The Multifactorial Role of PARP-1 in Tumor Microenvironment

 , ,
, ,
Abstract
1. Introduction
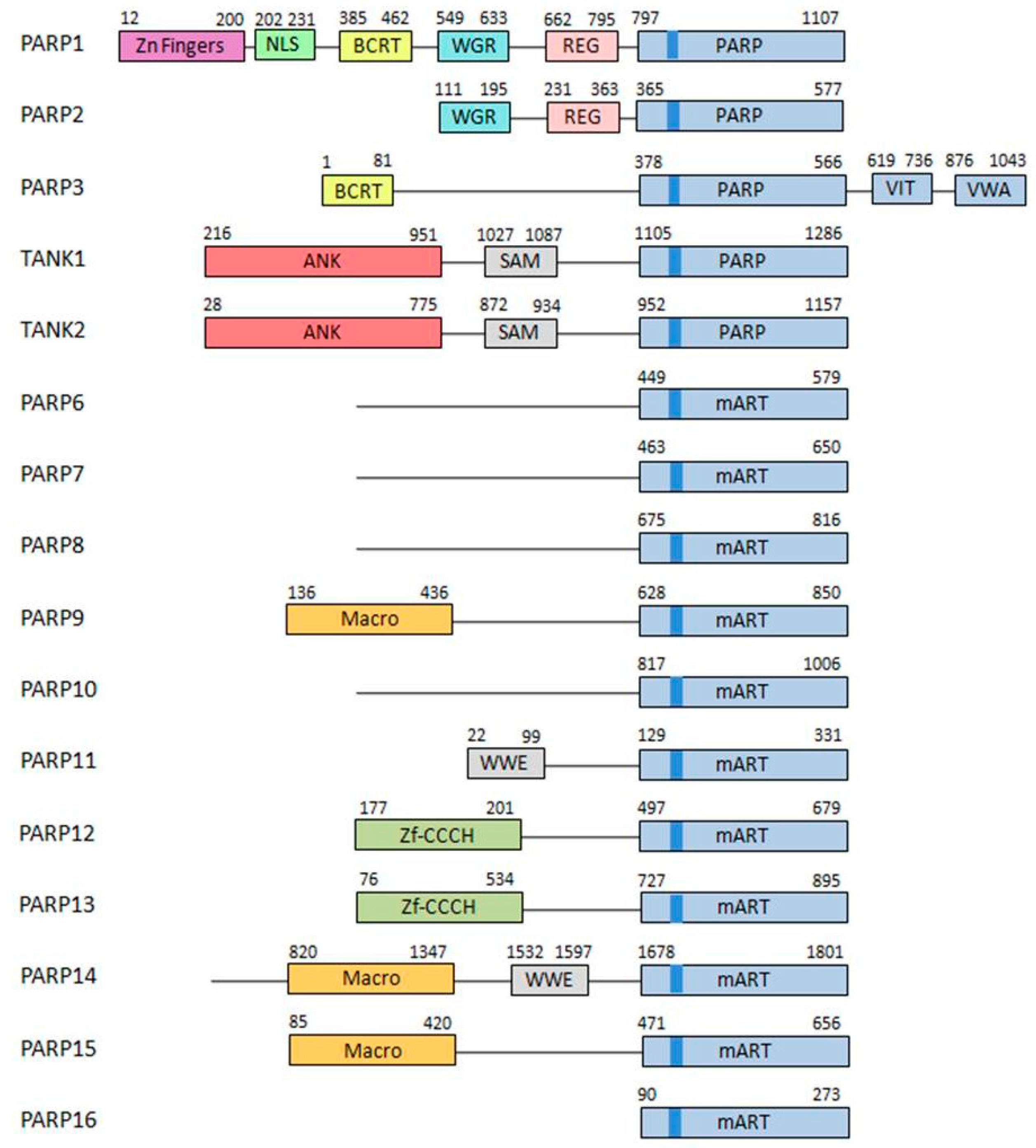
1.1. PARP Family of Proteins
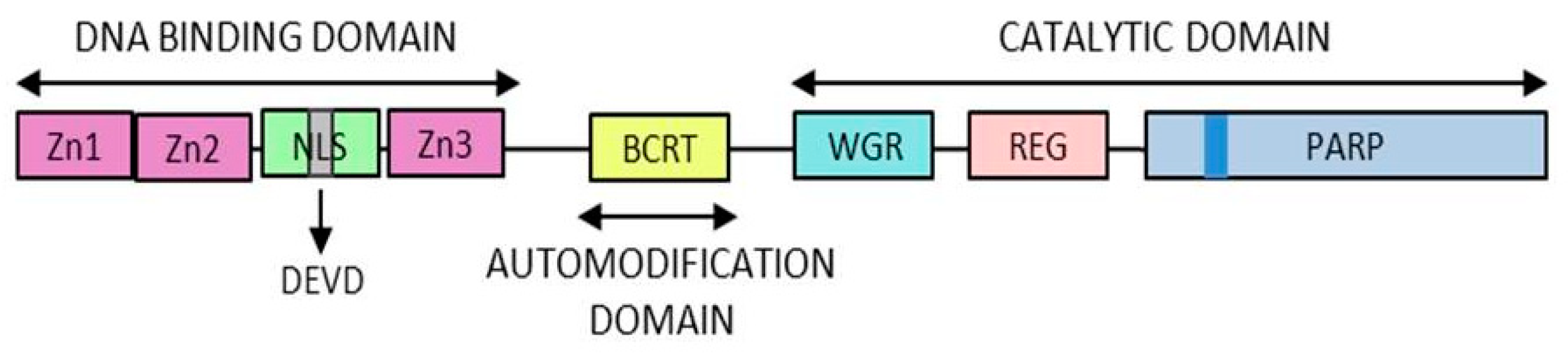
- DNA-dependent PARPs. Active during DNA damage thanks to their DNA-binding domain that consist in three zinc finger and a nuclear localization signal in the case of PARP-1 (ARTD1) [10]. Other members of this group are PARP-2 (ARTD2) and PARP-3 (ARTD3).
- Tankyrases. Containing Ankyrin-domain repeats responsible for protein-protein interactions. Very specific of this subfamily are the sterile α motifs (SAM), also related with protein-protein interactions: Tankyrase-1 (PARP-5A, ARTD5) and Tankyrase-2 (PARP-5B, ARTD6).
- CCCH PARPs. Containing CCCH motifs of the CX7–11CX3–9CX3H type, this domain is related with RNA-binding: TIPARP (PARP-7, ARTD7), PARP-12 (ARTD12) and PARP-13 (ARTD13).
- Macro-PARPs. Bearing macrodomain folds. They mediate the migration of the proteins to poly (and maybe also mono) ADP-ribosylation sites: BAL1 (PARP-9, ARTD9), BAL2 (PARP-14, ARTD8) and BAL3 (PARP-15, ARTD7).
- Other PARPS. Proteins that do not fit into any of the previous classifications [9]; PARP-4 (ARTD4), PARP-6 (ARTD17), PARP-8 (ARTD16), PARP-10 (ARTD10), PARP-11 (ARTD11) and PARP-16 (ARTD15).
- DNA-binding domain: Involved in DNA interaction, interdomain cooperation, chromatin condensation and protein–protein binding.
- NAD-binding domain: Serves as the catalytic domain, it contains the “PARP signature” sequence responsible for the PAR synthesis.
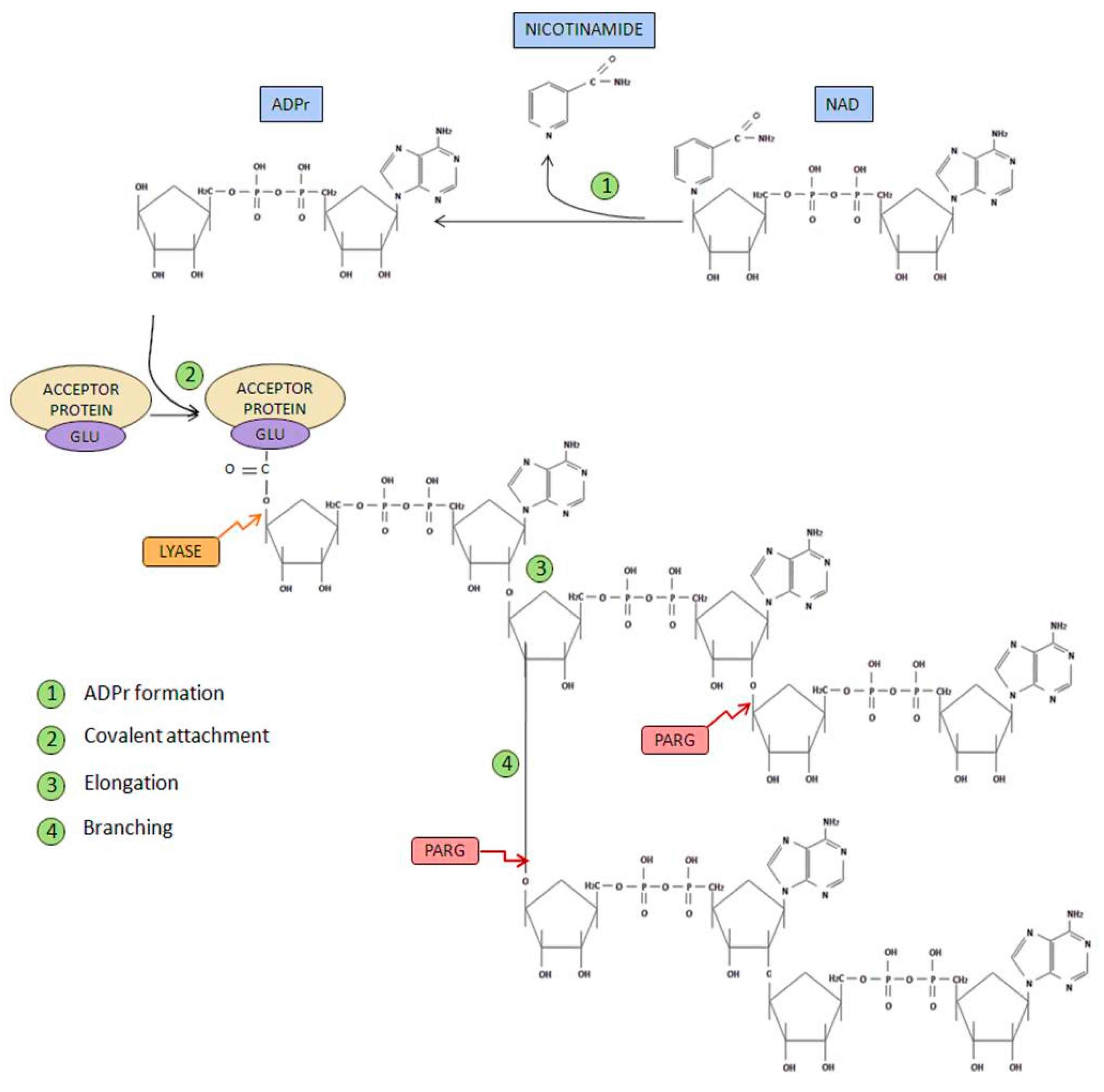
- Initiation phase: Firstly, poly(ADP-ribose) synthetase activity catalyzes the breakage of the glycosidic bond between nicotinamide and ribose on the NAD+ molecule. Through this process of oxidation, ADP-ribose is formed. Subsequently, ADP-ribose is covalently attached to different acceptor proteins via formation of an ester bond between the protein (through glutamate, aspartate or lysine residues) and ADP-ribose.
