Analysis of Dual Class I Histone Deacetylase and Lysine Demethylase Inhibitor Domatinostat (4SC-202) on Growth and Cellular and Genomic Landscape of Atypical Teratoid/Rhabdoid

 ,
,  and
and
Abstract
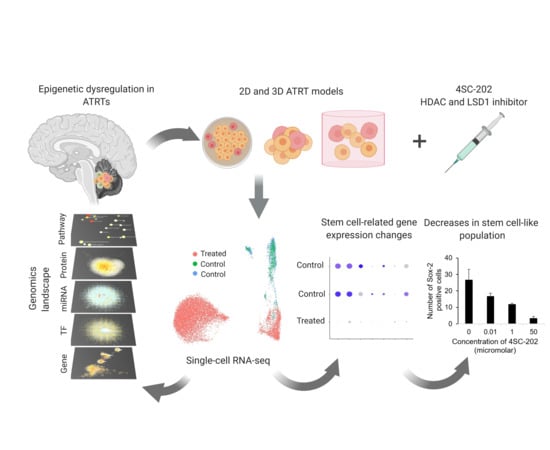
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
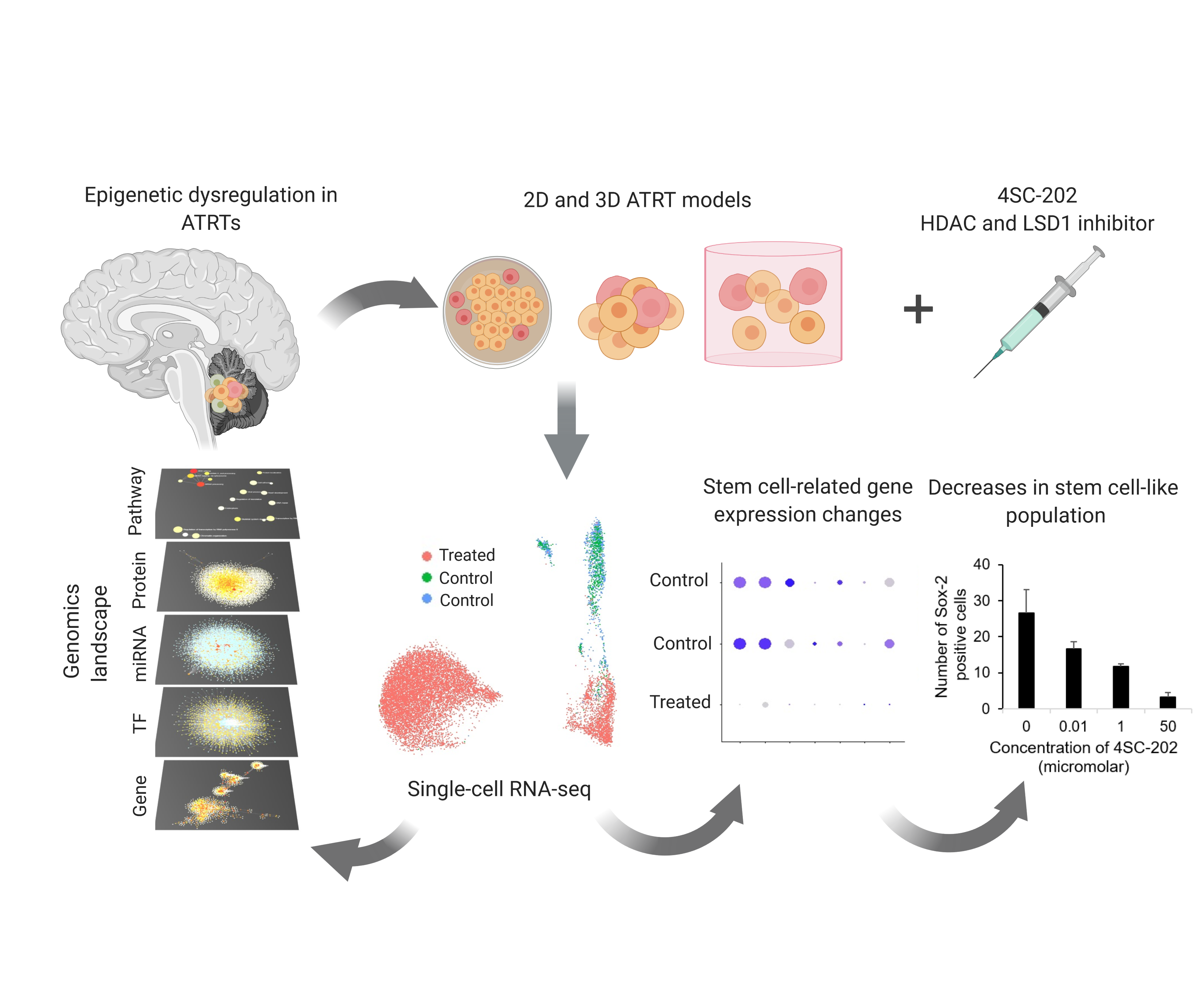
2.1. ATRTs Have Aberrant Expression of 4SC-202 Targets
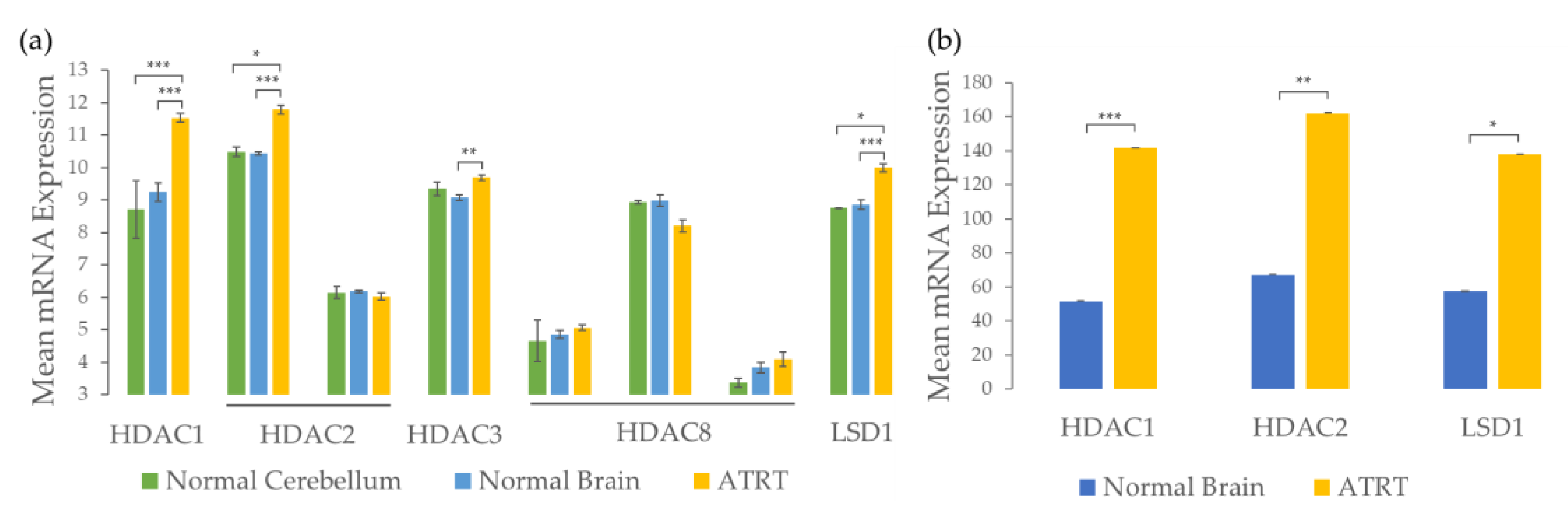
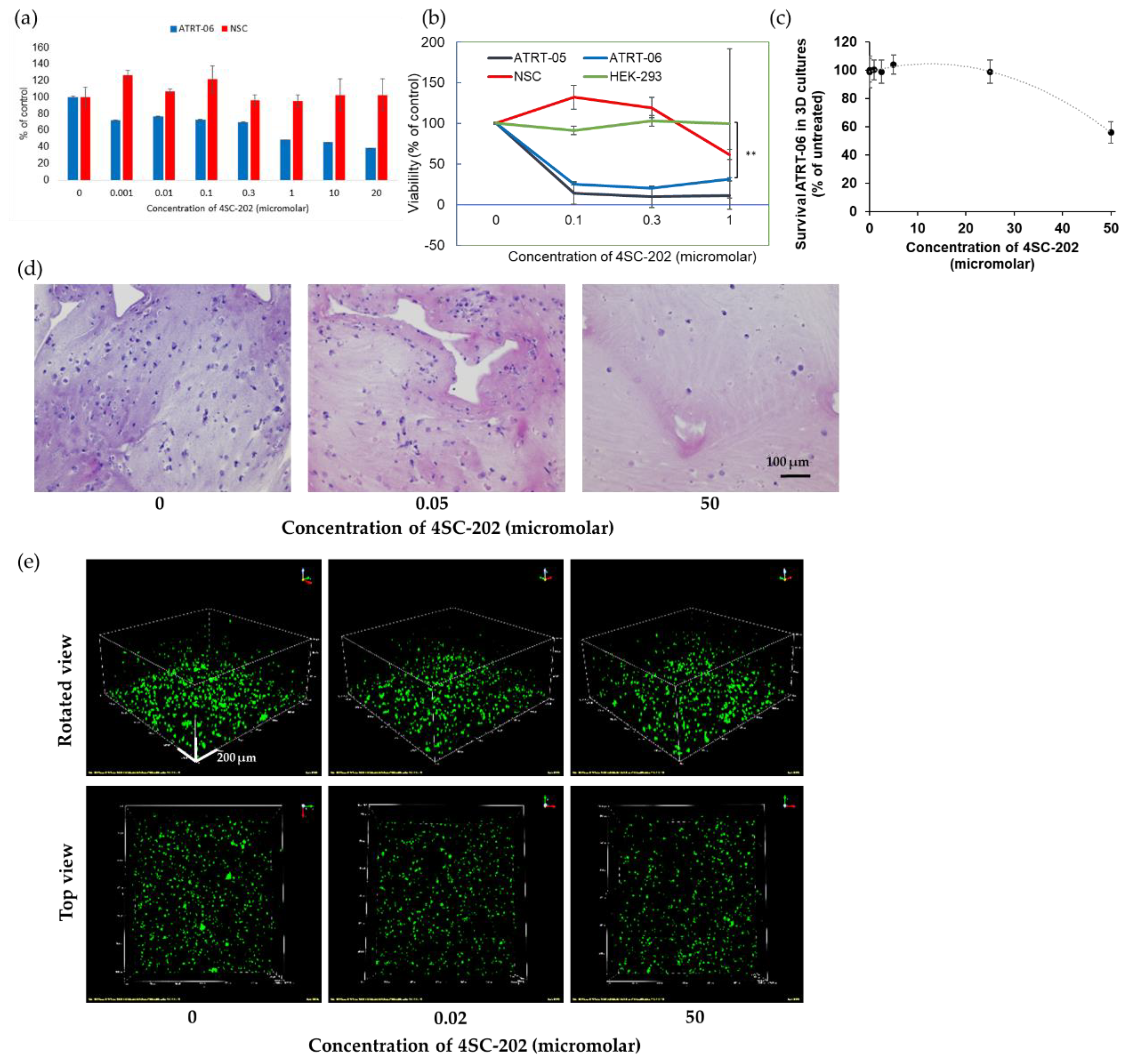
2.2. 4SC-202 Is Cytotoxic and Cytostatic to ATRT in Two- and Three- Dimensional Cell Culture
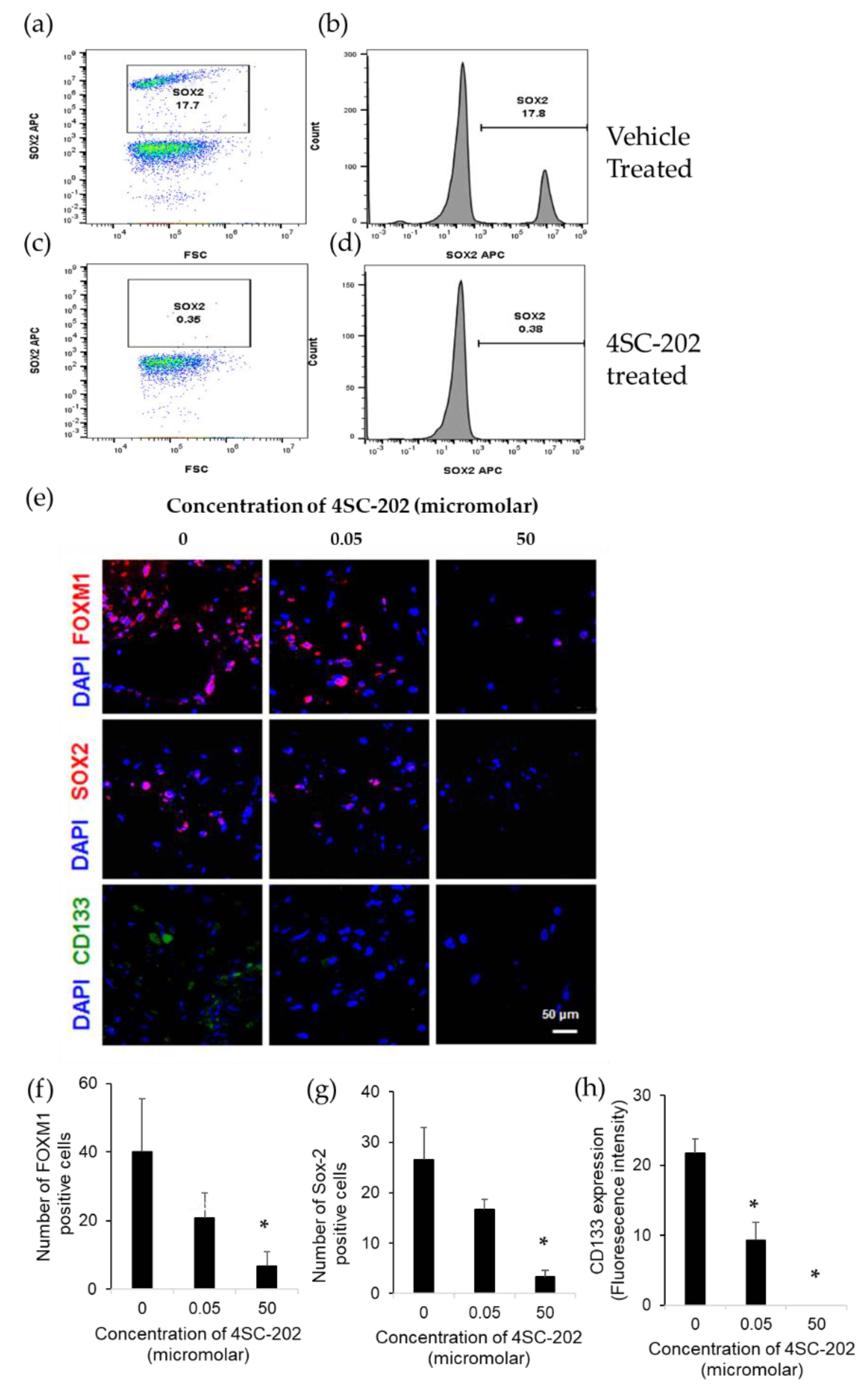
2.3. 4SC-202 Decreases the Population of Cells Overexpressing Stem Cell Markers
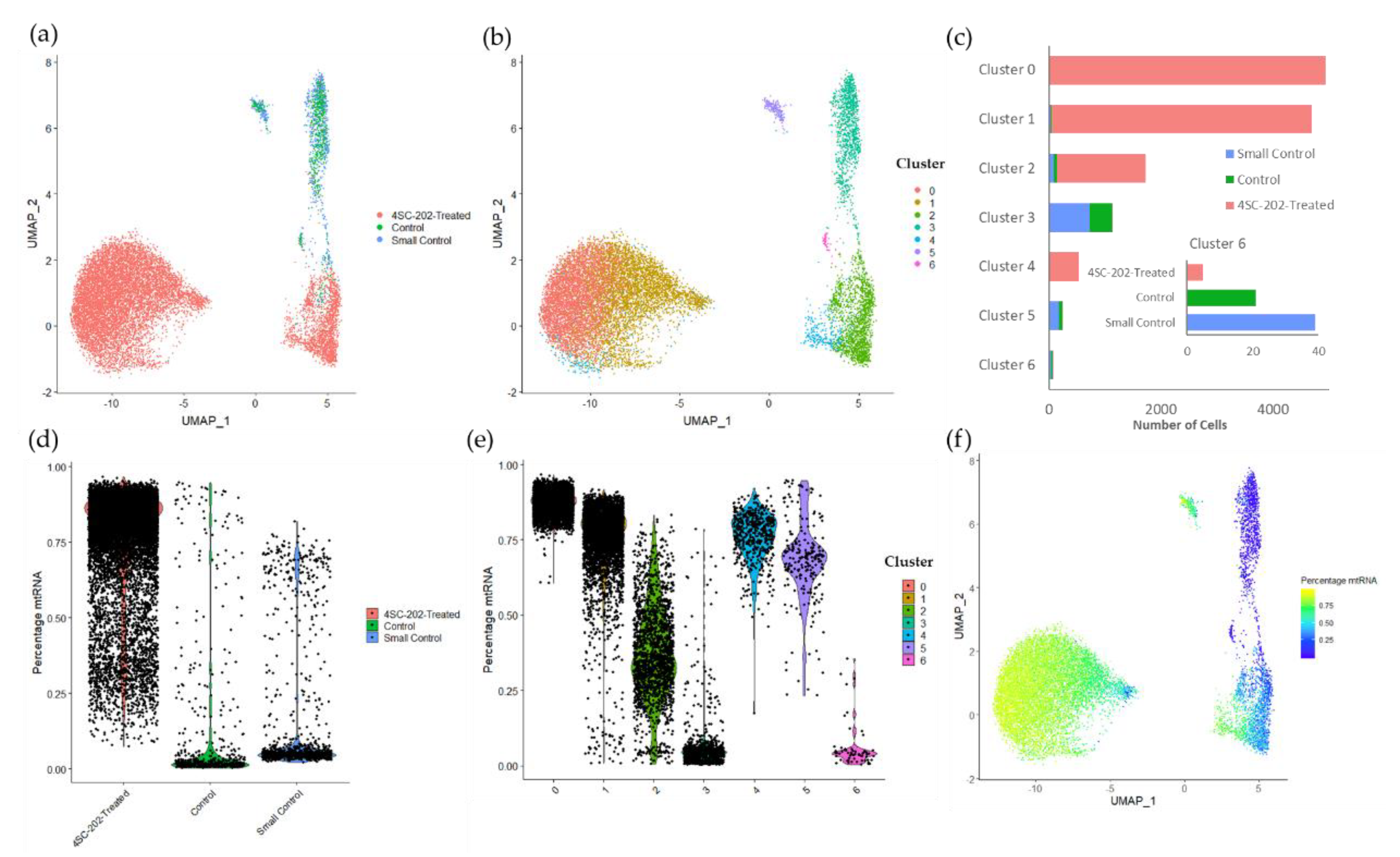
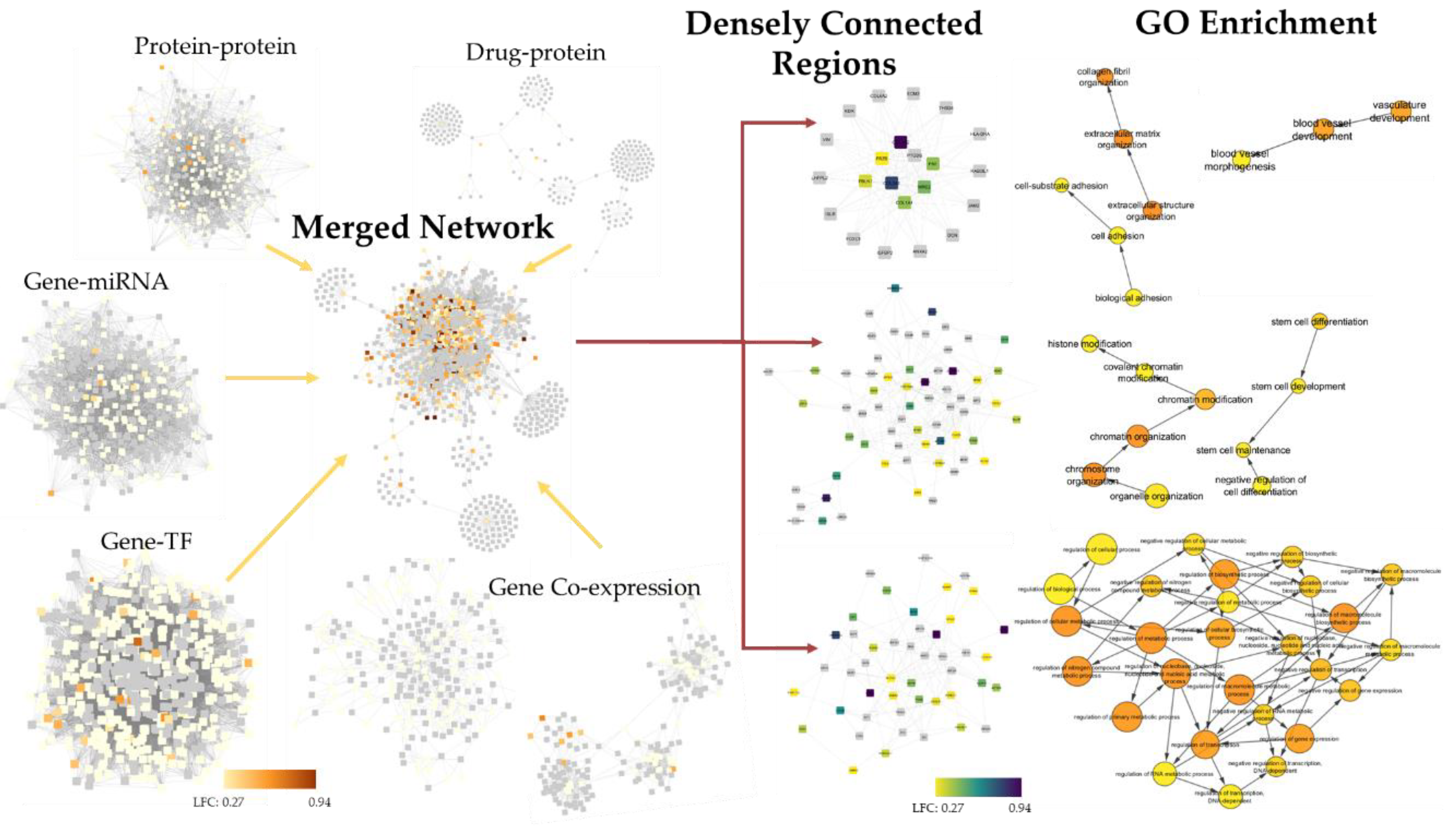
2.4. 4SC-202 Modulates Systems Biology Landscape in ATRT for Tumor Cell Population
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Spheroid Model
4.3. Development of 3D Scaffolds to Study the Effect of Domatinostat (4SC202) on ATRT-06 Cell Survival
4.4. Sytox Green Cell Proliferation Assay and Viability Assays in 2D
4.5. Flow Cytometry for Spheroid Analysis
4.6. Flow Cytometry for 3D Scaffold Analysis
4.7. Immunohistochemistry (IHC) and Immunofluorescence (IF) Staining of 3D Scaffolds
4.8. Confocal Imaging of 3D Scaffolds
4.9. Cell Clustering Analysis of Confocal Images From 3D Scaffolds
4.10. Single-Cell RNA-Sequencing Experimental Protocol
4.11. Single-Cell RNA-Sequencing Data Analysis
4.12. Bulk RNA-Sequencing
4.13. Microarray Analysis
4.14. NanoString Analysis
4.15. Biological Process-Level Systems Biology Analysis
4.16. Integrated Systems Biology Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Buscariollo, D.L.; Park, H.S.; Roberts, K.B.; Yu, J.B. Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a Surveillance, Epidemiology, and End Results analysis. Cancer 2012, 118, 4212–4219. [Google Scholar] [CrossRef]
- Ginn, K.F.; Gajjar, A. Atypical teratoid rhabdoid tumor: Current therapy and future directions. Front. Oncol. 2012, 2, 114. [Google Scholar] [CrossRef] [PubMed]
- Hilden, J.M.; Meerbaum, S.; Burger, P.; Finlay, J.; Janss, A.; Scheithauer, B.W.; Walter, A.W.; Rorke, L.B.; Biegel, J.A. Central nervous system atypical teratoid/rhabdoid tumor: Results of therapy in children enrolled in a registry. J. Clin. Oncol. 2004, 22, 2877–2884. [Google Scholar] [CrossRef] [PubMed]
- Lafay-Cousin, L.; Hawkins, C.; Carret, A.S.; Johnston, D.; Zelcer, S.; Wilson, B.; Jabado, N.; Scheinemann, K.; Eisenstat, D.; Fryer, C.; et al. Central nervous system atypical teratoid rhabdoid tumours: The Canadian Paediatric Brain Tumour Consortium experience. Eur. J. Cancer 2012, 48, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, D.A.; Gibson, S.; Brown, T.; Sebire, N.J.; Anderson, J. Clinical and pathological features of paediatric malignant rhabdoid tumours. Pediatr. Blood Cancer 2010, 54, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Tekautz, T.M.; Fuller, C.E.; Blaney, S.; Fouladi, M.; Broniscer, A.; Merchant, T.E.; Krasin, M.; Dalton, J.; Hale, G.; Kun, L.E.; et al. Atypical teratoid/rhabdoid tumors (ATRT): Improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J. Clin. Oncol. 2005, 23, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- von Hoff, K.; Hinkes, B.; Dannenmann-Stern, E.; von Bueren, A.O.; Warmuth-Metz, M.; Soerensen, N.; Emser, A.; Zwiener, I.; Schlegel, P.G.; Kuehl, J.; et al. Frequency, risk-factors and survival of children with atypical teratoid rhabdoid tumors (AT/RT) of the CNS diagnosed between 1988 and 2004, and registered to the German HIT database. Pediatr. Blood Cancer 2011, 57, 978–985. [Google Scholar] [CrossRef]
