImmunology 2017, 6, e1356964. [Google Scholar] [CrossRef]
- Mattarollo, S.R.; Loi, S.; Duret, H.; Ma, Y.; Zitvogel, L.; Smyth, M.J. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res. 2011, 71, 4809–4820. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Rubartelli, A.; Lotze, M.T. Inside, outside, upside down: Damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007, 28, 429–436. [Google Scholar] [CrossRef]
- Radogna, F.; Diederich, M. Stress-induced cellular responses in immunogenic cell death: Implications for cancer immunotherapy. Biochem. Pharmacol. 2018, 153, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Galluzzi, L.; Martins, I.; Schlemmer, F.; Adjemian, S.; Michaud, M.; Sukkurwala, A.Q.; Menger, L.; Zitvogel, L.; Kroemer, G. Molecular determinants of immunogenic cell death elicited by anticancer chemotherapy. Cancer Metastasis Rev. 2011, 30, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Dudek, A.M.; Garg, A.D.; Krysko, D.V.; De Ruysscher, D.; Agostinis, P. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev. 2013, 24, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Mesaeli, N.; Phillipson, C. Impaired p53 expression, function, and nuclear localization in calreticulin-deficient cells. Mol. Biol. Cell 2004, 15, 1862–1870. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Corbett, E.F.; Mesaeli, N.; Nakamura, K.; Michal, O. Calreticulin: One protein, one gene, many functions. Biochem. J. 1999, 344, 281–292. [Google Scholar] [CrossRef]
- Gold, L.I.; Eggleton, P.; Sweetwyne, M.T.; Van Duyn, L.B.; Greives, M.R.; Naylor, S.M.; Michalak, M.; Murphy-Ullrich, J.E. Calreticulin: Non-endoplasmic reticulum functions in physiology and disease. FASEB J. 2010, 24, 665–683. [Google Scholar] [CrossRef]
- Obeid, M.; Panaretakis, T.; Tesniere, A.; Joza, N.; Tufi, R.; Apetoh, L.; Ghiringhelli, F.; Zitvogel, L.; Kroemer, G. Leveraging the immune system during chemotherapy: Moving calreticulin to the cell surface converts apoptotic death from “silent” to immunogenic. Cancer Res. 2007, 67, 7941–7944. [Google Scholar] [CrossRef]
- Chao, M.P.; Jaiswal, S.; Weissman-Tsukamoto, R.; Alizadeh, A.A.; Gentles, A.J.; Volkmer, J.; Weiskopf, K.; Willingham, S.B.; Raveh, T.; Park, C.Y. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci. Transl. Med. 2010, 2, 63ra94. [Google Scholar] [CrossRef]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54. [Google Scholar] [CrossRef]
- Serrano-del Valle, A.; Anel, A.; Naval, J.; Marzo, I. Immunogenic cell death and immunotherapy of multiple myeloma. Front. Cell Dev. Biol. 2019, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Rufo, N.; Garg, A.D.; Agostinis, P. The unfolded protein response in immunogenic cell death and cancer immunotherapy. Trends Cancer 2017, 3, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; Van Endert, P.; et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Leclair, P.; Pedari, F.; Vieira, H.; Monajemi, M.; Sly, L.M.; Reid, G.S.; Lim, C.J. Integrins and ERp57 coordinate to regulate cell surface calreticulin in immunogenic cell death. Front. Oncol. 2019, 9, 411. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Kepp, O.; Schlemmer, F.; Adjemian, S.; Tailler, M.; Shen, S.; Michaud, M.; Menger, L.; Gdoura, A.; Tajeddine, N.; et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene 2011, 30, 1147. [Google Scholar] [CrossRef]
- Müller-Taubenberger, A.; Lupas, A.N.; Li, H.; Ecke, M.; Simmeth, E.; Gerisch, G. Calreticulin and calnexin in the endoplasmic reticulum are important for phagocytosis. EMBO J. 2001, 20, 6772–6782. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Panaretakis, T.; Tufi, R.; Joza, N.; Van Endert, P.; Ghiringhelli, F.; Apetoh, L.; Chaput, N.; Flament, C.; et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol. Rev. 2007, 220, 22–34. [Google Scholar] [CrossRef]
- Stoll, G.; Iribarren, K.; Michels, J.; Leary, A.; Zitvogel, L.; Cremer, I.; Kroemer, G. Calreticulin expression: Interaction with the immune infiltrate and impact on survival in patients with ovarian and non-small cell lung cancer. Oncoimmunology 2016, 5, e1177692. [Google Scholar] [CrossRef]
- Colangelo, T.; Polcaro, G.; Ziccardi, P.; Muccillo, L.; Galgani, M.; Pucci, B.; Milone, M.R.; Budillon, A.; Santopaolo, M.; Mazzoccoli, G.; et al. The miR-27a-calreticulin axis affects drug-induced immunogenic cell death in human colorectal cancer cells. Cell Death Dis. 2017, 7, e2108. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Shirjang, S.; Baradaran, B. MicroRNAs in the diagnosis and treatment of cancer. Immunol. Investig. 2017, 46, 880–897. [Google Scholar] [CrossRef]
- Burris, H.R.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Christine Cripps, M.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.; et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Martins, I.; Ma, Y.; Kepp, O.; Galluzzi, L.; Kroemer, G. Autophagy-dependent ATP release from dying cells via lysosomal exocytosis. Autophagy 2013, 9, 1624–1625. [Google Scholar] [CrossRef]
- Martins, I.; Wang, Y.; Michaud, M.; Ma, Y.; Sukkurwala, A.; Shen, S.; Kepp, O.; Metivier, D.; Galluzzi, L.; Perfettini, J.; et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 2014, 21, 79. [Google Scholar] [CrossRef]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β–dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170. [Google Scholar] [CrossRef]
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Catani, J.P.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013, 38, 729–741. [Google Scholar] [CrossRef]
- La Sala, A.; Sebastiani, S.; Ferrari, D.; Di Virgilio, F.; Idzko, M.; Norgauer, J.; Girolomoni, G. Dendritic cells exposed to extracellular adenosine triphosphate acquire the migratory properties of mature cells and show a reduced capacity to attract type 1 T lymphocytes. Blood 2002, 99, 1715–1722. [Google Scholar] [CrossRef]
- Zhang, Y.; Gong, L.H.; Zhang, H.Q.; Du, Q.; You, J.F.; Tian, X.X.; Fang, W.G. Extracellular ATP enhances in vitro invasion of prostate cancer cells by activating Rho GTPase and upregulating MMPs expression. Cancer Lett. 2010, 293, 189–197. [Google Scholar] [CrossRef]
- Idzko, M.; Hammad, H.; Van Nimwegen, M.; Kool, M.; Willart, M.A.; Muskens, F.; Hoogsteden, H.C.; Luttmann, W.; Ferrari, D.; Di Virgilio, F.; et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat. Med. 2007, 13, 913. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.G.; Yan, Z.; et al. HMGB1 in health and disease. Mol. Asp. Med. 2014, 40, 1–116. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Tang, D.; Kang, R. Oxidative stress-mediated HMGB1 biology. Front. Physiol. 2015, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, R.; Sampaolesi, M.; De Marchis, F.; Tonlorenzi, R.; Colombetti, S.; Mondino, A.; Cossu, G.; Bianchi, M.E. Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J. Cell Biol. 2004, 164, 441–449. [Google Scholar] [CrossRef]
- Guazzi, S.; Strangio, A.; Franzi, A.T.; Bianchi, M.E. HMGB1, an architectural chromatin protein and extracellular signalling factor, has a spatially and temporally restricted expression pattern in mouse brain. Gene Expr. Patterns 2003, 3, 29–33. [Google Scholar] [CrossRef]
- Vaccari, T.; Beltrame, M.; Ferrari, S.; Bianchi, M. Hmg4, a New Member of theHmg1/2Gene Family. Genomics 1998, 49, 247–252. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191. [Google Scholar] [CrossRef]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 2000, 192, 565–570. [Google Scholar] [CrossRef]