- Elongation and branching reaction: In addition, PARP-1-mediated poly(ADP-ribosyl) transferase activity is able to catalyze the reactions responsible of elongation and branching, using more ADP-ribose units obtained from NAD+.
- Ester bond breakage: Once PAR has been degraded, the firstly attached mono(ADP-ribosyl) moiety bond to the acceptor protein is removed by the ADP-ribosyl protein lyase [16].
- AMP and NAD recycling: Free poly(ADP-ribose) and ADP-ribose monomer are the final products of PAR degradation, this latter molecule can cause protein damage through glycation processes. ADP-ribose pyrophosphatase [17] converts this free ADP-ribose into AMP and ribose 5-phosphate, generating compounds much less reactive and more likely to be used in order to obtain new NAD+ [18].
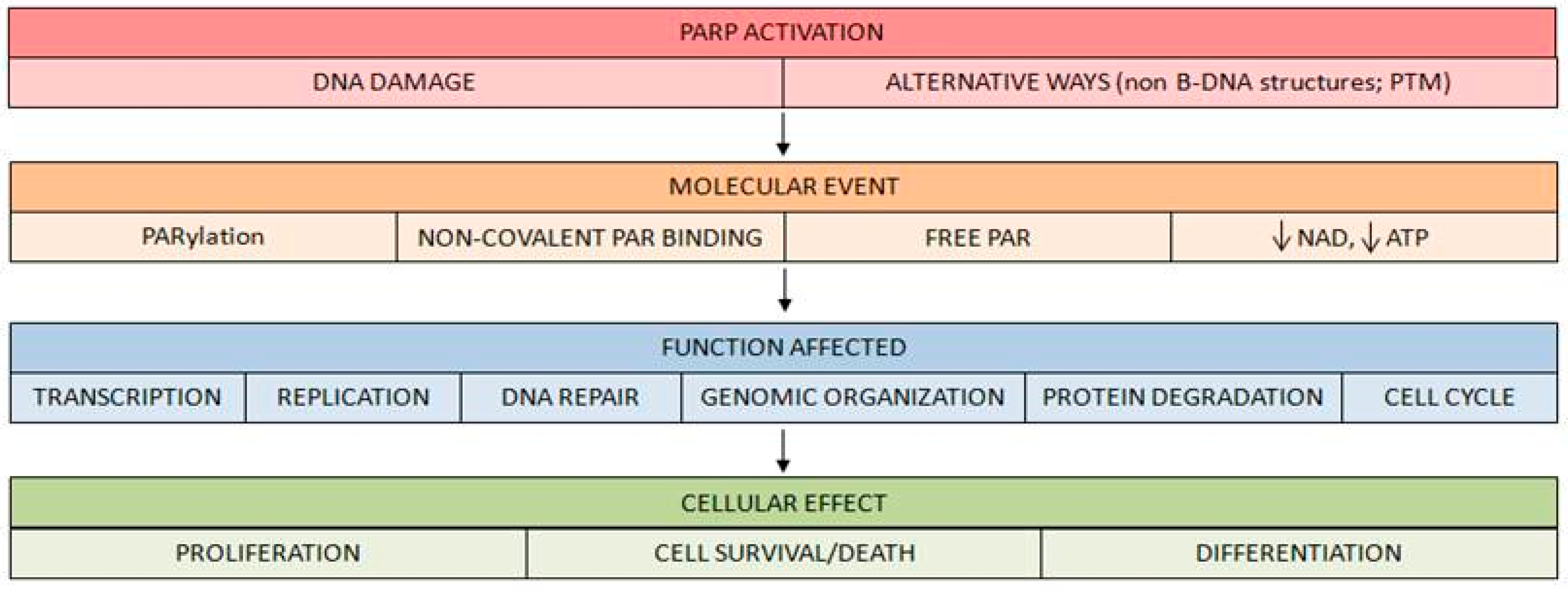
- PARP activation generates an important reduction of its substrate NAD+ after high DNA breakage accumulation. This depletion has important consequences on cell survival [28].
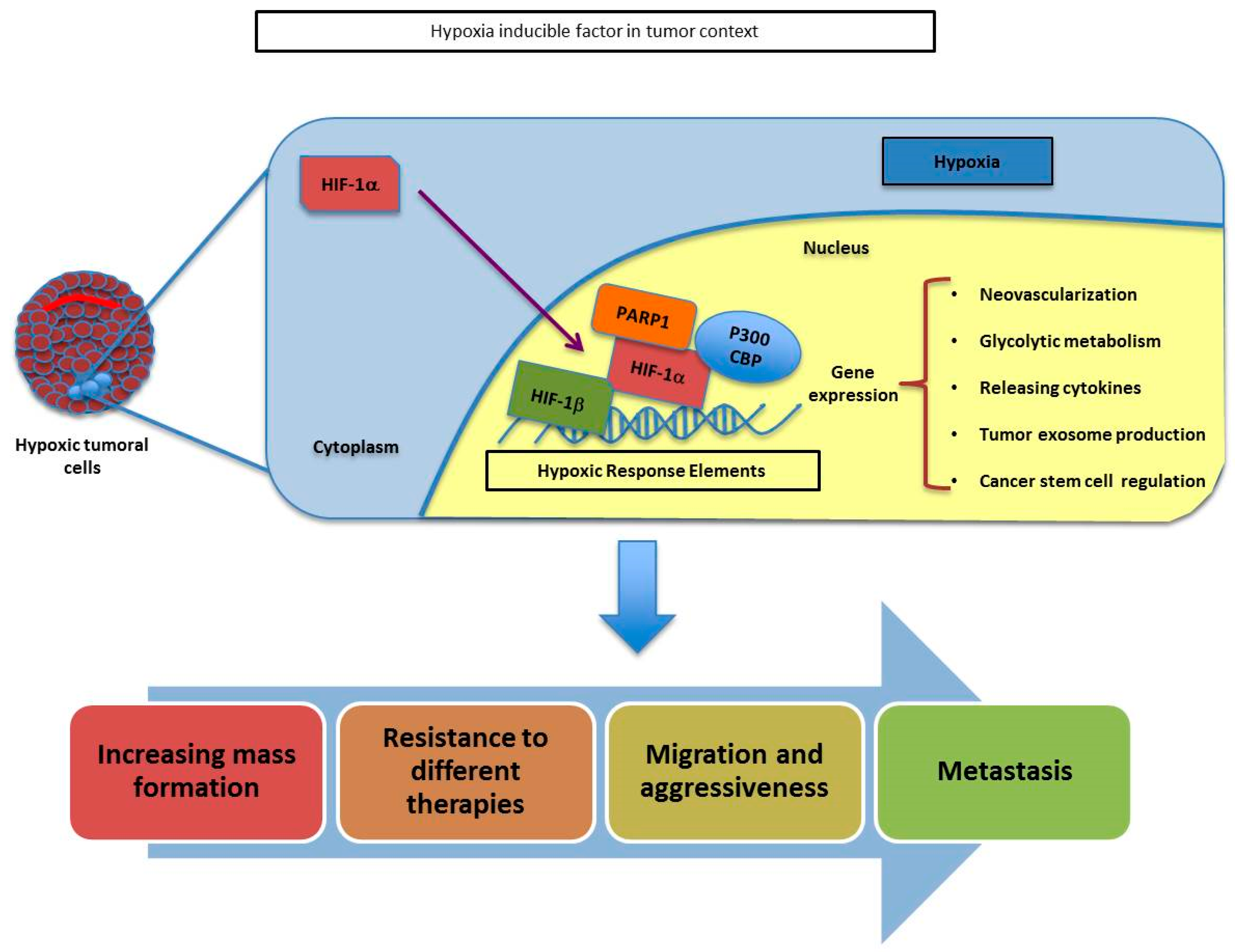
1.2. Tumor Hypoxic Response and PARP-1
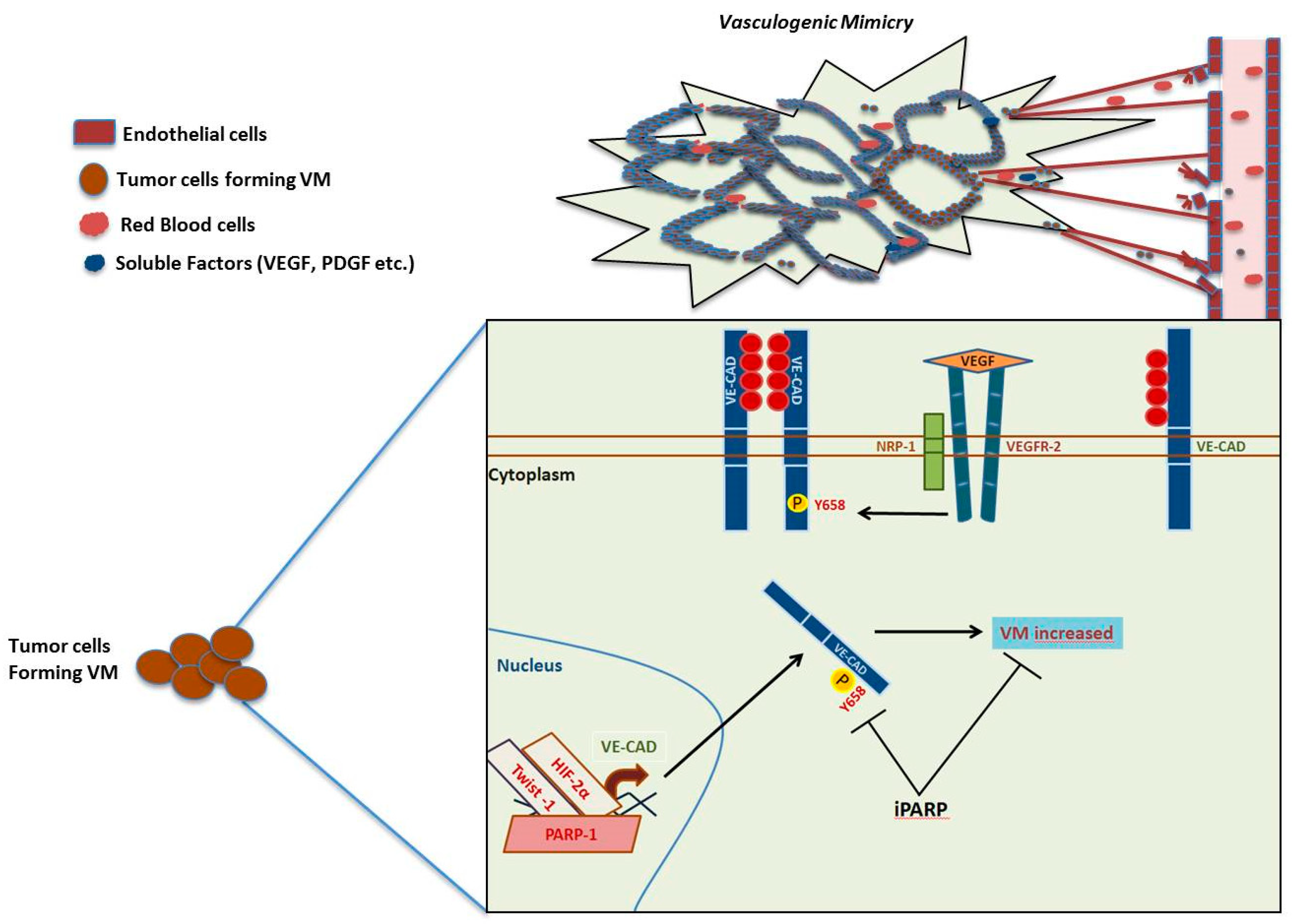
1.3. Angiogenesis, Vasculogenic Mimicry and PARP-1
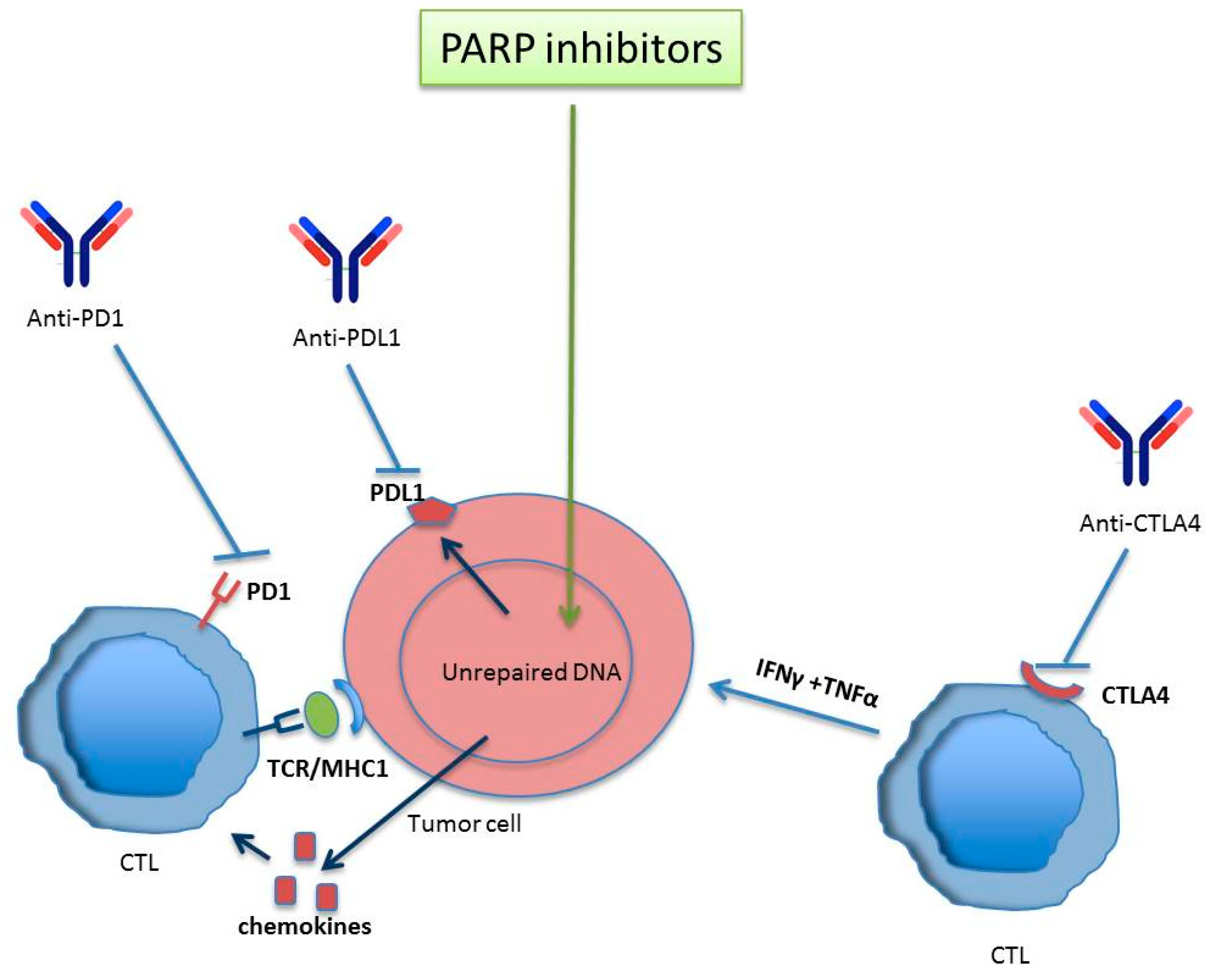
2. Immuno-Response Modulation by PARP
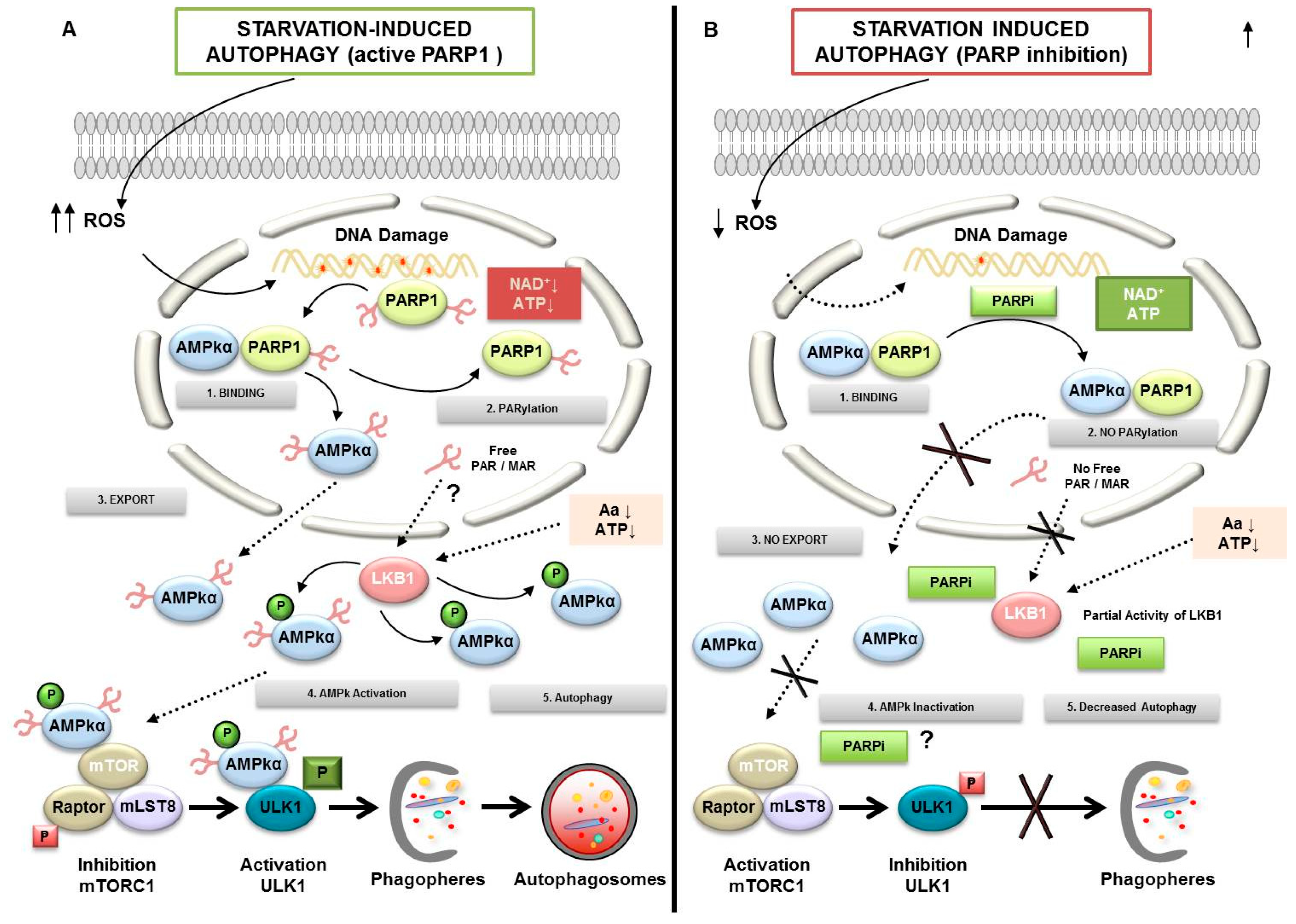
3. PARylation in Autophagy
4. Cancer Initiating Cells
5. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Hottiger, M.O.; Hassa, P.O.; Luscher, B.; Schuler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Peralta-Leal, A.; Rodriguez-Vargas, J.M.; Aguilar-Quesada, R.; Rodriguez, M.I.; Linares, J.L.; de Almodovar, M.R.; Oliver, F.J. PARP inhibitors: New partners in the therapy of cancer and inflammatory diseases. Free Radic. Biol. Med. 2009, 47, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Chambon, P.; Weill, J.D.; Mandel, P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 1963, 11, 39–43. [Google Scholar] [CrossRef]
- Meyer-Ficca, M.L.; Meyer, R.G.; Jacobson, E.L.; Jacobson, M.K. Poly (ADP-ribose) polymerases: Managing genome stability. Int. J. Biochem. Cell Biol. 2005, 37, 920–926. [Google Scholar] [CrossRef]
- Meyer-Ficca, M.L.; Meyer, R.G.; Coyle, D.L.; Jacobson, E.L.; Jacobson, M.K. Human poly (ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms that localize to different cell compartments. Exp. Cell Res. 2004, 297, 521–532. [Google Scholar] [CrossRef]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef]
- Hassa, P.O.; Hottiger, M.O. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 2008, 13, 3046–3082. [Google Scholar] [CrossRef]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly (ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly (ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Alemasova, E.E.; Lavrik, O.I. Poly (ADP-ribosyl) ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef]
- Kameshita, I.; Matsuda, Z.; Taniguchi, T.; Shizuta, Y. Poly (ADP-Ribose) synthetase. Separation and identification of three proteolytic fragments as the substrate-binding domain, the DNA-binding domain, and the automodification domain. J. Biol. Chem. 1984, 259, 4770–4776. [Google Scholar] [PubMed]
- Desmarais, Y.; Menard, L.; Lagueux, J.; Poirier, G.G. Enzymological properties of poly (ADP-ribose) polymerase: Characterization of automodification sites and NADase activity. Biochim. Biophys. Acta 1991, 1078, 179–186. [Google Scholar] [CrossRef]
- Bork, P.; Hofmann, K.; Bucher, P.; Neuwald, A.F.; Altschul, S.F.; Koonin, E.V. A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. FASEB J. 1997, 11, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, J.; Burkle, A. Introduction to poly (ADP-ribose) metabolism. Cell. Mol. Life Sci. 2005, 62, 721–730. [Google Scholar] [CrossRef]
- Miwa, M.; Sugimura, T. Splitting of the ribose-ribose linkage of poly (adenosine diphosphate-robose) by a calf thymus extract. J. Biol. Chem. 1971, 246, 6362–6364. [Google Scholar]
- Oka, J.; Ueda, K.; Hayaishi, O.; Komura, H.; Nakanishi, K. ADP-ribosyl protein lyase. Purification, properties, and identification of the product. J. Biol. Chem. 1984, 259, 986–995. [Google Scholar]
- Bernet, D.; Pinto, R.M.; Costas, M.J.; Canales, J.; Cameselle, J.C. Rat liver mitochondrial ADP-ribose pyrophosphatase in the matrix space with low Km for free ADP-ribose. Biochem. J. 1994, 299, 679–682. [Google Scholar] [CrossRef]
- Kim, M.Y.; Zhang, T.; Kraus, W.L. Poly (ADP-ribosyl) ation by PARP-1: PAR-laying NAD+ into a nuclear signal. Genes Dev. 2005, 19, 1951–1967. [Google Scholar] [CrossRef]
- Panzeter, P.L.; Realini, C.A.; Althaus, F.R. Noncovalent interactions of poly (adenosine diphosphate ribose) with histones. Biochemistry 1992, 31, 1379–1385. [Google Scholar] [CrossRef]
- Sauermann, G.; Wesierska-Gadek, J. Poly (ADP-ribose) effectively competes with DNA for histone H4 binding. Biochem. Biophys. Res. Commun. 1986, 139, 523–529. [Google Scholar] [CrossRef]
- Pleschke, J.M.; Kleczkowska, H.E.; Strohm, M.; Althaus, F.R. Poly (ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J. Biol. Chem. 2000, 275, 40974–40980. [Google Scholar] [CrossRef] [PubMed]