- Kleihues, P.; Louis, D.N.; Scheithauer, B.W.; Rorke, L.B.; Reifenberger, G.; Burger, P.C.; Cavenee, W.K. The WHO classification of tumors of the nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 215–225. [Google Scholar] [CrossRef]
- Benesch, M.; Nemes, K.; Neumayer, P.; Hasselblatt, M.; Timmermann, B.; Bison, B.; Ebetsberger-Dachs, G.; Bourdeaut, F.; Dufour, C.; Biassoni, V.; et al. Spinal cord atypical teratoid/rhabdoid tumors in children: Clinical, genetic, and outcome characteristics in a representative European cohort. Pediatr. Blood Cancer 2020, 67, e28022. [Google Scholar] [CrossRef]
- Packer, R.J.; Biegel, J.A.; Blaney, S.; Finlay, J.; Geyer, J.R.; Heideman, R.; Hilden, J.; Janss, A.J.; Kun, L.; Vezina, G.; et al. Atypical teratoid/rhabdoid tumor of the central nervous system: Report on workshop. J. Pediatr. Hematol. Oncol. 2002, 24, 337–342. [Google Scholar] [CrossRef]
- Woehrer, A.; Slavc, I.; Waldhoer, T.; Heinzl, H.; Zielonke, N.; Czech, T.; Benesch, M.; Hainfellner, J.A.; Haberler, C. Incidence of atypical teratoid/rhabdoid tumors in children. Cancer 2010, 116, 5725–5732. [Google Scholar] [CrossRef]
- Gruber, W.; Peer, E.; Elmer, D.P.; Sternberg, C.; Tesanovic, S.; Del Burgo, P.; Coni, S.; Canettieri, G.; Neureiter, D.; Bartz, R.; et al. Targeting class I histone deacetylases by the novel small molecule inhibitor 4SC-202 blocks oncogenic hedgehog-GLI signaling and overcomes smoothened inhibitor resistance. Int. J. Cancer 2018, 142, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Pinkerneil, M.; Hoffmann, M.J.; Kohlhof, H.; Schulz, W.A.; Niegisch, G. Evaluation of the therapeutic potential of the novel isotype specific HDAC inhibitor 4SC-202 in urothelial carcinoma cell lines. Target. Oncol. 2016, 11, 783–798. [Google Scholar] [CrossRef] [PubMed]
- Henning, S.W.; Doblhofer, R.; Kohlhof, H.; Jankowsky, R.; Maier, T.; Beckers, T.; Schmidt, M.; Hentsch, B. Preclinical characterization of 4SC-202. A novel isotype specific HDAC inhibitor. Eur. J. Cancer Suppl. 2010, 8, 61. [Google Scholar] [CrossRef]
- Zhijun, H.; Shusheng, W.; Han, M.; Jianping, L.; Li-Sen, Q.; Dechun, L. Pre-clinical characterization of 4SC-202, a novel class I HDAC inhibitor, against colorectal cancer cells. Tumour Biol. 2016, 37, 10257–10267. [Google Scholar] [CrossRef]
- Messerli, S.M.; Hoffman, M.M.; Gnimpieba, E.Z.; Kohlhof, H.; Bhardwaj, R.D. 4SC-202 as a potential treatment for the pediatric brain tumor medulloblastoma. Brain Sci. 2017, 7, 147. [Google Scholar] [CrossRef]
- Fu, M.; Wan, F.; Li, Z.; Zhang, F. 4SC-202 activates ASK1-dependent mitochondrial apoptosis pathway to inhibit hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2016, 471, 267–273. [Google Scholar] [CrossRef]