- Tesniere, A.; Apetoh, L.; Ghiringhelli, F.; Joza, N.; Panaretakis, T.; Kepp, O.; Schlemmer, F.; Zitvogel, L.; Kroemer, G. Immunogenic cancer cell death: A key-lock paradigm. Curr. Opin. Immunol. 2008, 20, 504–511. [Google Scholar] [CrossRef]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid–mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832. [Google Scholar] [CrossRef]
- Yang, H.; Hreggvidsdottir, H.S.; Palmblad, K.; Wang, H.; Ochani, M.; Li, J.; Lu, B.; Chavan, S.; Rosas-Ballina, M.; Al-Abed, Y.; et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. USA 2010, 107, 11942–11947. [Google Scholar] [CrossRef] [PubMed]
- Dumitriu, I.E.; Baruah, P.; Valentinis, B.; Voll, R.E.; Herrmann, M.; Nawroth, P.P.; Arnold, B.; Bianchi, M.E.; Manfredi, A.A.; Rovere-Querini, P. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J. Immunol. 2005, 174, 7506–7515. [Google Scholar] [CrossRef] [PubMed]
- Saenz, R.; Futalan, D.; Leutenez, L.; Eekhout, F.; Fecteau, J.F.; Sundelius, S.; Sundqvist, S.; Larsson, M.; Hayashi, T.; Minev, B. TLR4-dependent activation of dendritic cells by an HMGB1-derived peptide adjuvant. J. Transl. Med. 2014, 12, 211. [Google Scholar] [CrossRef] [PubMed]
- Lanneau, D.; Brunet, M.; Frisan, E.; Solary, E.; Fontenay, M.; Garrido, C. Heat shock proteins: Essential proteins for apoptosis regulation. J. Cell. Mol. Med. 2008, 12, 743–761. [Google Scholar] [CrossRef]
- Joly, A.L.; Wettstein, G.; Mignot, G.; Ghiringhelli, F.; Garrido, C. Dual role of heat shock proteins as regulators of apoptosis and innate immunity. J. Innate Immun. 2010, 2, 238–247. [Google Scholar] [CrossRef]
- Schlesinger, M.J. Heat shock proteins. J. Biol. Chem. 1990, 265, 12111–12114. [Google Scholar]
- Schmitt, E.; Gehrmann, M.; Brunet, M.; Multhoff, G.; Garrido, C. Intracellular and extracellular functions of heat shock proteins: Repercussions in cancer therapy. J. Leukoc. Biol. 2007, 81, 15–27. [Google Scholar] [CrossRef]
- Parcellier, A.; Gurbuxani, S.; Schmitt, E.; Solary, E.; Garrido, C. Heat shock proteins, cellular chaperones that modulate mitochondrial cell death pathways. Biochem. Biophys. Res. Commun. 2003, 304, 505–512. [Google Scholar] [CrossRef]
- Wang, R.; Town, T.; Gokarn, V.; Flavell, R.A.; Chandawarkar, R.Y. HSP70 enhances macrophage phagocytosis by interaction with lipid raft-associated TLR-7 and upregulating p38 MAPK and PI3K pathways. J. Surg. Res. 2006, 136, 58–69. [Google Scholar] [CrossRef]
- Lauber, K.; Brix, N.; Ernst, A.; Hennel, R.; Krombach, J.; Anders, H.; Belka, C. Targeting the heat shock response in combination with radiotherapy: Sensitizing cancer cells to irradiation-induced cell death and heating up their immunogenicity. Cancer Lett. 2015, 368, 209–229. [Google Scholar] [CrossRef]
- Spisek, R.; Dhodapkar, M. Towards a better way to die with chemotherapy: Role of heat shock protein exposure on dying tumor cells. Cell Cycle 2007, 6, 1962–1965. [Google Scholar] [CrossRef] [PubMed]
- Udono, H.; Srivastava, P.K. Comparison of tumor-specific immunogenicities of stress-induced proteins gp96, hsp90, and hsp70. J. Immunol. 1994, 152, 5398–5403. [Google Scholar] [PubMed]
- Bartůňková, J.; Špíšek, R. Impact of tumour cell death on the activation of anti-tumour immune response. In Phagocytosis of Dying Cells: From Molecular Mechanisms to Human Diseases; Springer: Berlin/Heidelberg, Germany, 2009; pp. 347–370. [Google Scholar]
- Schild, H.; Arnold-Schild, D.; Lammert, E.; Rammensee, H.G. Stress proteins and immunity mediated by cytotoxic T lymphocytes. Curr. Opin. Immunol. 1999, 11, 109–113. [Google Scholar] [CrossRef]
- Doody, A.D.; Kovalchin, J.T.; Mihalyo, M.A.; Hagymasi, A.T.; Drake, C.G.; Adler, A.J. Glycoprotein 96 can chaperone both MHC class I-and class II-restricted epitopes for in vivo presentation, but selectively primes CD8+ T cell effector function. J. Immunol. 2004, 172, 6087–6092. [Google Scholar] [CrossRef] [PubMed]
- Millward, M.; Underhill, C.; Lobb, S.; McBurnie, J.; Meech, S.; Gomez-Navarro, J.; Marshall, M.; Huang, B.; Mather, C. Phase I study of tremelimumab (CP-675 206) plus PF-3512676 (CPG 7909) in patients with melanoma or advanced solid tumours. Br. J. Cancer 2013, 108, 1998. [Google Scholar] [CrossRef] [PubMed]
- Tarhini, A.A.; Cherian, J.; Moschos, S.J.; Tawbi, H.A.; Shuai, Y.; Gooding, W.E.; Sander, C.; Kirkwood, J.M. Safety and efficacy of combination immunotherapy with interferon alfa-2b and tremelimumab in patients with stage IV melanoma. J. Clin. Oncol. 2012, 30, 322. [Google Scholar] [CrossRef]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer cell–autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301. [Google Scholar] [CrossRef]
- Vacchelli, E.; Sistigu, A.; Yamazaki, T.; Vitale, I.; Zitvogel, L.; Kroemer, G. Autocrine signaling of type 1 interferons in successful anticancer chemotherapy. Oncoimmunology 2015, 4, e988042. [Google Scholar]
- Melero, I.; Quetglas, J.I.; Reboredo, M.; Dubrot, J.; Rodriguez-Madoz, J.R.; Mancheño, U.; Casales, E.; Riezu-Boj, J.I.; Ruiz-Guillen, M.; Ochoa, M.C.; et al. Strict requirement for vector-induced type I interferon in efficacious antitumor responses to virally encoded IL12. Cancer Res. 2015, 75, 497–507. [Google Scholar] [CrossRef]
- Wang, X.; Schoenhals, J.E.; Li, A.; Valdecanas, D.R.; Ye, H.; Zang, F.; Tang, C.; Tang, M.; Liu, C.G.; Liu, X.; et al. Suppression of type I IFN signaling in tumors mediates resistance to anti-PD-1 treatment that can be overcome by radiotherapy. Cancer Res. 2017, 77, 839–850. [Google Scholar] [CrossRef]
- Perretti, M.; D’acquisto, F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 2009, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Teijeiro, S.; Menéndez, S.T.; Villaronga, M.Á.; Pena-Alonso, E.; Rodrigo, J.P.; Morgan, R.O.; Granda-Díaz, R.; Salom, C.; Fernandez, M.P.; García-Pedrero, J.M. Annexin A1 down-regulation in head and neck squamous cell carcinoma is mediated via transcriptional control with direct involvement of miR-196a/b. Sci. Rep. 2017, 7, 6790. [Google Scholar] [CrossRef] [PubMed]
- Pupjalis, D.; Goetsch, J.; Kottas, D.J.; Gerke, V.; Rescher, U. Annexin A1 released from apoptotic cells acts through formyl peptide receptors to dampen inflammatory monocyte activation via JAK/STAT/SOCS signalling. EMBO Mol. Med. 2011, 3, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, B.; Giacomantonio, M.; Marcato, P.; McFarland, S.; Pol, J.; Gujar, S. Dying to Be noticed: Epigenetic Regulation of immunogenic Cell Death for Cancer immunotherapy. Front. Immunol. 2018, 9, 654. [Google Scholar] [CrossRef]
- Vacchelli, E.; Ma, Y.; Baracco, E.E.; Sistigu, A.; Enot, D.P.; Pietrocola, F.; Yang, H.; Adjemian, S.; Chaba, K.; Semeraro, M.; et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 2015, 350, 972–978. [Google Scholar] [CrossRef]
- Tsai, K.W.; Hu, L.Y.; Wu, C.W.; Li, S.C.; Lai, C.H.; Kao, H.W.; Fang, W.L.; Lin, W.C. Epigenetic regulation of miR-196b expression in gastric cancer. GenesChromosomes Cancer 2010, 49, 969–980. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. The emergence of phox-ER stress induced immunogenic apoptosis. Oncoimmunology 2012, 1, 786–788. [Google Scholar] [CrossRef]
- Rojas-Rivera, D.; Rodriguez, D.A.; Sepulveda, D.; Hetz, C. ER stress sensing mechanism: Putting off the brake on UPR transducers. Oncotarget 2018, 9, 19461. [Google Scholar] [CrossRef]
- Mori, K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 2000, 101, 451–454. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. Signaling the unfolded protein response from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 25935–25938. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2007; pp. 716–731. [Google Scholar]