- Ahel, I.; Ahel, D.; Matsusaka, T.; Clark, A.J.; Pines, J.; Boulton, S.J.; West, S.C. Poly (ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature 2008, 451, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Timinszky, G.; Till, S.; Hassa, P.O.; Hothorn, M.; Kustatscher, G.; Nijmeijer, B.; Colombelli, J.; Altmeyer, M.; Stelzer, E.H.; Scheffzek, K.; et al. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 2009, 16, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.A.; Kim, N.S.; Yu, S.W.; Wang, H.; Koh, D.W.; Sasaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, R.C.; et al. Poly (ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA 2006, 103, 18308–18313. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kim, N.S.; Haince, J.F.; Kang, H.C.; David, K.K.; Andrabi, S.A.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly (ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal. 2011, 4, ra20. [Google Scholar] [CrossRef]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef]
- Yu, S.W.; Andrabi, S.A.; Wang, H.; Kim, N.S.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor mediates poly (ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319. [Google Scholar] [CrossRef]
- Berger, N.A.; Sims, J.L.; Catino, D.M.; Berger, S.J. Poly (ADP-ribose) polymerase mediates the suicide response to massive DNA damage: Studies in normal and DNA-repair defective cells. Princess Takamatsu Symp. 1983, 13, 219–226. [Google Scholar]
- de Murcia, G.; Schreiber, V.; Molinete, M.; Saulier, B.; Poch, O.; Masson, M.; Niedergang, C.; Menissier de Murcia, J. Structure and function of poly(ADP-ribose) polymerase. Mol. Cell. Biochem. 1994, 138, 15–24. [Google Scholar] [CrossRef]
- Burkle, A.; Virag, L. Poly (ADP-ribose): PARadigms and PARadoxes. Mol. Asp. Med. 2013, 34, 1046–1065. [Google Scholar] [CrossRef]
- Lonskaya, I.; Potaman, V.N.; Shlyakhtenko, L.S.; Oussatcheva, E.A.; Lyubchenko, Y.L.; Soldatenkov, V.A. Regulation of poly(ADP-ribose) polymerase-1 by DNA structure-specific binding. J. Biol. Chem. 2005, 280, 17076–17083. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, T.M.; Chan, W.Y.; Suh, S.W.; Wiggins, A.K.; Huang, E.J.; Swanson, R.A. Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proc. Natl. Acad. Sci. USA 2006, 103, 7136–7141. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Yamaguchi, H.; Wei, Y.; Hsu, J.L.; Wang, H.L.; Hsu, Y.H.; Lin, W.C.; Yu, W.H.; Leonard, P.G.; Lee, G.R.t.; et al. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat. Med. 2016, 22, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Armon, M. PARP-1 activation in the ERK signaling pathway. Trends Pharmacol. Sci. 2007, 28, 556–560. [Google Scholar] [CrossRef]
- Cohen-Armon, M.; Visochek, L.; Rozensal, D.; Kalal, A.; Geistrikh, I.; Klein, R.; Bendetz-Nezer, S.; Yao, Z.; Seger, R. DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: A link to histone acetylation. Mol. Cell 2007, 25, 297–308. [Google Scholar] [CrossRef]
- Gongol, B.; Marin, T.; Peng, I.C.; Woo, B.; Martin, M.; King, S.; Sun, W.; Johnson, D.A.; Chien, S.; Shyy, J.Y. AMPKalpha2 exerts its anti-inflammatory effects through PARP-1 and Bcl-6. Proc. Natl. Acad. Sci. USA 2013, 110, 3161–3166. [Google Scholar] [CrossRef]
- Beckert, S.; Farrahi, F.; Perveen Ghani, Q.; Aslam, R.; Scheuenstuhl, H.; Coerper, S.; Konigsrainer, A.; Hunt, T.K.; Hussain, M.Z. IGF-I-induced VEGF expression in HUVEC involves phosphorylation and inhibition of poly(ADP-ribose)polymerase. Biochem. Biophys. Res. Commun. 2006, 341, 67–72. [Google Scholar] [CrossRef]
- Boussios, S.; Karathanasi, A.; Cooke, D.; Neille, C.; Sadauskaite, A.; Moschetta, M.; Zakynthinakis-Kyriakou, N.; Pavlidis, N. PARP Inhibitors in Ovarian Cancer: The Route to “Ithaca”. Diagnostics 2019, 9, 55. [Google Scholar] [CrossRef]
- Hassa, P.O.; Haenni, S.S.; Buerki, C.; Meier, N.I.; Lane, W.S.; Owen, H.; Gersbach, M.; Imhof, R.; Hottiger, M.O. Acetylation of poly(ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-kappaB-dependent transcription. J. Biol. Chem. 2005, 280, 40450–40464. [Google Scholar] [CrossRef]
- Martin, N.; Schwamborn, K.; Schreiber, V.; Werner, A.; Guillier, C.; Zhang, X.D.; Bischof, O.; Seeler, J.S.; Dejean, A. PARP-1 transcriptional activity is regulated by sumoylation upon heat shock. EMBO J. 2009, 28, 3534–3548. [Google Scholar] [CrossRef]
- Loseva, O.; Jemth, A.S.; Bryant, H.E.; Schuler, H.; Lehtio, L.; Karlberg, T.; Helleday, T. PARP-3 is a mono-ADP-ribosylase that activates PARP-1 in the absence of DNA. J. Biol. Chem. 2010, 285, 8054–8060. [Google Scholar] [CrossRef] [PubMed]
- Thomlinson, R.H.; Gray, L.H. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Semenza, G.L. Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.W.; Tong, G.H.; Liu, Y. Cancer stem cells and hypoxia-inducible factors (Review). Int. J. Oncol. 2018, 53, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Yang, F.; Miao, S.; Liu, W.; Wang, C.; Shu, Y.; Shen, H. Role of hypoxia-induced exosomes in tumor biology. Mol. Cancer 2018, 17, 120. [Google Scholar] [CrossRef] [PubMed]
- Yehya, A.H.S.; Asif, M.; Petersen, S.H.; Subramaniam, A.V.; Kono, K.; Majid, A.; Oon, C.E. Angiogenesis: Managing the Culprits behind Tumorigenesis and Metastasis. Medicina 2018, 54, 8. [Google Scholar] [CrossRef]