- Mishra, V.K.; Wegwitz, F.; Kosinsky, R.L.; Sen, M.; Baumgartner, R.; Wulff, T.; Siveke, J.T.; Schildhaus, H.-U.; Najafova, Z.; Kari, V.; et al. Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4- and MYC-dependent manner. Nucleic Acids Res. 2017, 45, 6334–6349. [Google Scholar] [CrossRef]
- von Tresckow, B.; Gundermann, S.; Eichenauer, D.A.; Aulitzky, W.E.; Göbeler, M.; Sayehli, C.; Bacchus, L.; Hauns, B.; Mais, A.; Hentsch, B.; et al. First-in-human study of 4SC-202, a novel oral HDAC inhibitor in advanced hematologic malignancies (TOPAS study). J. Clin. Oncol. 2014, 32, 8559. [Google Scholar] [CrossRef]
- Kohlhof, H.; Prenzel, T.; Schrepfer, S.; Herz, T.; Regenbrecht, C.; Aberger, F.; Vitt, D.; Baumgartner, R. 4sc-202: A novel epigenetic modulator to target cancer stem cells. In Proceedings of the Keystone Symposia Stem Cells and Cancer, Banff, AB, Canada, 2–7 February 2014. [Google Scholar]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog signaling in the maintenance of cancer stem cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef]
- Li, Y.; Kong, D.; Ahmad, A.; Bao, B.; Sarkar, F.H. Pancreatic cancer stem cells: Emerging target for designing novel therapy. Cancer Lett. 2013, 338, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Yao, Z.; Jiang, Y.; Keller, E.T. Prostate cancer stem cell biology. Minerva Urol. Nefrol. 2012, 64, 19–33. [Google Scholar]
- Yu, Y.; Ramena, G.; Elble, R.C. The role of cancer stem cells in relapse of solid tumors. Front. Biosci. (Elite Ed) 2012, 4, 1528–1541. [Google Scholar] [CrossRef]
- Bao, B.; Ahmad, A.; Azmi, A.S.; Ali, S.; Sarkar, F.H. Overview of cancer stem cells (CSCs) and mechanisms of their regulation: Implications for cancer therapy. Curr. Protoc. Pharmacol. 2013, 61, 14.25.1–14.25.14. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Ruiz i Altaba, A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef]
- Peacock, C.D.; Wang, Q.; Gesell, G.S.; Corcoran-Schwartz, I.M.; Jones, E.; Kim, J.; Devereux, W.L.; Rhodes, J.T.; Huff, C.A.; Beachy, P.A.; et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl. Acad. Sci. USA 2007, 104, 4048–4053. [Google Scholar] [CrossRef]
- Birks, D.K.; Donson, A.M.; Patel, P.R.; Sufit, A.; Algar, E.M.; Dunham, C.; Kleinschmidt-DeMasters, B.K.; Handler, M.H.; Vibhakar, R.; Foreman, N.K. Pediatric rhabdoid tumors of kidney and brain show many differences in gene expression but share dysregulation of cell cycle and epigenetic effector genes. Pediatr. Blood Cancer 2013, 60, 1095–1102. [Google Scholar] [CrossRef]
- Chakravadhanula, M.; Ozols, V.V.; Hampton, C.N.; Zhou, L.; Catchpoole, D.; Bhardwaj, R.D. Expression of the HOX genes and HOTAIR in atypical teratoid rhabdoid tumors and other pediatric brain tumors. Cancer Genet. 2014, 207, 425–428. [Google Scholar] [CrossRef]
- Torchia, J.; Golbourn, B.; Feng, S.; Ho, K.C.; Sin-Chan, P.; Vasiljevic, A.; Norman, J.D.; Guilhamon, P.; Garzia, L.; Agamez, N.R.; et al. Integrated (epi)-genomic analyses identify subgroup-specific therapeutic targets in CNS rhabdoid tumors. Cancer Cell 2016, 30, 891–908. [Google Scholar] [CrossRef]