- Adkins, I.; Fucikova, J.; Garg, A.D.; Agostinis, P.; Špíšek, R. Physical modalities inducing immunogenic tumor cell death for cancer immunotherapy. Oncoimmunology 2014, 3, e968434. [Google Scholar] [CrossRef]
- Menger, L.; Vacchelli, E.; Adjemian, S.; Martins, I.; Ma, Y.; Shen, S.; Yamazaki, T.; Sukkurwala, A.Q.; Michaud, M.; Mignot, G.; et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci. Transl. Med. 2012, 4, 143ra99. [Google Scholar] [CrossRef] [PubMed]
- D’Eliseo, D.; Manzi, L.; Velotti, F. Capsaicin as an inducer of damage-associated molecular patterns (DAMPs) of immunogenic cell death (ICD) in human bladder cancer cells. Cell Stress Chaperones 2013, 18, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, H.; Mao, S.; Liu, J.; Li, S.; Wang, J. Reactive oxygen species involved in CT26 immunogenic cell death induced by Clostridium difficile toxin B. Immunol. Lett. 2015, 164, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.; Janko, C.; Ebel, N.; Meister, S.; Schlucker, E.; Meyer-Pittroff, R.; Fietkau, R.; Herrmann, M.; Gaipl, U.S. Cells under pressure-treatment of eukaryotic cells with high hydrostatic pressure, from physiologic aspects to pressure induced cell death. Curr. Med. Chem. 2008, 15, 2329–2336. [Google Scholar] [CrossRef]
- Brusa, D.; Migliore, E.; Garetto, S.; Simone, M.; Matera, L. Immunogenicity of 56 °C and UVC-treated prostate cancer is associated with release of HSP70 and HMGB1 from necrotic cells. Prostate 2009, 69, 1343–1352. [Google Scholar] [CrossRef]
- Wong, D.Y.Q.; Ong, W.W.F.; Ang, W.H. Induction of immunogenic cell death by chemotherapeutic platinum complexes. Angew. Chem. Int. Ed. 2015, 54, 6483–6487. [Google Scholar] [CrossRef]
- Sahin, T.T.; Kasuya, H.; Nomura, N.; Shikano, T.; Yamamura, K.; Gewen, T.; Kanzaki, A.; Fujii, T.; Sugae, T.; Imai, T.; et al. Impact of novel oncolytic virus HF10 on cellular components of the tumor microenviroment in patients with recurrent breast cancer. Cancer Gene Ther. 2012, 19, 229. [Google Scholar] [CrossRef]
- Miyamoto, S.; Inoue, H.; Nakamura, T.; Yamada, M.; Sakamoto, C.; Urata, Y.; Okazaki, T.; Marumoto, T.; Takahashi, A.; Takayama, K.; et al. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res. 2012, 72, 2609–2621. [Google Scholar] [CrossRef]
- Apetoh, L.; Mignot, G.; Panaretakis, T.; Kroemer, G.; Zitvogel, L. Immunogenicity of anthracyclines: Moving towards more personalized medicine. Trends Mol. Med. 2008, 14, 141–151. [Google Scholar] [CrossRef]
- Tewey, K.; Rowe, T.; Yang, L.; Halligan, B.; Liu, L. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984, 226, 466–468. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Matsushima, H.; Mizumoto, N.; Takashima, A. Classification of chemotherapeutic agents based on their differential in vitro effects on dendritic cells. Cancer Res. 2009, 69, 6978–6986. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Kaczmarek, A.; Krysko, O.; Heyndrickx, L.; Woznicki, J.; Bogaert, P.; Cauwels, A.; Takahashi, N.; Magez, S.; Bachert, C.; et al. TLR-2 and TLR-9 are sensors of apoptosis in a mouse model of doxorubicin-induced acute inflammation. Cell Death Differ. 2011, 18, 1316. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Reutelingsperger, C.; McGahon, A.J.; Rader, J.A.; Van Schie, R.; LaFace, D.M.; Green, D.R. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: Inhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 1995, 182, 1545–1556. [Google Scholar] [CrossRef]
- Martins, I.; Tesniere, A.; Kepp, O.; Michaud, M.; Schlemmer, F.; Senovilla, L.; Séror, C.; Métivier, D.; Perfettini, J.L.; Zitvogel, L.; et al. Chemotherapy induces ATP release from tumor cells. Cell Cycle 2009, 8, 3723–3728. [Google Scholar] [CrossRef]
- Ma, Y.; Aymeric, L.; Locher, C.; Mattarollo, S.R.; Delahaye, N.F.; Pereira, P.; Boucontet, L.; Apetoh, L.; Ghiringhelli, F.; Casares, N.; et al. Contribution of IL-17–producing γδ T cells to the efficacy of anticancer chemotherapy. J. Exp. Med. 2011, 208, 491–503. [Google Scholar] [CrossRef]
- Riganti, C.; Campia, I.; Kopecka, J.; Gazzano, E.; Doublier, S.; Aldieri, E.; Bosia, A.; Ghigo, D. Pleiotropic effects of cardioactive glycosides. Curr. Med. Chem. 2011, 18, 872–885. [Google Scholar] [CrossRef]
- Prassas, I.; Diamandis, E.P. Novel therapeutic applications of cardiac glycosides. Nat. Rev. Drug Discov. 2008, 7, 926. [Google Scholar] [CrossRef]
- Szallasi, A.; Blumberg, P.M. Vanilloid (capsaicin) receptors and mechanisms. Pharmacol. Rev. 1999, 51, 159–212. [Google Scholar] [PubMed]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816. [Google Scholar] [CrossRef] [PubMed]
- Bíró, T.; Brodie, C.; Modarres, S.; Lewin, N.E.; Ács, P.; Blumberg, P.M. Specific vanilloid responses in C6 rat glioma cells. Mol. Brain Res. 1998, 56, 89–98. [Google Scholar] [CrossRef]
- Bley, K.; Boorman, G.; Mohammad, B.; McKenzie, D.; Babbar, S. A comprehensive review of the carcinogenic and anticarcinogenic potential of capsaicin. Toxicol. Pathol. 2012, 40, 847–873. [Google Scholar] [CrossRef]
- Beltran, J.; Ghosh, A.K.; Basu, S. Immunotherapy of tumors with neuroimmune ligand capsaicin. J. Immunol. 2007, 178, 3260–3264. [Google Scholar] [CrossRef]
- Athanasiou, A.; Smith, P.A.; Vakilpour, S.; Kumaran, N.M.; Turner, A.E.; Bagiokou, D.; Layfield, R.; Ray, D.E.; Westwell, A.D.; Alexander, S.P.; et al. Vanilloid receptor agonists and antagonists are mitochondrial inhibitors: How vanilloids cause non-vanilloid receptor mediated cell death. Biochem. Biophys. Res. Commun. 2007, 354, 50–55. [Google Scholar] [CrossRef]
- Zhang, R.; Humphreys, I.; Sahu, R.P.; Shi, Y.; Srivastava, S.K. In vitro and in vivo induction of apoptosis by capsaicin in pancreatic cancer cells is mediated through ROS generation and mitochondrial death pathway. Apoptosis 2008, 13, 1465–1478. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Kee, K.H.; Suh, C.H.; Lim, S.C.; Oh, S.H. Capsaicin-induced apoptosis is regulated by endoplasmic reticulum stress-and calpain-mediated mitochondrial cell death pathways. Toxicology 2009, 264, 205–214. [Google Scholar] [CrossRef]
- Yang, K.M.; Pyo, J.O.; Kim, G.Y.; Yu, R.; Han, I.S.; Ju, S.A.; Kim, W.H.; Kim, B.S. Capsaicin induces apoptosis by generating reactive oxygen species and disrupting mitochondrial transmembrane potential in human colon cancer cell lines. Cell. Mol. Biol. Lett. 2009, 14, 497. [Google Scholar] [CrossRef]
- Buzzi, S.; Buzzi, L. Cancer immunity after treatment of Ehrlich tumor with diphtheria toxin. Cancer Res. 1974, 34, 3481–3486. [Google Scholar]
- Bekesi, J.G.; St-Arneault, G.; Holland, J. Increase of leukemia L1210 immunogenicity by Vibrio cholerae neuraminidase treatment. Cancer Res. 1971, 31, 2130–2132. [Google Scholar] [PubMed]
- Matarrese, P.; Falzano, L.; Fabbri, A.; Gambardella, L.; Frank, C.; Geny, B.; Popoff, M.R.; Malorni, W.; Fiorentini, C. Clostridium difficile toxin B causes apoptosis in epithelial cells by thrilling mitochondria involvement of ATP-sensitive mitochondrial potassium channels. J. Biol. Chem. 2007, 282, 9029–9041. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, R.; Nottrott, S.; Schoentaube, J.; Tatge, H.; Olling, A.; Just, I. Glucosylation of Rho GTPases by Clostridium difficile toxin A triggers apoptosis in intestinal epithelial cells. J. Med. Microbiol. 2008, 57, 765–770. [Google Scholar] [CrossRef]