- Sooriakumaran, P.; Kaba, R. Angiogenesis and the tumour hypoxia response in prostate cancer: A review. Int. J. Surg. 2005, 3, 61–67. [Google Scholar] [CrossRef][Green Version]
- Terry, S.; Faouzi Zaarour, R.; Hassan Venkatesh, G.; Francis, A.; El-Sayed, W.; Buart, S.; Bravo, P.; Thiery, J.; Chouaib, S. Role of Hypoxic Stress in Regulating Tumor Immunogenicity, Resistance and Plasticity. Int. J. Mol. Sci. 2018, 19, 44. [Google Scholar] [CrossRef]
- Rofstad, E.K.; Danielsen, T. Hypoxia-induced metastasis of human melanoma cells: Involvement of vascular endothelial growth factor-mediated angiogenesis. Br. J. Cancer 1999, 80, 1697–1707. [Google Scholar] [CrossRef]
- Graham, K.; Unger, E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomed. 2018, 13, 6049–6058. [Google Scholar] [CrossRef]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 transcription factors--similar but not identical. Mol. Cells 2010, 29, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Martin-Oliva, D.; Aguilar-Quesada, R.; O’Valle, F.; Munoz-Gamez, J.A.; Martinez-Romero, R.; Garcia Del Moral, R.; Ruiz de Almodovar, J.M.; Villuendas, R.; Piris, M.A.; Oliver, F.J. Inhibition of poly(ADP-ribose) polymerase modulates tumor-related gene expression, including hypoxia-inducible factor-1 activation, during skin carcinogenesis. Cancer Res. 2006, 66, 5744–5756. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Romero, R.; Canuelo, A.; Martinez-Lara, E.; Javier Oliver, F.; Cardenas, S.; Siles, E. Poly(ADP-ribose) polymerase-1 modulation of in vivo response of brain hypoxia-inducible factor-1 to hypoxia/reoxygenation is mediated by nitric oxide and factor inhibiting HIF. J. Neurochem. 2009, 111, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Elser, M.; Borsig, L.; Hassa, P.O.; Erener, S.; Messner, S.; Valovka, T.; Keller, S.; Gassmann, M.; Hottiger, M.O. Poly(ADP-ribose) polymerase 1 promotes tumor cell survival by coactivating hypoxia-inducible factor-1-dependent gene expression. Mol. Cancer Res. 2008, 6, 282–290. [Google Scholar] [CrossRef]
- Canuelo, A.; Martinez-Romero, R.; Martinez-Lara, E.; Sanchez-Alcazar, J.A.; Siles, E. The hypoxic preconditioning agent deferoxamine induces poly (ADP-ribose) polymerase-1-dependent inhibition of the mitochondrial respiratory chain. Mol. Cell. Biochem. 2012, 363, 101–108. [Google Scholar] [CrossRef]
- Liu, S.K.; Coackley, C.; Krause, M.; Jalali, F.; Chan, N.; Bristow, R.G. A novel poly (ADP-ribose) polymerase inhibitor, ABT-888, radiosensitizes malignant human cell lines under hypoxia. Radiother. Oncol. 2008, 88, 258–268. [Google Scholar] [CrossRef]
- Holmquist-Mengelbier, L.; Fredlund, E.; Lofstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, A.; Gradin, K.; et al. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423. [Google Scholar] [CrossRef]
- Raval, R.R.; Lau, K.W.; Tran, M.G.; Sowter, H.M.; Mandriota, S.J.; Li, J.L.; Pugh, C.W.; Maxwell, P.H.; Harris, A.L.; Ratcliffe, P.J. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol. Cell. Biol. 2005, 25, 5675–5686. [Google Scholar] [CrossRef]
- Aguilar-Quesada, R.; Munoz-Gamez, J.A.; Martin-Oliva, D.; Peralta-Leal, A.; Quiles-Perez, R.; Rodriguez-Vargas, J.M.; Ruiz de Almodovar, M.; Conde, C.; Ruiz-Extremera, A.; Oliver, F.J. Modulation of transcription by PARP-1: Consequences in carcinogenesis and inflammation. Curr. Med. Chem. 2007, 14, 1179–1187. [Google Scholar] [CrossRef]
- Gonzalez-Flores, A.; Aguilar-Quesada, R.; Siles, E.; Pozo, S.; Rodriguez-Lara, M.I.; Lopez-Jimenez, L.; Lopez-Rodriguez, M.; Peralta-Leal, A.; Villar, D.; Martin-Oliva, D.; et al. Interaction between PARP-1 and HIF-2alpha in the hypoxic response. Oncogene 2014, 33, 891–898. [Google Scholar] [CrossRef]
- Natale, G.; Bocci, G.; Lenzi, P. Looking for the Word “Angiogenesis” in the History of Health Sciences: From Ancient Times to the First Decades of the Twentieth Century. World J. Surg. 2017, 41, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Dome, B.; Hendrix, M.J.; Paku, S.; Tovari, J.; Timar, J. Alternative vascularization mechanisms in cancer: Pathology and therapeutic implications. Am. J. Pathol. 2007, 170. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Bilecz, A.; Daley, F.; Kostaras, E.; Nathan, M.R.; Wan, E.; Frentzas, S.; Schweiger, T.; et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J. Pathol. 2017, 241, 362–374. [Google Scholar] [CrossRef]
- Oza, A.M.; Cook, A.D.; Pfisterer, J.; Embleton, A.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; et al. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): Overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015, 16, 928–936. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Boussios, S.; Karihtala, P.; Moschetta, M.; Abson, C.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Ryan, J.E.; Sheriff, M.; Rassy, E.; Pavlidis, N. Veliparib in ovarian cancer: A new synthetically lethal therapeutic approach. Investig. New Drugs 2020, 38, 181–193. [Google Scholar] [CrossRef]