- Ho, B.; Johann, P.D.; Grabovska, Y.; De Dieu Andrianteranagna, M.J.; Yao, F.; Frühwald, M.; Hasselblatt, M.; Bourdeaut, F.; Williamson, D.; Huang, A.; et al. Molecular subgrouping of Atypical Teratoid/Rhabdoid Tumors (ATRT)—A reinvestigation and current consensus. Neuro Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, M.; Krämer, O.H.; Heinzel, T. HDACi—Targets beyond chromatin. Cancer Lett. 2009, 280, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Ilicic, T.; Kim, J.K.; Kolodziejczyk, A.A.; Bagger, F.O.; McCarthy, D.J.; Marioni, J.C.; Teichmann, S.A. Classification of low quality cells from single-cell RNA-seq data. Genome Biol. 2016, 17, 29. [Google Scholar] [CrossRef]
- Ng, J.M.Y.; Martinez, D.; Marsh, E.D.; Zhang, Z.; Rappaport, E.; Santi, M.; Curran, T. Generation of a mouse model of atypical teratoid/rhabdoid tumor of the central nervous system through combined deletion of Snf5 and p53. Cancer Res. 2015, 75, 4629–4639. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive integration of single-cell data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Gong, W.; Kwak, I.-Y.; Pota, P.; Koyano-Nakagawa, N.; Garry, D.J. DrImpute: Imputing dropout events in single cell RNA sequencing data. BMC Bioinform. 2018, 19, 220. [Google Scholar] [CrossRef]
- Kao, C.-L.; Huang, P.-I.; Tsai, P.-H.; Tsai, M.-L.; Lo, J.-F.; Lee, Y.-Y.; Chen, Y.-J.; Chen, Y.-W.; Chiou, S.-H. Resveratrol-induced apoptosis and increased radiosensitivity in CD133-positive cells derived from atypical teratoid/rhabdoid tumor. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 219–228. [Google Scholar] [CrossRef]
- Chiou, S.-H.; Kao, C.-L.; Chen, Y.-W.; Chien, C.-S.; Hung, S.-C.; Lo, J.-F.; Chen, Y.-J.; Ku, H.-H.; Hsu, M.-T.; Wong, T.-T. Identification of CD133-positive radioresistant cells in atypical teratoid/rhabdoid tumor. PLoS ONE 2008, 3, e2090. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Gill, E.E.; Hancock, R.E.W. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823–844. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wobser, M.; Weber, A.; Glunz, A.; Tauch, S.; Seitz, K.; Butelmann, T.; Hesbacher, S.; Goebeler, M.; Bartz, R.; Kohlhof, H.; et al. Elucidating the mechanism of action of domatinostat (4SC-202) in cutaneous T cell lymphoma cells. J. Hematol. Oncol. 2019, 12, 30. [Google Scholar] [CrossRef]
- Duval, K.; Grover, H.; Han, L.-H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef]
- Jaroch, K.; Jaroch, A.; Bojko, B. Cell cultures in drug discovery and development: The need of reliable in vitro-in vivo extrapolation for pharmacodynamics and pharmacokinetics assessment. J. Pharm. Biomed. Anal. 2018, 147, 297–312. [Google Scholar] [CrossRef]
- Fang, Y.; Eglen, R.M. Three-Dimensional cell cultures in drug discovery and development. SLAS Discov. Adv. Life Sci. R D 2017, 22, 456–472. [Google Scholar]
- Holle, A.W.; Young, J.L.; Spatz, J.P. In vitro cancer cell-ECM interactions inform in vivo cancer treatment. Adv. Drug Deliv. Rev. 2016, 97, 270–279. [Google Scholar] [CrossRef]
- Muranen, T.; Selfors, L.M.; Worster, D.T.; Iwanicki, M.P.; Song, L.; Morales, F.C.; Gao, S.; Mills, G.B.; Brugge, J.S. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 2012, 21, 227–239. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Stretching the boundaries of extracellular matrix research. Nat. Rev. Mol. Cell Biol. 2014, 15, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; del Río Hernández, A. Matrix stiffness induces epithelial–mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef] [PubMed]