- Huang, T.; Li, S.; Li, G.; Tian, Y.; Wang, H.; Shi, L.; Perez-Cordon, G.; Mao, L.; Wang, X.; Wang, J. Utility of Clostridium difficile toxin B for inducing anti-tumor immunity. PLoS ONE 2014, 9, e110826. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.; Dreger, S.; Gerhard, R.; Barth, H.; Just, I.; Genth, H. Difference in the cytotoxic effects of toxin B from Clostridium difficile strain VPI 10463 and toxin B from variant Clostridium difficile strain 1470. Infect. Immun. 2007, 75, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Qa’Dan, M.; Ramsey, M.; Daniel, J.; Spyres, L.M.; Safiejko-Mroczka, B.; Ortiz-Leduc, W.; Ballard, J.D. Clostridium difficile toxin B activates dual caspase-dependent and caspase-independent apoptosis in intoxicated cells. Cell. Microbiol. 2002, 4, 425–434. [Google Scholar] [CrossRef]
- Farrow, M.A.; Chumbler, N.M.; Lapierre, L.A.; Franklin, J.L.; Rutherford, S.A.; Goldenring, J.R.; Lacy, D.B. Clostridium difficile toxin B-induced necrosis is mediated by the host epithelial cell NADPH oxidase complex. Proc. Natl. Acad. Sci. USA 2013, 110, 18674–18679. [Google Scholar] [CrossRef]
- Just, I.; Selzer, J.; Wilm, M.; Von Eichel-Streiber, C.; Mann, M.; Aktories, K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 1995, 375, 500. [Google Scholar] [CrossRef]
- Apodaca, G. Physiology of the urothelium Lori A Birder, William C de Groat. In Textbook of the Neurogenic Bladder; CRC Press: Boca Raton, FL, USA, 2008; pp. 31–51. [Google Scholar]
- Diehl, P.; Schauwecker, J.; Mittelmeier, W.; Schmitt, M. High hydrostatic pressure, a novel approach in orthopedic surgical oncology to disinfect bone, tendons and cartilage. Anticancer Res. 2008, 28, 3877–3883. [Google Scholar]
- Helmstein, K. Treatment of bladder carcinoma by a hydrostatic pressure technique. Report on 76 cases. Panminerva Med. 1976, 18, 194. [Google Scholar]
- Eisenthal, A.; Ramakrishna, V.; Skornick, Y.; Shinitzky, M. Induction of cell-mediated immunity against B16-BL6 melanoma in mice vaccinated with cells modified by hydrostatic pressure and chemical crosslinking. Cancer Immunol. Immunother. 1993, 36, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Goldman, Y.; Shinitzky, M. Effective elimination of lung metastases induced by tumor cells treated with hydrostatic pressure and N-acetyl-L-cysteine. Cancer Res. 2000, 60, 350–358. [Google Scholar] [PubMed]
- Frey, B.; Franz, S.; Sheriff, A.; Korn, A.; Bluemelhuber, G.; Gaipl, U.; Voll, R.; Meyer-Pittroff, R.; Herrmann, M. Hydrostatic pressure induced death of mammalian cells engages pathways related to apoptosis or necrosis. Cell. Mol. Biol. Paris Wegmann 2004, 50, 459–468. [Google Scholar]
- Fucikova, J.; Moserova, I.; Truxova, I.; Hermanova, I.; Vancurova, I.; Partlova, S.; Fialova, A.; Sojka, L.; Cartron, P.F.; Houska, M.; et al. High hydrostatic pressure induces immunogenic cell death in human tumor cells. Int. J. Cancer 2014, 135, 1165–1177. [Google Scholar] [CrossRef]
- Rožková, D.; Tišerová, H.; Fučíková, J.; Lašt’ovička, J.; Podrazil, M.; Ulčová, H.; Budínský, V.; Prausová, J.; Linke, Z.; Minárik, I.; et al. FOCUS on FOCIS: Combined chemo-immunotherapy for the treatment of hormone-refractory metastatic prostate cancer. Clin. Immunol. 2009, 131, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Begovič, M.; Herberman, R.B.; Gorelik, E. Ultraviolet light-induced increase in tumor cell susceptibility to TNF-dependent and TNF-independent natural cell-mediated cytotoxicity. Cell. Immunol. 1991, 138, 349–359. [Google Scholar] [CrossRef]
- Maverakis, E.; Miyamura, Y.; Bowen, M.P.; Correa, G.; Ono, Y.; Goodarzi, H. Light, including ultraviolet. J. Autoimmun. 2010, 34, J247–J257. [Google Scholar] [CrossRef]
- Widel, M.; Krzywon, A.; Gajda, K.; Skonieczna, M.; Rzeszowska-Wolny, J. Induction of bystander effects by UVA, UVB, and UVC radiation in human fibroblasts and the implication of reactive oxygen species. Free Radic. Biol. Med. 2014, 68, 278–287. [Google Scholar] [CrossRef]
- Zhong, J.L.; Yang, L.; Lü, F.; Xiao, H.; Xu, R.; Wang, L.; Zhu, F.; Zhang, Y. UVA, UVB and UVC induce differential response signaling pathways converged on the eIF2α phosphorylation. Photochem. Photobiol. 2011, 87, 1092–1104. [Google Scholar] [CrossRef]
- Bensasson, R.V. Primary Photo-Processes in Biology and Medicine; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013; Volume 85. [Google Scholar]
- Dolmans, D.E.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef]
- Henderson, B.W.; Dougherty, T.J. How does photodynamic therapy work? Photochem. Photobiol. 1992, 55, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Shumaker, B.; Hetzel, F. Clinical laser photodynamic therapy in the treatment of bladder carcinoma. Photochem. Photobiol. 1987, 46, 899–901. [Google Scholar] [CrossRef]
- Gollnick, S.O.; Liu, X.; Owczarczak, B.; Musser, D.A.; Henderson, B.W. Altered expression of interleukin 6 and interleukin 10 as a result of photodynamic therapy in vivo. Cancer Res. 1997, 57, 3904–3909. [Google Scholar]
- Korbelik, M.; Krosl, G.; Krosl, J.; Dougherty, G.J. The role of host lymphoid populations in the response of mouse EMT6 tumor to photodynamic therapy. Cancer Res. 1996, 56, 5647–5652. [Google Scholar]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol. Immunother. 2012, 61, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Korbelik, M.; Zhang, W.; Merchant, S. Involvement of damage-associated molecular patterns in tumor response to photodynamic therapy: Surface expression of calreticulin and high-mobility group box-1 release. Cancer Immunol. Immunother. 2011, 60, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Karioti, A.; Bilia, A.R. Hypericins as potential leads for new therapeutics. Int. J. Mol. Sci. 2010, 11, 562–594. [Google Scholar] [CrossRef]
- Buytaert, E.; Callewaert, G.; Hendrickx, N.; Scorrano, L.; Hartmann, D.; Missiaen, L.; Vandenheede, J.R.; Heirman, I.; Grooten, J.; Agostinis, P. Role of endoplasmic reticulum depletion and multidomain proapoptotic BAX and BAK proteins in shaping cell death after hypericin-mediated photodynamic therapy. FASEB J. 2006, 20, 756–758. [Google Scholar] [CrossRef]
- Du, H.Y.; Olivo, M.; Mahendran, R.; Huang, Q.; Shen, H.M.; Ong, C.N.; Bay, B.H. Hypericin photoactivation triggers down-regulation of matrix metalloproteinase-9 expression in well-differentiated human nasopharyngeal cancer cells. Cell. Mol. Life Sci. 2007, 64, 979. [Google Scholar] [CrossRef]
- Hu, C.; Li, X.; Wang, W.; Zhang, R.; Deng, L. Metal-N-heterocyclic carbene complexes as anti-tumor agents. Curr. Med. Chem. 2014, 21, 1220–1230. [Google Scholar] [CrossRef]
- Zou, T. Luminescent Organoplatinum (II) Complexes Containing Bis (N-Heterocyclic Carbene) Ligands Selectively Target Endoplasmic Reticulum and Induce Potent Phototoxicity. In Anti-Cancer N-Heterocyclic Carbene Complexes of Gold (III), Gold (I) and Platinum (II); Springer: Berlin/Heidelberg, Germany, 2016; pp. 135–162. [Google Scholar]
- Takasu, A.; Masui, A.; Hamada, M.; Imai, T.; Iwai, S.; Yura, Y. Immunogenic cell death by oncolytic herpes simplex virus type 1 in squamous cell carcinoma cells. Cancer Gene Ther. 2016, 23, 107. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, O.G.; Errington-Mais, F.; Steele, L.; Hadac, E.; Jennings, V.; Scott, K.; Peach, H.; Phillips, R.M.; Bond, J.; Pandha, H.; et al. Measles virus causes immunogenic cell death in human melanoma. Gene Ther. 2013, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- Koks, C.A.; Garg, A.D.; Ehrhardt, M.; Riva, M.; Vandenberk, L.; Boon, L.; Vleeschouwer, S.D.; Agostinis, P.; Graf, N.; Van Gool, S.W. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int. J. Cancer 2015, 136, E313–E325. [Google Scholar] [CrossRef] [PubMed]
- Kimball, S.R. Eukaryotic initiation factor eIF2. Int. J. Biochem. Cell Biol. 1999, 31, 25–29. [Google Scholar] [CrossRef]