- Tentori, L.; Lacal, P.M.; Muzi, A.; Dorio, A.S.; Leonetti, C.; Scarsella, M.; Ruffini, F.; Xu, W.; Min, W.; Stoppacciaro, A.; et al. Poly (ADP-ribose) polymerase (PARP) inhibition or PARP-1 gene deletion reduces angiogenesis. Eur. J. Cancer 2007, 43, 2124–2133. [Google Scholar] [CrossRef]
- Pyriochou, A.; Olah, G.; Deitch, E.A.; Szabo, C.; Papapetropoulos, A. Inhibition of angiogenesis by the poly (ADP-ribose) polymerase inhibitor PJ-34. Int. J. Mol. Med. 2008, 22, 113–118. [Google Scholar] [CrossRef]
- Lacal, P.M.; Tentori, L.; Muzi, A.; Ruffini, F.; Dorio, A.S.; Xu, W.; Arcelli, D.; Zhang, J.; Graziani, G. Pharmacological inhibition of poly(ADP-ribose) polymerase activity down-regulates the expression of syndecan-4 and Id-1 in endothelial cells. Int. J. Oncol. 2009, 34, 861–872. [Google Scholar]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Seftor, E.A.; Meltzer, P.S.; Kirschmann, D.A.; Pe’er, J.; Maniotis, A.J.; Trent, J.M.; Folberg, R.; Hendrix, M.J. Molecular determinants of human uveal melanoma invasion and metastasis. Clin. Exp. Metastasis 2002, 19, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Bittner, M.; Meltzer, P.; Chen, Y.; Jiang, Y.; Seftor, E.; Hendrix, M.; Radmacher, M.; Simon, R.; Yakhini, Z.; Ben-Dor, A.; et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 2000, 406, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, M.J.; Seftor, E.A.; Meltzer, P.S.; Gardner, L.M.; Hess, A.R.; Kirschmann, D.A.; Schatteman, G.C.; Seftor, R.E. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: Role in vasculogenic mimicry. Proc. Natl. Acad. Sci. USA 2001, 98, 8018–8023. [Google Scholar] [CrossRef] [PubMed]
- Ruffini, F.; Graziani, G.; Levati, L.; Tentori, L.; D’Atri, S.; Lacal, P.M. Cilengitide downmodulates invasiveness and vasculogenic mimicry of neuropilin 1 expressing melanoma cells through the inhibition of alphavbeta5 integrin. Int. J. Cancer 2015, 136, E545–E558. [Google Scholar] [CrossRef] [PubMed]
- Pagani, E.; Ruffini, F.; Antonini Cappellini, G.C.; Scoppola, A.; Fortes, C.; Marchetti, P.; Graziani, G.; D’Atri, S.; Lacal, P.M. Placenta growth factor and neuropilin-1 collaborate in promoting melanoma aggressiveness. Int. J. Oncol. 2016, 48, 1581–1589. [Google Scholar] [CrossRef]
- Schnegg, C.I.; Yang, M.H.; Ghosh, S.K.; Hsu, M.Y. Induction of Vasculogenic Mimicry Overrides VEGF-A Silencing and Enriches Stem-like Cancer Cells in Melanoma. Cancer Res. 2015, 75, 1682–1690. [Google Scholar] [CrossRef]
- Rodriguez, M.I.; Peralta-Leal, A.; O’Valle, F.; Rodriguez-Vargas, J.M.; Gonzalez-Flores, A.; Majuelos-Melguizo, J.; Lopez, L.; Serrano, S.; de Herreros, A.G.; Rodriguez-Manzaneque, J.C.; et al. PARP-1 regulates metastatic melanoma through modulation of vimentin-induced malignant transformation. PLoS Genet. 2013, 9, e1003531. [Google Scholar] [CrossRef]
- Crawford, Y.; Ferrara, N. VEGF inhibition: Insights from preclinical and clinical studies. Cell Tissue Res. 2009, 335, 261–269. [Google Scholar] [CrossRef]
- Nakasone, E.S.; Hurvitz, S.A.; McCann, K.E. Harnessing the immune system in the battle against breast cancer. Drugs Context 2018, 7, 212520. [Google Scholar] [CrossRef]
- Bai, P.; Virag, L. Role of poly (ADP-ribose) polymerases in the regulation of inflammatory processes. FEBS Lett. 2012, 586, 3771–3777. [Google Scholar] [CrossRef] [PubMed]
- Stilmann, M.; Hinz, M.; Arslan, S.C.; Zimmer, A.; Schreiber, V.; Scheidereit, C. A nuclear poly (ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol. Cell 2009, 36, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Oliver, F.J.; Menissier-de Murcia, J.; Nacci, C.; Decker, P.; Andriantsitohaina, R.; Muller, S.; de la Rubia, G.; Stoclet, J.C.; de Murcia, G. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999, 18, 4446–4454. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Stilmann, M.; Arslan, S.C.; Khanna, K.K.; Dittmar, G.; Scheidereit, C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-kappaB activation. Mol. Cell 2010, 40, 63–74. [Google Scholar] [CrossRef]
- Ha, H.C.; Hester, L.D.; Snyder, S.H. Poly (ADP-ribose) polymerase-1 dependence of stress-induced transcription factors and associated gene expression in glia. Proc. Natl. Acad. Sci. USA 2002, 99, 3270–3275. [Google Scholar] [CrossRef]
- Olabisi, O.A.; Soto-Nieves, N.; Nieves, E.; Yang, T.T.; Yang, X.; Yu, R.Y.; Suk, H.Y.; Macian, F.; Chow, C.W. Regulation of transcription factor NFAT by ADP-ribosylation. Mol. Cell. Biol. 2008, 28, 2860–2871. [Google Scholar] [CrossRef]
- Bai, P.; Canto, C.; Brunyanszki, A.; Huber, A.; Szanto, M.; Cen, Y.; Yamamoto, H.; Houten, S.M.; Kiss, B.; Oudart, H.; et al. PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab. 2011, 13, 450–460. [Google Scholar] [CrossRef]
- Szanto, M.; Brunyanszki, A.; Kiss, B.; Nagy, L.; Gergely, P.; Virag, L.; Bai, P. Poly (ADP-ribose) polymerase-2: Emerging transcriptional roles of a DNA-repair protein. Cell. Mol. Life Sci. 2012, 69, 4079–4092. [Google Scholar] [CrossRef]
- Mehrotra, P.; Riley, J.P.; Patel, R.; Li, F.; Voss, L.; Goenka, S. PARP-14 functions as a transcriptional switch for Stat6-dependent gene activation. J. Biol. Chem. 2011, 286, 1767–1776. [Google Scholar] [CrossRef]