- de la Puente, P.; Muz, B.; Gilson, R.C.; Azab, F.; Luderer, M.; King, J.; Achilefu, S.; Vij, R.; Azab, A.K. 3D tissue-engineered bone marrow as a novel model to study pathophysiology and drug resistance in multiple myeloma. Biomaterials 2015, 73, 70–84. [Google Scholar] [CrossRef]
- Tannock, I.F.; Lee, C.M.; Tunggal, J.K.; Cowan, D.S.M.; Egorin, M.J. Limited penetration of anticancer drugs through tumor tissue: A potential cause of resistance of solid tumors to chemotherapy. Clin. Cancer Res. 2002, 8, 878–884. [Google Scholar]
- Wang, W.; Zhang, Z.; Kuang, X.; Wang, J. LSD1 inhibitor 4SC-202 inducing apoptosis in myelodysplastic syndrome cells via NF-κb-HO-1 pathway. Blood 2019, 134, 5415. [Google Scholar] [CrossRef]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Wicinski, J.; Cervera, N.; Finetti, P.; Hur, M.H.; Diebel, M.E.; Monville, F.; Dutcher, J.; et al. Breast cancer cell lines contain functional cancer stem scells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009, 69, 1302–1313. [Google Scholar] [CrossRef]
- Yeung, T.M.; Gandhi, S.C.; Wilding, J.L.; Muschel, R.; Bodmer, W.F. Cancer stem cells from colorectal cancer-derived cell lines. Proc. Natl. Acad. Sci. USA 2010, 107, 3722–3727. [Google Scholar] [CrossRef]
- Huang, D.; Gao, Q.; Guo, L.; Zhang, C.; Jiang, W.; Li, H.; Wang, J.; Han, X.; Shi, Y.; Lu, S.H. Isolation and identification of cancer stem-like cells in esophageal carcinoma cell lines. Stem Cells Dev. 2009, 18, 465–473. [Google Scholar] [CrossRef]
- Wang, J.; Wu, Y.; Gao, W.; Li, F.; Bo, Y.; Zhu, M.; Fu, R.; Liu, Q.; Wen, S.; Wang, B. Identification and characterization of CD133+CD44+ cancer stem cells from human laryngeal squamous cell carcinoma cell lines. J. Cancer 2017, 8, 497–506. [Google Scholar] [CrossRef]
- Liao, G.B.; Li, X.Z.; Zeng, S.; Liu, C.; Yang, S.M.; Yang, L.; Hu, C.J.; Bai, J.Y. Regulation of the master regulator FOXM1 in cancer. Cell Commun. Signal. 2018, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Joshi, K.; Banasavadi-Siddegowda, Y.; Mo, X.; Kim, S.H.; Mao, P.; Kig, C.; Nardini, D.; Sobol, R.W.; Chow, L.M.L.; Kornblum, H.I.; et al. MELK-dependent FOXM1 phosphorylation is essential for proliferation of glioma stem cells. Stem Cells 2013, 31, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Wang, C.; Wang, Z.; Zona, S.; Lin, S.X.; Wang, X.; Yan, M.; Zheng, F.M.; Li, S.S.; Xu, B.; et al. FOXM1 recruits nuclear Aurora kinase A to participate in a positive feedback loop essential for the self-renewal of breast cancer stem cells. Oncogene 2017, 36, 3428–3440. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Gao, F.; Li, S.; Liu, L. FOXM1 promotes cell proliferation, invasion, and stem cell properties in nasopharyngeal carcinoma. Front. Oncol. 2018, 8, 483. [Google Scholar] [CrossRef] [PubMed]