- Balachandran, S.; Barber, G.N. PKR in innate immunity, cancer, and viral oncolysis. In Cancer Genomics and Proteomics; Springer: Berlin/Heidelberg, Germany, 2007; pp. 277–301. [Google Scholar]
- He, B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006, 13, 393. [Google Scholar] [CrossRef] [PubMed]
- Brand, S.R.; Kobayashi, R.; Mathews, M.B. The Tat protein of human immunodeficiency virus type 1 is a substrate and inhibitor of the interferon-induced, virally activated protein kinase, PKR. J. Biol. Chem. 1997, 272, 8388–8395. [Google Scholar] [CrossRef]
- Ramelot, T.A.; Cort, J.R.; Yee, A.A.; Liu, F.; Goshe, M.B.; Edwards, A.M.; Smith, R.D.; Arrowsmith, C.H.; Dever, T.E.; Kennedy, M.A. Myxoma virus immunomodulatory protein M156R is a structural mimic of eukaryotic translation initiation factor eIF2α. J. Mol. Biol. 2002, 322, 943–954. [Google Scholar] [CrossRef]
- Pavio, N.; Romano, P.R.; Graczyk, T.M.; Feinstone, S.M.; Taylor, D.R. Protein synthesis and endoplasmic reticulum stress can be modulated by the hepatitis C virus envelope protein E2 through the eukaryotic initiation factor 2α kinase PERK. J. Virol. 2003, 77, 3578–3585. [Google Scholar] [CrossRef]
- Kawagishi-Kobayashi, M.; Cao, C.; Lu, J.; Ozato, K.; Dever, T.E. Pseudosubstrate inhibition of protein kinase PKR by swine pox virus C8L gene product. Virology 2000, 276, 424–434. [Google Scholar] [CrossRef]
- Romano, P.R.; Zhang, F.; Tan, S.L.; Garcia-Barrio, M.T.; Katze, M.G.; Dever, T.E.; Hinnebusch, A.G. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: Role of complex formation and the E3 N-terminal domain. Mol. Cell. Biol. 1998, 18, 7304–7316. [Google Scholar] [CrossRef]
- Mulvey, M.; Arias, C.; Mohr, I. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J. Virol. 2007, 81, 3377–3390. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Min, J.Y.; Krug, R.M.; Sen, G.C. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 2006, 349, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Poppers, J.; Mulvey, M.; Perez, C.; Khoo, D.; Mohr, I. Identification of a lytic-cycle Epstein-Barr virus gene product that can regulate PKR activation. J. Virol. 2003, 77, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Burýšek, L.; Pitha, P.M. Latently expressed human herpesvirus 8-encoded interferon regulatory factor 2 inhibits double-stranded RNA-activated protein kinase. J. Virol. 2001, 75, 2345–2352. [Google Scholar] [CrossRef]
- Hakki, M.; Marshall, E.E.; De Niro, K.L.; Geballe, A.P. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J. Virol. 2006, 80, 11817–11826. [Google Scholar] [CrossRef] [PubMed]
- Hebner, C.M.; Wilson, R.; Rader, J.; Bidder, M.; Laimins, L.A. Human papillomaviruses target the double-stranded RNA protein kinase pathway. J. Gen. Virol. 2006, 87, 3183–3193. [Google Scholar] [CrossRef] [PubMed]
- Child, S.J.; Geballe, A.P. Binding and relocalization of protein kinase R by murine cytomegalovirus. J. Virol. 2009, 83, 1790–1799. [Google Scholar] [CrossRef]
- Child, S.J.; Hanson, L.K.; Brown, C.E.; Janzen, D.M.; Geballe, A.P. Double-stranded RNA binding by a heterodimeric complex of murine cytomegalovirus m142 and m143 proteins. J. Virol. 2006, 80, 10173–10180. [Google Scholar] [CrossRef]
- Goodman, A.G.; Smith, J.A.; Balachandran, S.; Perwitasari, O.; Proll, S.C.; Thomas, M.J.; Korth, M.J.; Barber, G.N.; Schiff, L.A.; Katze, M.G. The cellular protein P58IPK regulates influenza virus mRNA translation and replication through a PKR-mediated mechanism. J. Virol. 2007, 81, 2221–2230. [Google Scholar] [CrossRef]
- Clerzius, G.; Gélinas, J.F.; Daher, A.; Bonnet, M.; Meurs, E.F.; Gatignol, A. ADAR1 interacts with PKR during human immunodeficiency virus infection of lymphocytes and contributes to viral replication. J. Virol. 2009, 83, 10119–10128. [Google Scholar] [CrossRef]
- Panaretakis, T.; Joza, N.; Modjtahedi, N.; Tesniere, A.; Vitale, I.; Durchschlag, M.; Fimia, G.; Kepp, O.; Piacentini, M.; Froehlich, K.; et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ. 2008, 15, 1499. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Senovilla, L.; Galluzzi, L.; Panaretakis, T.; Schlemmer, F.; Madeo, F.; Zitvogel, L.; Kroemer, G. Viral subversion of immunogenic cell death. Cell Cycle 2009, 8, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Morselli, E.; Vitale, I.; Senovilla, L.; Pinti, M.; Zitvogel, L.; Kroemer, G. Viral strategies for the evasion of immunogenic cell death. J. Intern. Med. 2010, 267, 526–542. [Google Scholar] [CrossRef] [PubMed]
- Eletto, D.; Russo, G.; Passiatore, G.; Del Valle, L.; Giordano, A.; Khalili, K.; Gualco, E.; Peruzzi, F. Inhibition of SNAP25 expression by HIV-1 Tat involves the activity of mir-128a. J. Cell. Physiol. 2008, 216, 764–770. [Google Scholar] [CrossRef]
- Ng, F.W.; Nguyen, M.; Kwan, T.; Branton, P.E.; Nicholson, D.W.; Cromlish, J.A.; Shore, G.C. p28 Bap31, a Bcl-2/Bcl-XL-and procaspase-8–associated protein in the endoplasmic reticulum. J. Cell Biol. 1997, 139, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Breckenridge, D.G.; Stojanovic, M.; Marcellus, R.C.; Shore, G.C. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 2003, 160, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Büttner, S.; Eisenberg, T.; Herker, E.; Carmona-Gutierrez, D.; Kroemer, G.; Madeo, F. Why yeast cells can undergo apoptosis: Death in times of peace, love, and war. J. Cell Biol. 2006, 175, 521–525. [Google Scholar] [CrossRef]
- Zhou, Q.; Snipas, S.; Orth, K.; Muzio, M.; Dixit, V.M.; Salvesen, G.S. Target protease specificity of the viral serpin CrmA Analysis of five caspases. J. Biol. Chem. 1997, 272, 7797–7800. [Google Scholar] [CrossRef]
- Komiyama, T.; Ray, C.A.; Pickup, D.J.; Howard, A.D.; Thornberry, N.A.; Peterson, E.P.; Salvesen, G. Inhibition of interleukin-1 beta converting enzyme by the cowpox virus serpin CrmA. An example of cross-class inhibition. J. Biol. Chem. 1994, 269, 19331–19337. [Google Scholar]
- Muzio, M.; Salvesen, G.S.; Dixit, V.M. FLICE induced apoptosis in a cell-free system cleavage of caspase zymogens. J. Biol. Chem. 1997, 272, 2952–2956. [Google Scholar] [CrossRef]
- MacNeill, A.L.; Turner, P.C.; Moyer, R.W. Mutation of the Myxoma virus SERP2 P1-site to prevent proteinase inhibition causes apoptosis in cultured RK-13 cells and attenuates disease in rabbits, but mutation to alter specificity causes apoptosis without reducing virulence. Virology 2006, 356, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Filippova, M.; Johnson, M.M.; Bautista, M.; Filippov, V.; Fodor, N.; Tungteakkhun, S.S.; Williams, K.; Duerksen-Hughes, P.J. The large and small isoforms of human papillomavirus type 16 E6 bind to and differentially affect procaspase 8 stability and activity. J. Virol. 2007, 81, 4116–4129. [Google Scholar] [CrossRef] [PubMed]
- Skaletskaya, A.; Bartle, L.M.; Chittenden, T.; McCormick, A.L.; Mocarski, E.S.; Goldmacher, V.S. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Natl. Acad. Sci. USA 2001, 98, 7829–7834. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ghebranious, N.; Donehower, L.A. Mouse models in tumor suppression. Oncogene 1998, 17, 3385. [Google Scholar] [CrossRef]
- Yamazaki, T.; Hannani, D.; Poirier-Colame, V.; Ladoire, S.; Locher, C.; Sistigu, A.; Prada, N.; Adjemian, S.; Catani, J.P.; Freudenberg, M.; et al. Defective immunogenic cell death of HMGB1-deficient tumors: Compensatory therapy with TLR4 agonists. Cell Death Differ. 2014, 21, 69. [Google Scholar] [CrossRef]
- Marshak-Rothstein, A. Toll-like receptors in systemic autoimmune disease. Nat. Rev. Immunol. 2006, 6, 823. [Google Scholar] [CrossRef]