- Rosado, M.M.; Bennici, E.; Novelli, F.; Pioli, C. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology 2013, 139, 428–437. [Google Scholar] [CrossRef]
- Yelamos, J.; Monreal, Y.; Saenz, L.; Aguado, E.; Schreiber, V.; Mota, R.; Fuente, T.; Minguela, A.; Parrilla, P.; de Murcia, G.; et al. PARP-2 deficiency affects the survival of CD4+CD8+ double-positive thymocytes. EMBO J. 2006, 25, 4350–4360. [Google Scholar] [CrossRef] [PubMed]
- Sambucci, M.; Laudisi, F.; Novelli, F.; Bennici, E.; Rosado, M.M.; Pioli, C. Effects of PARP-1 deficiency on Th1 and Th2 cell differentiation. Sci. World J. 2013, 2013, 375024. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Rohl, I.; Hopfner, K.P.; Ludwig, J.; Hornung, V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef]
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef]
- Muthuswamy, R.; Berk, E.; Junecko, B.F.; Zeh, H.J.; Zureikat, A.H.; Normolle, D.; Luong, T.M.; Reinhart, T.A.; Bartlett, D.L.; Kalinski, P. NF-kappaB hyperactivation in tumor tissues allows tumor-selective reprogramming of the chemokine microenvironment to enhance the recruitment of cytolytic T effector cells. Cancer Res. 2012, 72, 3735–3743. [Google Scholar] [CrossRef]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Linnemann, C.; van Buuren, M.M.; Bies, L.; Verdegaal, E.M.; Schotte, R.; Calis, J.J.; Behjati, S.; Velds, A.; Hilkmann, H.; Atmioui, D.E.; et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med. 2015, 21, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Yao, X.; Crystal, J.S.; Li, Y.F.; El-Gamil, M.; Gross, C.; Davis, L.; Dudley, M.E.; Yang, J.C.; Samuels, Y.; et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin. Cancer Res. 2014, 20, 3401–3410. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef]
- Li, M.; Yu, X. The role of poly (ADP-ribosyl) ation in DNA damage response and cancer chemotherapy. Oncogene 2015, 34, 3349–3356. [Google Scholar] [CrossRef]
- Lee, J.M.; Gulley, J.L. Checkpoint and PARP inhibitors, for whom and when. Oncotarget 2017, 8, 95036–95037. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Wu, C.T.; Chen, W.C.; Chang, Y.H.; Lin, W.Y.; Chen, M.F. The role of PD-L1 in the radiation response and clinical outcome for bladder cancer. Sci. Rep. 2016, 6, 19740. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.X. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, L.; Cong, Z.; Amoozgar, Z.; Kiner, E.; Xing, D.; Orsulic, S.; Matulonis, U.; Goldberg, M.S. The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1 (-/-) murine model of ovarian cancer. Biochem. Biophys. Res. Commun. 2015, 463, 551–556. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Vargas, J.M.; Ruiz-Magana, M.J.; Ruiz-Ruiz, C.; Majuelos-Melguizo, J.; Peralta-Leal, A.; Rodriguez, M.I.; Munoz-Gamez, J.A.; de Almodovar, M.R.; Siles, E.; Rivas, A.L.; et al. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res. 2012, 22, 1181–1198. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Alexander, A.; Cai, S.L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar] [CrossRef]
- Tripathi, D.N.; Chowdhury, R.; Trudel, L.J.; Tee, A.R.; Slack, R.S.; Walker, C.L.; Wogan, G.N. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc. Natl. Acad. Sci. USA 2013, 110, E2950–E2957. [Google Scholar] [CrossRef]
- Munoz-Gamez, J.A.; Rodriguez-Vargas, J.M.; Quiles-Perez, R.; Aguilar-Quesada, R.; Martin-Oliva, D.; de Murcia, G.; Menissier de Murcia, J.; Almendros, A.; Ruiz de Almodovar, M.; Oliver, F.J. PARP-1 is involved in autophagy induced by DNA damage. Autophagy 2009, 5, 61–74. [Google Scholar] [CrossRef]
- Chen, Z.T.; Zhao, W.; Qu, S.; Li, L.; Lu, X.D.; Su, F.; Liang, Z.G.; Guo, S.Y.; Zhu, X.D. PARP-1 promotes autophagy via the AMPK/mTOR pathway in CNE-2 human nasopharyngeal carcinoma cells following ionizing radiation, while inhibition of autophagy contributes to the radiation sensitization of CNE-2 cells. Mol. Med. Rep. 2015, 12, 1868–1876. [Google Scholar] [CrossRef]
- Rodriguez-Vargas, J.M.; Rodriguez, M.I.; Majuelos-Melguizo, J.; Garcia-Diaz, A.; Gonzalez-Flores, A.; Lopez-Rivas, A.; Virag, L.; Illuzzi, G.; Schreiber, V.; Dantzer, F.; et al. Autophagy requires poly(adp-ribosyl)ation-dependent AMPK nuclear export. Cell Death Differ. 2016, 23, 2007–2018. [Google Scholar] [CrossRef] [PubMed]
- Shang, F.; Zhang, J.; Li, Z.; Zhang, J.; Yin, Y.; Wang, Y.; Marin, T.L.; Gongol, B.; Xiao, H.; Zhang, Y.Y.; et al. Cardiovascular Protective Effect of Metformin and Telmisartan: Reduction of PARP1 Activity via the AMPK-PARP1 Cascade. PLoS ONE 2016, 11, e0151845. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef]
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; Van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005, 65, 9328–9337. [Google Scholar] [CrossRef]
- Kim, C.F.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef]
- Bertolini, G.; Roz, L.; Perego, P.; Tortoreto, M.; Fontanella, E.; Gatti, L.; Pratesi, G.; Fabbri, A.; Andriani, F.; Tinelli, S.; et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc. Natl. Acad. Sci. USA 2009, 106, 16281–16286. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Chan, K.W.; Hu, L.; Lee, T.K.; Wo, J.Y.; Ng, I.O.; Zheng, B.J.; Guan, X.Y. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 2007, 132, 2542–2556. [Google Scholar] [CrossRef]