- Kopanja, D.; Pandey, A.; Kiefer, M.; Wang, Z.; Chandan, N.; Carr, J.R.; Franks, R.; Yu, D.Y.; Guzman, G.; Maker, A.; et al. Essential roles of FoxM1 in Ras-induced liver cancer progression and in cancer cells with stem cell features. J. Hepatol. 2015, 63, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, K.H.; Kim, D.G.; Cho, H.J.; Kim, Y.; Rheey, J.; Shin, K.; Seo, Y.J.; Choi, Y.-S.; Lee, J.-I.; et al. FoxM1 promotes stemness and radio-resistance of glioblastoma by regulating the master stem cell regulator Sox2. PLoS ONE 2015, 10, e0137703. [Google Scholar] [CrossRef]
- Liu, K.; Lin, B.; Zhao, M.; Yang, X.; Chen, M.; Gao, A.; Liu, F.; Que, J.; Lan, X. The multiple roles for Sox2 in stem cell maintenance and tumorigenesis. Cell. Signal. 2013, 25, 1264–1271. [Google Scholar] [CrossRef]
- Boumahdi, S.; Driessens, G.; Lapouge, G.; Rorive, S.; Nassar, D.; Le Mercier, M.; Delatte, B.; Caauwe, A.; Lenglez, S.; Nkusi, E.; et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 2014, 511, 246–250. [Google Scholar] [CrossRef]
- Herreros-Villanueva, M.; Zhang, J.S.; Koenig, A.; Abel, E.V.; Smyrk, T.C.; Bamlet, W.R.; De Narvajas, A.A.M.; Gomez, T.S.; Simeone, D.M.; Bujanda, L.; et al. SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis 2013, 2, e61. [Google Scholar] [CrossRef]
- Wang, Z.; Park, H.J.; Carr, J.R.; Chen, Y.J.; Zheng, Y.; Li, J.; Tyner, A.L.; Costa, R.H.; Bagchi, S.; Raychaudhuri, P. FoxM1 in tumorigenicity of the neuroblastoma cells and renewal of the neural progenitors. Cancer Res. 2011, 71, 4292–4302. [Google Scholar] [CrossRef]
- Pal, S.; Kozono, D.; Yang, X.; Fendler, W.; Fitts, W.; Ni, J.; Alberta, J.A.; Zhao, J.; Liu, K.X.; Bian, J.; et al. Dual HDAC and PI3K inhibition abrogates NFkB- and FOXM1-mediated DNA damage response to radiosensitize pediatric high-grade gliomas. Cancer Res. 2018, 78, 4007–4021. [Google Scholar] [CrossRef] [PubMed]
- Alimova, I.; Pierce, A.; Danis, E.; Donson, A.; Birks, D.K.; Griesinger, A.; Foreman, N.K.; Santi, M.; Soucek, L.; Venkataraman, S.; et al. Inhibition of MYC attenuates tumor cell self-renewal and promotes senescence in SMARCB1-deficient Group 2 atypical teratoid rhabdoid tumors to suppress tumor growth in vivo. Int. J. Cancer 2019, 144, 1983–1995. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Zhu, Y.; Wei, W.; Shao, S.; Rui, X. Transcription factor FoxM1 is the downstream target of c-Myc and contributes to the development of prostate cancer. World J. Surg. Oncol. 2018, 16, 59. [Google Scholar] [CrossRef]
- Johann, P.D.; Erkek, S.; Zapatka, M.; Kerl, K.; Buchhalter, I.; Hovestadt, V.; Jones, D.T.W.; Sturm, D.; Hermann, C.; Segura Wang, M.; et al. Atypical Teratoid/Rhabdoid Tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell 2016, 29, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Carafa, V.; Conte, M.; Tambaro, F.P.; Ciro, A.; Martens, J.; Nees, M.; Benedetti, R.; Pallavicini, I.; Minucci, S.; et al. C-Myc modulation and acetylation is a key HDAC inhibitor target in cancer. Clin. Cancer Res. 2017, 23, 2542–2555. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.M.; Hiebert, S.W.; Eischen, C.M. Myc Induces miRNA-mediated apoptosis in response to HDAC inhibition in hematologic malignancies. Cancer Res. 2016, 76, 736–748. [Google Scholar] [CrossRef]