- Limagne, E.; Thibaudin, M.; Nuttin, L.; Spill, A.; Dérangère, V.; Fumet, J.-D.; Amellal, N.; Peranzoni, E.; Cattan, V.; Ghiringhelli, F. Trifluridine/Tipiracil plus Oxaliplatin Improves PD-1 Blockade in Colorectal Cancer by Inducing Immunogenic Cell Death and Depleting Macrophages. Cancer Immunol. Res. 2019, 7, 1958–1969. [Google Scholar] [CrossRef]
- Chan, S.R.; Vermi, W.; Luo, J.; Lucini, L.; Rickert, C.; Fowler, A.M.; Lonardi, S.; Arthur, C.; Young, L.J.; Levy, D.E. STAT1-deficient mice spontaneously develop estrogen receptor α-positive luminal mammary carcinomas. Breast Cancer Res. 2012, 14, R16. [Google Scholar] [CrossRef]
- Peter, C.; Wesselborg, S.; Kirsten, L. Role of attraction and danger signals in the uptake of apoptotic and necrotic cells and its immunological outcome. In Phagocytosis of Dying Cells: From Molecular Mechanisms to Human Diseases; Springer: Berlin/Heidelberg, Germany, 2009; pp. 63–101. [Google Scholar]
- Suzuki, Y.; Mimura, K.; Yoshimoto, Y.; Watanabe, M.; Ohkubo, Y.; Izawa, S.; Murata, K.; Fujii, H.; Nakano, T.; Kono, K. Immunogenic tumor cell death induced by chemoradiotherapy in patients with esophageal squamous cell carcinoma. Cancer Res. 2012, 72, 3967–3976. [Google Scholar] [CrossRef] [PubMed]
- Derakhshani, A.; Rezaei, Z.; Safarpour, H.; Sabri, M.; Mir, A.; Sanati, M.A.; Vahidian, F.; Moghadam, A.G.; Aghadoukht, A.; Hajiasgharzadeh, K. Overcoming trastuzumab resistance in HER2-positive breast cancer using combination therapy. J. Cell. Physiol. 2020, 235, 3142–3156. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.; Vacchelli, E.; Aranda, F.; Castoldi, F.; Eggermont, A.; Cremer, I.; Sautes-Fridman, C.; Fucikova, J.; Galon, J.; Spisek, R.; et al. Trial Watch: Immunogenic cell death inducers for anticancer chemotherapy. Oncoimmunology 2015, 4, e1008866. [Google Scholar] [CrossRef] [PubMed]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and cancer: Golden anniversary. Nat. Rev. Clin. Oncol. 2009, 6, 638. [Google Scholar] [CrossRef]
- Nakahara, T.; Uchi, H.; Lesokhin, A.M.; Avogadri, F.; Rizzuto, G.A.; Hirschhorn-Cymerman, D.; Panageas, K.S.; Merghoub, T.; Wolchok, J.D.; Houghton, A.N. Cyclophosphamide enhances immunity by modulating the balance of dendritic cell subsets in lymphoid organs. Blood 2010, 115, 4384–4392. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Zitvogel, L.; Kroemer, G. The secret ally: Immunostimulation by anticancer drugs. Nat. Rev. Drug Discov. 2012, 11, 215. [Google Scholar] [CrossRef]
- Vacchelli, E.; Senovilla, L.; Eggermont, A.; Fridman, W.H.; Galon, J.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Chemotherapy with immunogenic cell death inducers. Oncoimmunology 2013, 2, e23510. [Google Scholar] [CrossRef]
- Weiss, R.B. The anthracyclines: Will we ever find a better doxorubicin? Semin. Oncol. 1992, 19, 670–686. [Google Scholar]
- Medrano, R.F.; Mendonca, S.A.; Ribeiro, A.H.; Catani, J.P.; Feitosa, V.A.; Rodrigues, E.G.; Strauss, B.E. Abstract B39: Potentiation of Doxorubicin Low-Dose Efficacy through Its Association with p19Arf/Interferon-beta Immunotherapy: Combining Two Immunogenic Cell Death Inducers for the Treatment of Cancer; AACR: Philadelphia, PA, USA, 2018. [Google Scholar]
- Zhu, S.; Waguespack, M.; Barker, S.A.; Li, S. Doxorubicin directs the accumulation of interleukin-12 induced IFN gamma into tumors for enhancing STAT1 dependent antitumor effect. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 4252–4260. [Google Scholar] [CrossRef]
- Alagkiozidis, I.; Facciabene, A.; Carpenito, C.; Benencia, F.; Jonak, Z.; Adams, S.; Carroll, R.G.; Gimotty, P.A.; Hammond, R.; Danet-Desnoyers, G.Ä.; et al. Increased immunogenicity of surviving tumor cells enables cooperation between liposomal doxorubicin and IL-18. J. Transl. Med. 2009, 7, 104. [Google Scholar] [CrossRef]
- Kawano, M.; Tanaka, K.; Itonaga, I.; Iwasaki, T.; Miyazaki, M.; Ikeda, S.; Tsumura, H. Dendritic cells combined with doxorubicin induces immunogenic cell death and exhibits antitumor effects for osteosarcoma. Oncol. Lett. 2016, 11, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Reshetnikov, V.; Arkhypov, A.; Julakanti, P.R.; Mokhir, A. A cancer specific oxaliplatin-releasing Pt (iv)-prodrug. Dalton Trans. 2018, 47, 6679–6682. [Google Scholar] [CrossRef] [PubMed]
- Stordal, B.; Pavlakis, N.; Davey, R. Oxaliplatin for the treatment of cisplatin-resistant cancer: A systematic review. Cancer Treat. Rev. 2007, 33, 347–357. [Google Scholar] [CrossRef]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010, 29, 482. [Google Scholar] [CrossRef]
- Bezu, L.; Gomes-da-Silva, L.C.; Dewitte, H.; Breckpot, K.; Fucikova, J.; Spisek, R.; Galluzzi, L.; Kepp, O.; Kroemer, G. Combinatorial strategies for the induction of immunogenic cell death. Front. Immunol. 2015, 6, 187. [Google Scholar] [CrossRef] [PubMed]
- Cirone, M.; Garufi, A.; Di Renzo, L.; Granato, M.; Faggioni, A.; D’Orazi, G. Zinc supplementation is required for the cytotoxic and immunogenic effects of chemotherapy in chemoresistant p53-functionally deficient cells. Oncoimmunology 2013, 2, e26198. [Google Scholar] [CrossRef]
- Li, L.; Li, Y.; Yang, C.H.; Radford, D.C.; Wang, J.; Janát-Amsbury, M.; Kopeček, J.; Yang, J. Inhibition of Immunosuppressive Tumors by Polymer-Assisted Inductions of Immunogenic Cell Death and Multivalent PD-L1 Crosslinking. Adv. Funct. Mater. 2020, 30, 1908961. [Google Scholar] [CrossRef]
- Tu, K.; Deng, H.; Kong, L.; Wang, Y.; Yang, T.; Hu, Q.; Hu, M.; Yang, C.; Zhang, Z. Reshaping tumor immune microenvironment through acidity-responsive nanoparticles featured with CRISPR/Cas9-mediated PD-L1 attenuation and chemotherapeutics-induced immunogenic cell death. ACS Appl. Mater. Interfaces 2020, 12, 16018–16030. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Nair, V.S.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Kepp, O.; Zitvogel, L.; Kroemer, G. Clinical Evidence That Immunogenic Cell Death Sensitizes to PD-1/PD-L1 Blockade. OncoImmunology 2019, 8, e1637188. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Forveille, S.; Iribarren, K.; Sauvat, A.; Senovilla, L.; Wang, Y.; Humeau, J.; Perez-Lanzon, M.; Zhou, H.; Martínez-Leal, J.F.; et al. Lurbinectedin synergizes with immune checkpoint blockade to generate anticancer immunity. OncoImmunology 2019, 8, e1656502. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wen, X.; Li, T.; Melancon, M.; Gupta, S.; Peng, W.; Li, C. Irreversible Electroporation Induces Immunogenic Cell Death and Mediates Durable Response in Orthotopic PDAC Model in Combination with anti-PD-1. AACR 2019. [Google Scholar] [CrossRef]
- Lantos, J.; Young, R.J.; Miranda, P.C.; Wenger, C.; Wong, E.T. TTFields therapy: Preclinical and clinical data. In Handbook of Neuro-Oncology Neuroimaging; Elsevier: Amsterdam, The Netherlands, 2016; pp. 243–256. [Google Scholar]
- Giladi, M.; Schneiderman, R.S.; Voloshin, T.; Porat, Y.; Munster, M.; Blat, R.; Sherbo, S.; Bomzon, Z.; Urman, N.; Itzhaki, A.; et al. Mitotic spindle disruption by alternating electric fields leads to improper chromosome segregation and mitotic catastrophe in cancer cells. Sci. Rep. 2015, 5, 18046. [Google Scholar] [CrossRef]
- Gera, N.; Yang, A.; Holtzman, T.S.; Lee, S.X.; Wong, E.T.; Swanson, K.D. Tumor treating fields perturb the localization of septins and cause aberrant mitotic exit. PLoS ONE 2015, 10, e0125269. [Google Scholar] [CrossRef]
- Voloshin, T.; Kaynan, N.; Davidi, S.; Porat, Y.; Shteingauz, A.; Schneiderman, R.S.; Zeevi, E.; Munster, M.; Blat, R.; Brami, C.T.; et al. Tumor-treating fields (TTFields) induce immunogenic cell death resulting in enhanced antitumor efficacy when combined with anti-PD-1 therapy. Cancer Immunol. Immunother. 2020, 1–14. [Google Scholar] [CrossRef]