- Bussolati, B.; Bruno, S.; Grange, C.; Ferrando, U.; Camussi, G. Identification of a tumor-initiating stem cell population in human renal carcinomas. FASEB J. 2008, 22, 3696–3705. [Google Scholar] [CrossRef]
- Islam, F.; Qiao, B.; Smith, R.A.; Gopalan, V.; Lam, A.K. Cancer stem cell: Fundamental experimental pathological concepts and updates. Exp. Mol. Pathol. 2015, 98, 184–191. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Friedmann-Morvinski, D.; Verma, I.M. Dedifferentiation and reprogramming: Origins of cancer stem cells. EMBO Rep. 2014, 15, 244–253. [Google Scholar] [CrossRef]
- Kumar, S.M.; Liu, S.; Lu, H.; Zhang, H.; Zhang, P.J.; Gimotty, P.A.; Guerra, M.; Guo, W.; Xu, X. Acquired cancer stem cell phenotypes through Oct4-mediated dedifferentiation. Oncogene 2012, 31, 4898–4911. [Google Scholar] [CrossRef]
- Corominas-Faja, B.; Cufi, S.; Oliveras-Ferraros, C.; Cuyas, E.; Lopez-Bonet, E.; Lupu, R.; Alarcon, T.; Vellon, L.; Iglesias, J.M.; Leis, O.; et al. Nuclear reprogramming of luminal-like breast cancer cells generates Sox2-overexpressing cancer stem-like cellular states harboring transcriptional activation of the mTOR pathway. Cell Cycle 2013, 12, 3109–3124. [Google Scholar] [CrossRef]
- Chiou, S.H.; Jiang, B.H.; Yu, Y.L.; Chou, S.J.; Tsai, P.H.; Chang, W.C.; Chen, L.K.; Chen, L.H.; Chien, Y.; Chiou, G.Y. Poly (ADP-ribose) polymerase 1 regulates nuclear reprogramming and promotes iPSC generation without c-Myc. J. Exp. Med. 2013, 210, 85–98. [Google Scholar] [CrossRef]
- Weber, F.A.; Bartolomei, G.; Hottiger, M.O.; Cinelli, P. Artd1/Parp1 regulates reprogramming by transcriptional regulation of Fgf4 via Sox2 ADP-ribosylation. Stem Cells 2013, 31, 2364–2373. [Google Scholar] [CrossRef]
- Gao, F.; Kwon, S.W.; Zhao, Y.; Jin, Y. PARP1 poly (ADP-ribosyl) ates Sox2 to control Sox2 protein levels and FGF4 expression during embryonic stem cell differentiation. J. Biol. Chem. 2009, 284, 22263–22273. [Google Scholar] [CrossRef]
- Doege, C.A.; Inoue, K.; Yamashita, T.; Rhee, D.B.; Travis, S.; Fujita, R.; Guarnieri, P.; Bhagat, G.; Vanti, W.B.; Shih, A.; et al. Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature 2012, 488, 652–655. [Google Scholar] [CrossRef]
- Hsieh, M.H.; Chen, Y.T.; Chen, Y.T.; Lee, Y.H.; Lu, J.; Chien, C.L.; Chen, H.F.; Ho, H.N.; Yu, C.J.; Wang, Z.Q.; et al. PARP1 controls KLF4-mediated telomerase expression in stem cells and cancer cells. Nucleic Acids Res. 2017, 45, 10492–10503. [Google Scholar] [CrossRef]
- Marion, R.M.; Strati, K.; Li, H.; Tejera, A.; Schoeftner, S.; Ortega, S.; Serrano, M.; Blasco, M.A. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 2009, 4, 141–154. [Google Scholar] [CrossRef]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef]
- Piccirillo, S.G.; Reynolds, B.A.; Zanetti, N.; Lamorte, G.; Binda, E.; Broggi, G.; Brem, H.; Olivi, A.; Dimeco, F.; Vescovi, A.L. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006, 444, 761–765. [Google Scholar] [CrossRef]
- Watanabe, Y.; Papoutsoglou, P.; Maturi, V.; Tsubakihara, Y.; Hottiger, M.O.; Heldin, C.H.; Moustakas, A. Regulation of Bone Morphogenetic Protein Signaling by ADP-ribosylation. J. Biol. Chem. 2016, 291, 12706–12723. [Google Scholar] [CrossRef]
- Lonn, P.; van der Heide, L.P.; Dahl, M.; Hellman, U.; Heldin, C.H.; Moustakas, A. PARP-1 attenuates Smad-mediated transcription. Mol. Cell 2010, 40, 521–532. [Google Scholar] [CrossRef]
- Penuelas, S.; Anido, J.; Prieto-Sanchez, R.M.; Folch, G.; Barba, I.; Cuartas, I.; Garcia-Dorado, D.; Poca, M.A.; Sahuquillo, J.; Baselga, J.; et al. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell 2009, 15, 315–327. [Google Scholar] [CrossRef]
- Faraoni, I.; Graziani, G. Role of BRCA Mutations in Cancer Treatment with Poly (ADP-ribose) Polymerase (PARP) Inhibitors. Cancers 2018, 10, 487. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
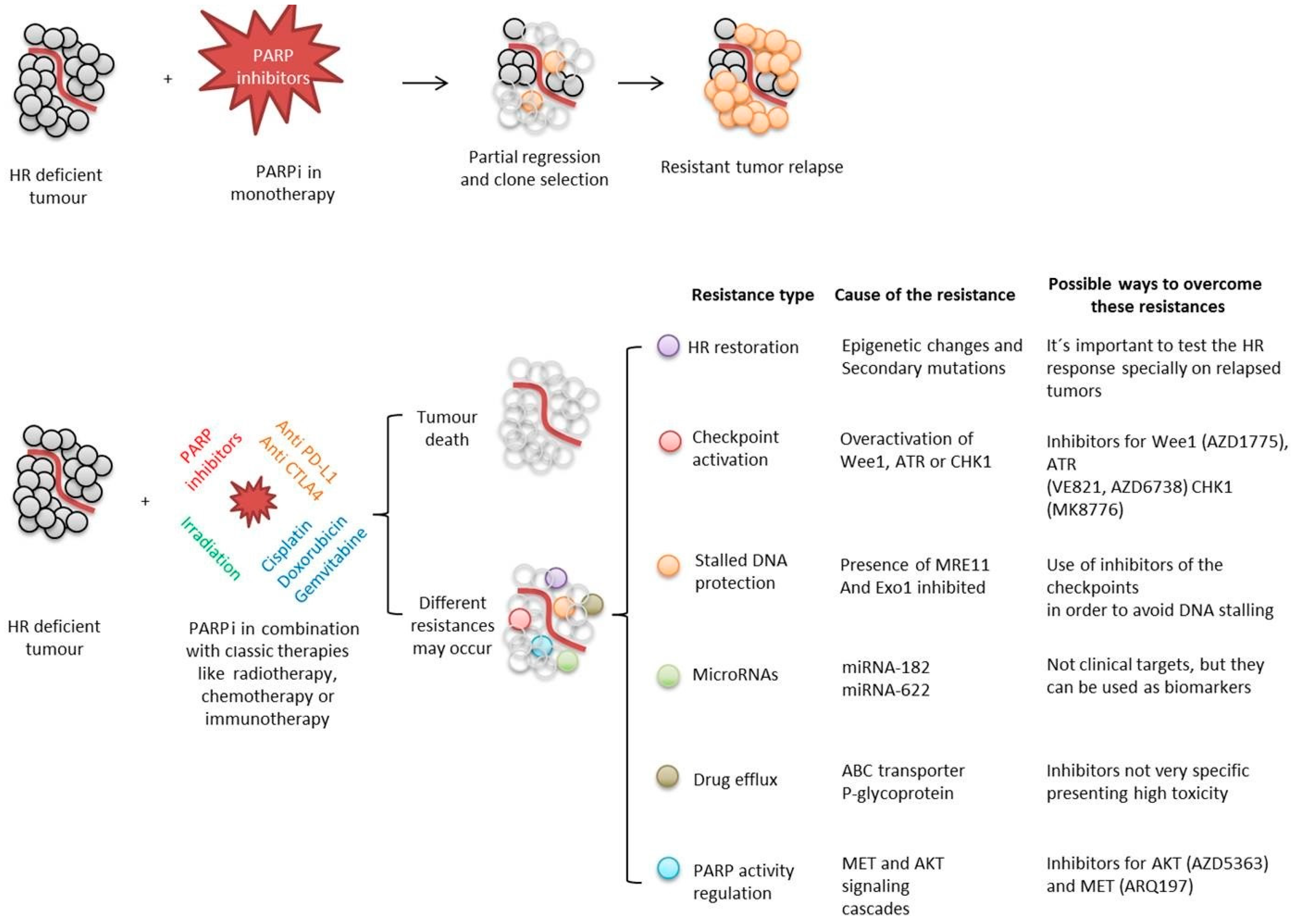{kind=link}
| Status | Study Title | Conditions | Interventions | Phase | Number Enrolled | NCT Number |
|---|---|---|---|---|---|---|
| Active, not recruiting | A Phase 3 Randomized, Placebo-controlled Trial of Carboplatin and Paclitaxel With or Without Veliparib (ABT-888) in HER2-negative Metastatic or Locally Advanced Unresectable BRCA-associated Breast Cancer | Metastatic Breast Cancer | Drug: Paclitaxel Drug: Veliparib Drug: Carboplatin Other: Placebo | Phase3 | 513 | NCT02163694 |
| Active, not recruiting | Veliparib With Carboplatin and Paclitaxel and as Continuation Maintenance Therapy in Subjects With Newly Diagnosed Stage III or IV, High-grade Serous, Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cancer | Ovarian Cancer Ovarian Neoplasm | Drug: Veliparib Drug: Paclitaxel Drug: Carboplatin Other: Placebo | Phase 3 | 1140 | NCT02470585 |
| Active, not recruiting (Has results) | Olaparib Treatment in BRCA Mutated Ovarian Cancer Patients After Complete or Partial Response to Platinum Chemotherapy | Platinum Sensitive BRCA Mutated Relapsed Ovarian Cancer Following Complete or Partial Response to Platinum Based Chemotherapy | Drug: Olaparib 300mg tablets Drug: Placebo to match olaparib 300mg | Phase 3 | 327 | NCT01874353 |