- Neve, A.; Cantatore, F.P.; Maruotti, N.; Corrado, A.; Ribatti, D. Extracellular matrix modulates angiogenesis in physiological and pathological conditions. Biomed Res. Int. 2014, 2014, 756078. [Google Scholar] [CrossRef]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef]
- Lin, K.T.; Wang, Y.W.; Chen, C.T.; Ho, C.M.; Su, W.H.; Jou, Y.S. HDAC inhibitors augmented cell migration and metastasis through induction of PKCs leading to identification of low toxicity modalities for combination cancer therapy. Clin. Cancer Res. 2012, 18, 4691–4701. [Google Scholar] [CrossRef]
- Abe, F.; Kitadate, A.; Ikeda, S.; Yamashita, J.; Nakanishi, H.; Takahashi, N.; Asaka, C.; Teshima, K.; Miyagaki, T.; Sugaya, M.; et al. Histone deacetylase inhibitors inhibit metastasis by restoring a tumor suppressive microRNA-150 in advanced cutaneous T-cell lymphoma. Oncotarget 2017, 8, 7572–7585. [Google Scholar] [CrossRef]
- Xu, W.; Liu, H.; Liu, Z.G.; Wang, H.S.; Zhang, F.; Wang, H.; Zhang, J.; Chen, J.J.; Huang, H.J.; Tan, Y.; et al. Histone deacetylase inhibitors upregulate Snail via Smad2/3 phosphorylation and stabilization of Snail to promote metastasis of hepatoma cells. Cancer Lett. 2018, 420, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Elshafae, S.M.; Kohart, N.A.; Altstadt, L.A.; Dirksen, W.P.; Rosol, T.J. The effect of a histone deacetylase inhibitor (AR-42) on canine prostate cancer growth and metastasis. Prostate 2017, 77, 776–793. [Google Scholar] [CrossRef] [PubMed]
- de la Puente, P.; Ludeña, D.; López, M.; Ramos, J.; Iglesias, J. Differentiation within autologous fibrin scaffolds of porcine dermal cells with the mesenchymal stem cell phenotype. Exp. Cell Res. 2013, 319, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Calar, K.; Evans, C.; Petrasko, M.; de la Puente, P. Bioengineering a novel 3D in-vitro model to recreate physiological oxygen levels and tumor-immune interactions. bioRxiv 2019. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- Linkert, M.; Rueden, C.T.; Allan, C.; Burel, J.-M.; Moore, W.; Patterson, A.; Loranger, B.; Moore, J.; Neves, C.; Macdonald, D.; et al. Metadata matters: Access to image data in the real world. J. Cell Biol. 2010, 189, 777–782. [Google Scholar] [CrossRef]
- Nolan, R.; Padilla-Parra, S. ijtiff: An R package providing TIFF I/O for ImageJ users. J. Open Source Softw. 2018, 3, 633. [Google Scholar] [CrossRef]
- Wickham, H. The split-apply-combine strategy for data analysis. J. Stat. Softw. 2011, 40, 1–29. [Google Scholar] [CrossRef]
- Wickham, H. Reshaping Data with the reshape Package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Kassambara, A. Ggpubr: ’ggplot2’ based Publication Ready Plots. R package version 0.2.5. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 26 January 2020).
- Hoaglin, D.C.; John, W. Tukey and data analysis. Stat. Sci. 2003, 18, 311–318. [Google Scholar] [CrossRef]
- Landini, G.; Randell, D.A.; Fouad, S.; Galton, A. Automatic thresholding from the gradients of region boundaries. J. Microsc. 2017, 265, 185–195. [Google Scholar] [CrossRef]