- Yamazaki, T.; Buqué, A.; Ames, T.D.; Galluzzi, L. PT-112 induces immunogenic cell death and synergizes with immune checkpoint blockers in mouse tumor models. OncoImmunology 2020, 9, 1721810. [Google Scholar] [CrossRef]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef]
- Rodríguez-Salazar, M.d.C.; Franco-Molina, M.A.; Mendoza-Gamboa, E.; Martínez-Torres, A.C.; Zapata-Benavides, P.; López-González, J.S.; Coronado-Cerda, E.E.; Alcocer-González, J.M.; Tamez-Guerra, R.S.; Rodríguez-Padilla, C. The novel immunomodulator IMMUNEPOTENT CRP combined with chemotherapy agent increased the rate of immunogenic cell death and prevented melanoma growth. Oncol. Lett. 2017, 14, 844–852. [Google Scholar] [CrossRef][Green Version]
- Gebremeskel, S.; Johnston, B. Concepts and mechanisms underlying chemotherapy induced immunogenic cell death: Impact on clinical studies and considerations for combined therapies. Oncotarget 2015, 6, 41600. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Immunogenic Cell Death in Radiation Therapy; Taylor & Francis: Oxfordshire, UK, 2013. [Google Scholar]
- Golden, E.B.; Apetoh, L. Radiotherapy and immunogenic cell death. In Seminars in Radiation Oncology; WB Saunders: Philadelphia, PA, USA, 2015; pp. 11–17. [Google Scholar]
- Golden, E.; Formenti, S. Is tumor (R) ejection by the immune system the “5th R” of radiobiology? Oncoimmunology 2014, 3, e28133. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.; Rubner, Y.; Wunderlich, R.; Weiss, E.M.; Pockley, A.G.; Fietkau, R.; Gaipl, U.S. Induction of abscopal anti-tumor immunity and immunogenic tumor cell death by ionizing irradiation-implications for cancer therapies. Curr. Med. Chem. 2012, 19, 1751–1764. [Google Scholar] [CrossRef] [PubMed]
- Stangl, S.; Themelis, G.; Friedrich, L.; Ntziachristos, V.; Sarantopoulos, A.; Molls, M.; Skerra, A.; Multhoff, G. Detection of irradiation-induced, membrane heat shock protein 70 (Hsp70) in mouse tumors using Hsp70 Fab fragment. Radiother. Oncol. 2011, 99, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Criollo, A.; Ortiz, C.; Lidereau, R.; Mariette, C.; Chaput, N.; Mira, J.P.; Delaloge, S.; et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol. Rev. 2007, 220, 47–59. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kepp, O.; Senovilla, L.; Menger, L.; Chaput, N.; Kroemer, G. Immunogenic tumor cell death for optimal anticancer therapy: The calreticulin exposure pathway. Clin. Cancer Res. 2010, 16, 2100–2104. [Google Scholar] [CrossRef]
- Chakraborty, M.; Abrams, S.I.; Coleman, C.N.; Camphausen, K.; Schlom, J.; Hodge, J.W. External beam radiation of tumors alters phenotype of tumor cells to render them susceptible to vaccine-mediated T-cell killing. Cancer Res. 2004, 64, 4328–4337. [Google Scholar] [CrossRef]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef]
- Kachikwu, E.L.; Iwamoto, K.S.; Liao, Y.P.; DeMarco, J.J.; Agazaryan, N.; Economou, J.S.; McBride, W.H.; Schaue, D. Radiation enhances regulatory T cell representation. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1128–1135. [Google Scholar] [CrossRef]
- Gorin, J.B.; Ménager, J.; Gouard, S.; Maurel, C.; Guilloux, Y.; Faivre-Chauvet, A.; Morgenstern, A.; Bruchertseifer, F.; Chérel, M.; Davodeau, F.; et al. Antitumor immunity induced after α irradiation. Neoplasia 2014, 16, 319–328. [Google Scholar] [CrossRef]
- Golden, E.B.; Frances, D.; Pellicciotta, I.; Demaria, S.; Helen Barcellos-Hoff, M.; Formenti, S.C. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology 2014, 3, e28518. [Google Scholar] [CrossRef]
- Golden, E.; Pellicciotta, I.; Demaria, S.; Barcellos-Hoff, M.H.; Formenti, S.C. The convergence of radiation and immunogenic cell death signaling pathways. Front. Oncol. 2012, 2, 88. [Google Scholar] [CrossRef]
- Demaria, S.; Kawashima, N.; Yang, A.M.; Devitt, M.L.; Babb, J.S.; Allison, J.P.; Formenti, S.C. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. 2005, 11, 728–734. [Google Scholar] [PubMed]
- Park, S.S.; Dong, H.; Liu, X.; Harrington, S.M.; Krco, C.J.; Grams, M.P.; Mansfield, A.S.; Furutani, K.M.; Olivier, K.R.; Kwon, E.D. PD-1 restrains radiotherapy-induced abscopal effect. Cancer Immunol. Res. 2015, 3, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.X. Irradiation and anti–PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef]
- Chajon, E.; Castelli, J.; Marsiglia, H.; De Crevoisier, R. The synergistic effect of radiotherapy and immunotherapy: A promising but not simple partnership. Crit. Rev. Oncol. Hematol. 2017, 111, 124–132. [Google Scholar] [CrossRef]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373. [Google Scholar] [CrossRef]
- Zegers, C.M.; Rekers, N.H.; Quaden, D.H.; Lieuwes, N.G.; Yaromina, A.; Germeraad, W.T.; Wieten, L.; Biessen, E.A.; Boon, L.; Neri, D.; et al. Radiotherapy combined with the immunocytokine L19-IL2 provides long-lasting antitumor effects. Clin. Cancer Res. 2015, 21, 1151–1160. [Google Scholar] [CrossRef]
- Gough, M.J.; Crittenden, M.R.; Sarff, M.; Pang, P.; Seung, S.K.; Vetto, J.T.; Hu, H.M.; Redmond, W.L.; Holland, J.; Weinberg, A.D.; et al. Adjuvant therapy with agonistic antibodies to CD134 (OX40) increases local control following surgical or radiation therapy of cancer in mice. J. Immunother. 2010, 33, 798. [Google Scholar] [CrossRef]
- Honeychurch, J.; Glennie, M.J.; Johnson, P.W.; Illidge, T.M. Anti-CD40 monoclonal antibody therapy in combination with irradiation results in a CD8 T-cell–dependent immunity to B-cell lymphoma. Blood 2003, 102, 1449–1457. [Google Scholar] [CrossRef]
- Garg, A.D.; Nowis, D.; Golab, J.; Agostinis, P. Photodynamic therapy: Illuminating the road from cell death towards anti-tumour immunity. Apoptosis 2010, 15, 1050–1071. [Google Scholar] [CrossRef]
- Cecic, I.; Korbelik, M. Deposition of complement proteins on cells treated by photodynamic therapy in vitro. J. Environ. Pathol. Toxicol. Oncol. 2006, 25, 189–204. [Google Scholar] [PubMed]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part one—photosensitizers, photochemistry and cellular localization. Photodiagnosis Photodyn. Ther. 2004, 1, 279–293. [Google Scholar] [CrossRef]
- Cecic, I.; Sun, J.; Korbelik, M. Role of complement anaphylatoxin C3a in photodynamic therapy-elicited engagement of host neutrophils and other immune cells. Photochem. Photobiol. 2006, 82, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Kousis, P.C.; Henderson, B.W.; Maier, P.G.; Gollnick, S.O. Photodynamic therapy enhancement of antitumor immunity is regulated by neutrophils. Cancer Res. 2007, 67, 10501–10510. [Google Scholar] [CrossRef]
- Jalili, A.; Makowski, M.; Świtaj, T.; Nowis, D.; Wilczyński, G.M.; Wilczek, E.; Chorąży-Massalska, M.; Radzikowska, A.; Maśliński, W.; Biały, Ł.; et al. Effective photoimmunotherapy of murine colon carcinoma induced by the combination of photodynamic therapy and dendritic cells. Clin. Cancer Res. 2004, 10, 4498–4508. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.R.; Castano, A.P.; Mroz, P. Combination immunotherapy and photodynamic therapy for cancer. In Proceedings of the Light-Activated Tissue Regeneration and Therapy Conference; Springer: Boston, MA, USA, 2008; pp. 99–113. [Google Scholar]
- Korbelik, M. Complement upregulation in photodynamic therapy-treated tumors: Role of Toll-like receptor pathway and NFκB. Cancer Lett. 2009, 281, 232–238. [Google Scholar] [CrossRef]