| Active, not recruiting | A Phase III Trial of Niraparib Versus Physician’s Choice in HER2 Negative, Germline BRCA Mutation-positive Breast Cancer Patients | Carcinoma of Breast Human Epidermal Growth Factor 2 Negative Carcinoma of Breast BRCA1 Gene Mutation BRCA2 Gene Mutation | Drug: niraparib Drug: Physician’s choice | Phase 3 | 306 | NCT01905592 |
| Active, not recruiting | A Study of Niraparib Maintenance Treatment in Patients With Advanced Ovarian Cancer Following Response on Front-Line Platinum-Based Chemotherapy | Ovarian Cancer | Drug: Niraparib Drug: Placebo | Phase 3 | 620 | NCT02655016 |
| Active, not recruiting (Has results) | A Study of Rucaparib as Switch Maintenance Following Platinum-Based Chemotherapy in Patients With Platinum-Sensitive, High-Grade Serous or Endometrioid Epithelial Ovarian, Primary Peritoneal or Fallopian Tube Cancer | Ovarian Cancer Fallopian Tube Cancer Peritoneal Cancer | Drug: Rucaparib Drug: Placebo | Phase 3 | 564 | NCT01968213 |
| Active, not recruiting (Has results) | Assessment of the Efficacy and Safety of Olaparib Monotherapy Versus Physicians Choice Chemotherapy in the Treatment of Metastatic Breast Cancer Patients With Germline BRCA1/2 Mutations. | Breast Cancer Metastatic BRCA 1 Gene Mutation BRCA 2 Gene Mutation | Drug: Olaparib Drug: Physician’s choice chemotherapy | Phase 3 | 302 | NCT02000622 |
| Active, not recruiting | Olaparib Maintenance Monotherapy in Patients With BRCA Mutated Ovarian Cancer Following First Line Platinum Based Chemotherapy. | Newly Diagnosed Advanced Ovarian Cancer FIGO Stage III-IV(and 4 more) | Drug: Olaparib 300mg tablets | Phase 3 | 451 | NCT01844986 |
| Active, not recruiting | Olaparib or Cediranib Maleate and Olaparib Compared With Standard Platinum-Based Chemotherapy in Treating Patients With Recurrent Platinum-Sensitive Ovarian, Fallopian Tube, or Primary Peritoneal Cancer | BRCA Rearrangement Deleterious BRCA1 Gene Mutation Deleterious BRCA2 Gene Mutation (and 13 more) | Drug: Carboplatin Drug: Cediranib Maleate Drug: Gemcitabine Hydrochloride (and 6 more) | Phase 3 | 549 | NCT02446600 |
| Status | Study Title | Conditions | Interventions | Phase | Number Enrolled | NCT Number |
|---|---|---|---|---|---|---|
| Recruiting | Phase 2, A Study of Niraparib Combined With Bevacizumab Maintenance Treatment in Patients With Advanced Ovarian Cancer Following Response on Front-Line Platinum-Based Chemotherapy | Ovarian Cancer Fallopian Tube Cancer Primary Peritoneal Carcinoma | Drug: Niraparib Biological: Bevacizumab | Phase 2 | 90 | NCT03326193 |
| Recruiting | A Study of Cediranib and Olaparib at Disease Worsening in Ovarian Cancer | Ovarian Cancer | Drug: Cediranib Drug: Olaparib | Not Applicable | 30 | NCT02681237 |
| Recruiting | A Study of Fluzoparib Given in Combination With Apatinib in Ovarian or Breast Cancer Patients | Ovarian Cancer Triple Negative Breast Cancer | Drug: Fluzoparib Drug: Apatinib | Phase 1 | 76 | NCT03075462 |
| Recruiting | Phase 2 Multicohort Study to Evaluate the Safety and Efficacy of Novel Treatment Combinations in Patients With Recurrent Ovarian Cancer | Ovarian Cancer | Drug: Niraparib Drug: TSR-042 Drug: Bevacizumab | Phase 2 | 40 | NCT03574779 |
| Recruiting | Mesothelioma Stratified Therapy (MiST): A Multi-drug Phase II Trial in Malignant Mesothelioma | Mesothelioma, Malignant | Drug: Rucaparib Drug: Abemaciclib Drug: pembrolizumab & bemcentinib Drug: Atezolizumab & Bevacizumab | Phase 2 | 120 | NCT03654833 |
| Recruiting | Study Evaluating the Efficacy of Maintenance Olaparib and Cediranib or Olaparib Alone in Ovarian Cancer Patients | Ovarian Cancer | Drug: Olaparib Drug: Cediranib | Phase 3 | 618 | NCT03278717 |
| Completed | A Study of Cediranib and Olaparib at the Time Ovarian Cancer Worsens on Olaparib | Ovarian Cancer | Drug: Olaparib Drug: Cediranib | Phase 2 | 4 | NCT02340611 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martí, J.M.; Fernández-Cortés, M.; Serrano-Sáenz, S.; Zamudio-Martinez, E.; Delgado-Bellido, D.; Garcia-Diaz, A.; Oliver, F.J. The Multifactorial Role of PARP-1 in Tumor Microenvironment. Cancers 2020, 12, 739. https://doi.org/10.3390/cancers12030739
Martí JM, Fernández-Cortés M, Serrano-Sáenz S, Zamudio-Martinez E, Delgado-Bellido D, Garcia-Diaz A, Oliver FJ. The Multifactorial Role of PARP-1 in Tumor Microenvironment. Cancers. 2020; 12(3):739. https://doi.org/10.3390/cancers12030739
Chicago/Turabian StyleMartí, Juan Manuel, Mónica Fernández-Cortés, Santiago Serrano-Sáenz, Esteban Zamudio-Martinez, Daniel Delgado-Bellido, Angel Garcia-Diaz, and Francisco Javier Oliver. 2020. "The Multifactorial Role of PARP-1 in Tumor Microenvironment" Cancers 12, no. 3: 739. https://doi.org/10.3390/cancers12030739
APA StyleMartí, J. M., Fernández-Cortés, M., Serrano-Sáenz, S., Zamudio-Martinez, E., Delgado-Bellido, D., Garcia-Diaz, A., & Oliver, F. J. (2020). The Multifactorial Role of PARP-1 in Tumor Microenvironment. Cancers, 12(3), 739. https://doi.org/10.3390/cancers12030739