- Otsu, N. A Threshold Selection Method from Gray-Level Histograms. IEEE Trans. Syst. Man. Cybern. 1979, 9, 62–66. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelières, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- Schmid, B.; Schindelin, J.; Cardona, A.; Longair, M.; Heisenberg, M. A high-level 3D visualization API for Java and ImageJ. BMC Bioinform. 2010, 11, 274. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Hoffman, M.M.; Minette, C.J.; Messerli, S.M.; Bhardwaj, R.D.; Gnimpieba, E.Z. NanoStringBioNet: Integrated R framework for bioscience knowledge discovery from NanoString nCounter data. In Proceedings of the 2017 IEEE International Conference on Bioinformatics and Biomedicine, BIBM 2017, Kansas City, MO, USA, 13–16 November 2017; pp. 2157–2162. [Google Scholar]
- Nickles, D.; Sandmann, T.; Ziman, R.; Bourgon, R. NanoStringQCPro: Quality Metrics and Data Processing Methods for NanoString mRNA Gene. R package version 1.16.0.
- Wang, H.; Horbinski, C.; Wu, H.; Liu, Y.; Sheng, S.; Liu, J.; Weiss, H.; Stromberg, A.J.; Wang, C. NanoStringDiff: A novel statistical method for differential expression analysis based on NanoString nCounter data. Nucleic Acids Res. 2016, 44, e151. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef]
- Basha, O.; Shpringer, R.; Argov, C.M.; Yeger-Lotem, E. The DifferentialNet database of differential protein-protein interactions in human tissues. Nucleic Acids Res. 2018, 46, D522–D526. [Google Scholar] [CrossRef]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef]
- Lee, S.; Zhang, C.; Arif, M.; Liu, Z.; Benfeitas, R.; Bidkhori, G.; Deshmukh, S.; Al Shobky, M.; Lovric, A.; Boren, J.; et al. TCSBN: A database of tissue and cancer specific biological networks. Nucleic Acids Res. 2018, 46, D595–D600. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffman, M.M.; Zylla, J.S.; Bhattacharya, S.; Calar, K.; Hartman, T.W.; Bhardwaj, R.D.; Miskimins, W.K.; de la Puente, P.; Gnimpieba, E.Z.; Messerli, S.M. Analysis of Dual Class I Histone Deacetylase and Lysine Demethylase Inhibitor Domatinostat (4SC-202) on Growth and Cellular and Genomic Landscape of Atypical Teratoid/Rhabdoid. Cancers 2020, 12, 756. https://doi.org/10.3390/cancers12030756
Hoffman MM, Zylla JS, Bhattacharya S, Calar K, Hartman TW, Bhardwaj RD, Miskimins WK, de la Puente P, Gnimpieba EZ, Messerli SM. Analysis of Dual Class I Histone Deacetylase and Lysine Demethylase Inhibitor Domatinostat (4SC-202) on Growth and Cellular and Genomic Landscape of Atypical Teratoid/Rhabdoid. Cancers. 2020; 12(3):756. https://doi.org/10.3390/cancers12030756
Chicago/Turabian StyleHoffman, Mariah M., Jessica S. Zylla, Somshuvra Bhattacharya, Kristin Calar, Timothy W. Hartman, Ratan D. Bhardwaj, W. Keith Miskimins, Pilar de la Puente, Etienne Z. Gnimpieba, and Shanta M. Messerli. 2020. "Analysis of Dual Class I Histone Deacetylase and Lysine Demethylase Inhibitor Domatinostat (4SC-202) on Growth and Cellular and Genomic Landscape of Atypical Teratoid/Rhabdoid" Cancers 12, no. 3: 756. https://doi.org/10.3390/cancers12030756
APA StyleHoffman, M. M., Zylla, J. S., Bhattacharya, S., Calar, K., Hartman, T. W., Bhardwaj, R. D., Miskimins, W. K., de la Puente, P., Gnimpieba, E. Z., & Messerli, S. M. (2020). Analysis of Dual Class I Histone Deacetylase and Lysine Demethylase Inhibitor Domatinostat (4SC-202) on Growth and Cellular and Genomic Landscape of Atypical Teratoid/Rhabdoid. Cancers, 12(3), 756. https://doi.org/10.3390/cancers12030756