- Garg, A.D.; Agostinis, P. ER stress, autophagy and immunogenic cell death in photodynamic therapy-induced anti-cancer immune responses. Photochem. Photobiol. Sci. 2014, 13, 474–487. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Enlightening the impact of immunogenic cell death in photodynamic cancer therapy. EMBO J. 2012, 31, 1055–1057. [Google Scholar] [CrossRef]
- Sazgarnia, A.; Montazerabadi, A.R.; Bahreyni-Toosi, M.H.; Ahmadi, A.; Aledavood, A. In vitro survival of MCF-7 breast cancer cells following combined treatment with ionizing radiation and mitoxantrone-mediated photodynamic therapy. Photodiagnosis Photodyn. Ther. 2013, 10, 72–78. [Google Scholar] [CrossRef]
- Ahn, J.C.; Biswas, R.; Mondal, A.; Lee, Y.K.; Chung, P.S. Cisplatin enhances the efficacy of 5-aminolevulinic acid mediated photodynamic therapy in human head and neck squamous cell carcinoma. Gen. Physiol. Biophys. 2014, 33, 53–62. [Google Scholar] [CrossRef]
- Brodin, N.P.; Guha, C.; Tomé, W.A. Photodynamic therapy and its role in combined modality anticancer treatment. Technol. Cancer Res. Treat. 2015, 14, 355–368. [Google Scholar] [CrossRef]
- Castano, A.P.; Mroz, P.; Wu, M.X.; Hamblin, M.R. Photodynamic therapy plus low-dose cyclophosphamide generates antitumor immunity in a mouse model. Proc. Natl. Acad. Sci. USA 2008, 105, 5495–5500. [Google Scholar] [CrossRef] [PubMed]
- Korbelik, M.; Banáth, J.; Saw, K.M.; Zhang, W.; Čiplys, E. Calreticulin as Cancer Treatment Adjuvant: Combination with Photodynamic Therapy and Photodynamic Therapy-Generated Vaccines. Front. Oncol. 2015, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Doix, B.; Trempolec, N.; Riant, O.; Feron, O. Low photosensitizer dose and early radiotherapy enhance antitumor immune response of photodynamic therapy-based dendritic cell vaccination. Front. Oncol. 2019, 9, 811. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; He, X.; Yang, Z.; Yang, X.; Xiao, W.; Liu, R.; Xie, R.; Qin, L.; Gao, H. Sequentially responsive biomimetic nanoparticles with optimal size in combination with checkpoint blockade for cascade synergetic treatment of breast cancer and lung metastasis. Biomaterials 2019, 217, 119309. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ICD Inducers | Molecular Mechanism(s) of Action | Mechanism(s) of ICD Induction | Ref. |
|---|---|---|---|
| Type I | |||
| Cardiac glycosides | Targeting Na+/K+ pump, upregulation of DR-4 and -5, TIAM1 and PAR4, inhibition of DNA topoisomerase | CRT exposure, ATP secretion and passive release of HMGB1 | [117] |
| Capsaicin (CPS) | TRPV1-mediated signaling | Ectopic expression of CRT and HSP90, release of ATP, HMGB1, HSP70 and HSP90 | [118] |
| Clostridium difficile toxin B | Modification of Rho protein activity | CRT exposure, secretion of ATP, HSP70, HSP90 and HMGB1 release and autophagy | [119] |
| High hydrostatic pressure (HHP) | - | CRT, HSP70 and HSP90 exposure, ATP release, increased expression of maturation markers on DCs, increased cytokine production in DC | [120] |
| Ultraviolet light (UV) C | DNA damage | CRT exposure, HSP70 and HMGB1 release, strong phagocytosis activity of DCs, proinflammatory cytokine production in DCs. | [52,121] |
| Type II | |||
| Pt-N-heterocyclic carbene (NHC) | ER associated production of ROS | Surface-exposed CRT and the extracellular release of ATP and HMGB1 | [122] |
| Coxsackievirus B3 | Accumulation of viral proteins in the ER and ER stress | Increasing the CD8+ lymphocytes, development of tumor-nourishing blood vessels | [123,124] |
| Therapeutic Agents | Mechanism | Cancer Type |
|---|---|---|
| Therapies combining chemotherapy-induced ICD | ||
| Doxorubicin plus IL-18 | increased expression of MHC class I and Fas by ID8 murine ovarian cancer cells, sensitization to CTL and Fas-mediated killing | ovarian cancer |
| Doxorubicin plus DCs | increased frequency of CD8+ T cells, serum interferon-γ levels | osteosarcoma tumors |
| 5-fluorouracil plus folinic acid, oxaliplatin | induction of ICD suppressed the expression of PD-L2 | colorectal cancer |
| Trifluridine/tipiracil plus oxaliplatin | stimulation of ICD and antitumor CD8 cells, depletion of TAM | colorectal cancer |
| LTX-401 plus double checkpoint inhibition of PD-1 and CTLA-4 | reduce the growth of the tumor | fibrosarcomas |
| Lurbinectedin plus CTLA-4/PD-1 dual checkpoint blockade | extended life expectancy, tumor clearance | osteosarcoma |
| IRE plus anti-PD-1 | activation of DCs, alleviation of immunosuppressive tumor microenvironment | PDAC |
| TTFields plus the anti-PD-1 | decrease in tumor volume, increases in CD45+ tumor infiltrating cells | Lung cancer |
| CRISPR/Cas9-Mediated Knockout of the Cdk5 Gene plus paclitaxel | reduce regulatory T lymphocytes, repolarize tumor-associated macrophages, enhance antitumor immunity | colorectal cancer cells (CT26), murine melanoma cells (B16F10) and murine fibrosarcoma |
| ICRP plus Oxaliplatin | inhibit melanoma tumor development and growth | melanoma |
| oxaliplatin plus IL-12 | inhibit tumor recurrence, enhance the ratio of CD8 + T lymphocyte /MDSCs and CD8+ T lymphocyte /Tregs within the tumors | colorectal cancer |
| Therapies combining radiotherapy-induced ICD | ||
| CTLA-4/PD-1 dual checkpoint blockade plus radiotherapy | heighten CD8+/Treg ratio, complete response rate of eighty percent | metastatic melanoma |
| L19-IL2 plus radiotherapy | increases in the CTL population in murine models of cancer stimulation of cytotoxic CD8+ T lymphocyte responses | CT26 colon tumors |
| Radiotherapy plus an adjuvant agonist antibody against CD134 (OX40) | improved antitumor immune responses and tumor control | lung cancer |
| Anti-CD40 plus radiotherapy antibody | stimulate CD8+ T-cell mediated immunity | B-cell lymphoma |
| Therapies combining photodynamic therapy-induced ICD | ||
| Cisplatin plus PDT | improved cancer cell death, diminishing the toxicity of antineoplastic drugs | cervical cancer |
| Cyclophosphamide plus PDT | decrease Tregs population, inhibit the tumor recurrence | reticulum cell sarcoma |
| PDT-based DC vaccination plus radiotherapy | significant tumor growth delay | squamous cell carcinoma |
| pheophorbide A plus PXTK and dPPA | activation of CD4+, CD8+ T cells and NK cells, enhances secretion of cytokines (TNF-α and IL-12), tumor growth inhibition | breast cancer |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asadzadeh, Z.; Safarzadeh, E.; Safaei, S.; Baradaran, A.; Mohammadi, A.; Hajiasgharzadeh, K.; Derakhshani, A.; Argentiero, A.; Silvestris, N.; Baradaran, B. Current Approaches for Combination Therapy of Cancer: The Role of Immunogenic Cell Death. Cancers 2020, 12, 1047. https://doi.org/10.3390/cancers12041047
Asadzadeh Z, Safarzadeh E, Safaei S, Baradaran A, Mohammadi A, Hajiasgharzadeh K, Derakhshani A, Argentiero A, Silvestris N, Baradaran B. Current Approaches for Combination Therapy of Cancer: The Role of Immunogenic Cell Death. Cancers. 2020; 12(4):1047. https://doi.org/10.3390/cancers12041047
Chicago/Turabian StyleAsadzadeh, Zahra, Elham Safarzadeh, Sahar Safaei, Ali Baradaran, Ali Mohammadi, Khalil Hajiasgharzadeh, Afshin Derakhshani, Antonella Argentiero, Nicola Silvestris, and Behzad Baradaran. 2020. "Current Approaches for Combination Therapy of Cancer: The Role of Immunogenic Cell Death" Cancers 12, no. 4: 1047. https://doi.org/10.3390/cancers12041047
APA StyleAsadzadeh, Z., Safarzadeh, E., Safaei, S., Baradaran, A., Mohammadi, A., Hajiasgharzadeh, K., Derakhshani, A., Argentiero, A., Silvestris, N., & Baradaran, B. (2020). Current Approaches for Combination Therapy of Cancer: The Role of Immunogenic Cell Death. Cancers, 12(4), 1047. https://doi.org/10.3390/cancers12041047