Pathogenesis and Clinical Management of Uterine Serous Carcinoma

 ,
, 
Abstract
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Type I | Type II | References |
|---|---|---|---|
| Typical patient age, years | 50–69 | ≥70 | [4,8,11] |
| Hormone sensitivity | Yes | No | [4,8,11] |
| Precursor lesions | Atypical endometrial hyperplasia | Less defined | [4,8,11] |
| Subtypes | Endometrioid carcinoma and its variants | Uterine serous carcinoma and its variants | [4] |
| Behavior | Favorable/localized | Aggressive/prone to metastasis | [4] |
| Molecular alterations | MSI with MMR defects (20%) | TP53 mutation (90%) | [1,8,11,12] |
| PTEN deletion (80%) | HER2 overexpression (45%) | ||
| CTNNB1 (40%) | HER2 amplification (70%) | ||
| PI3K alteration (39%) | |||
| Five-year survival | 85% | 43% | [4] |
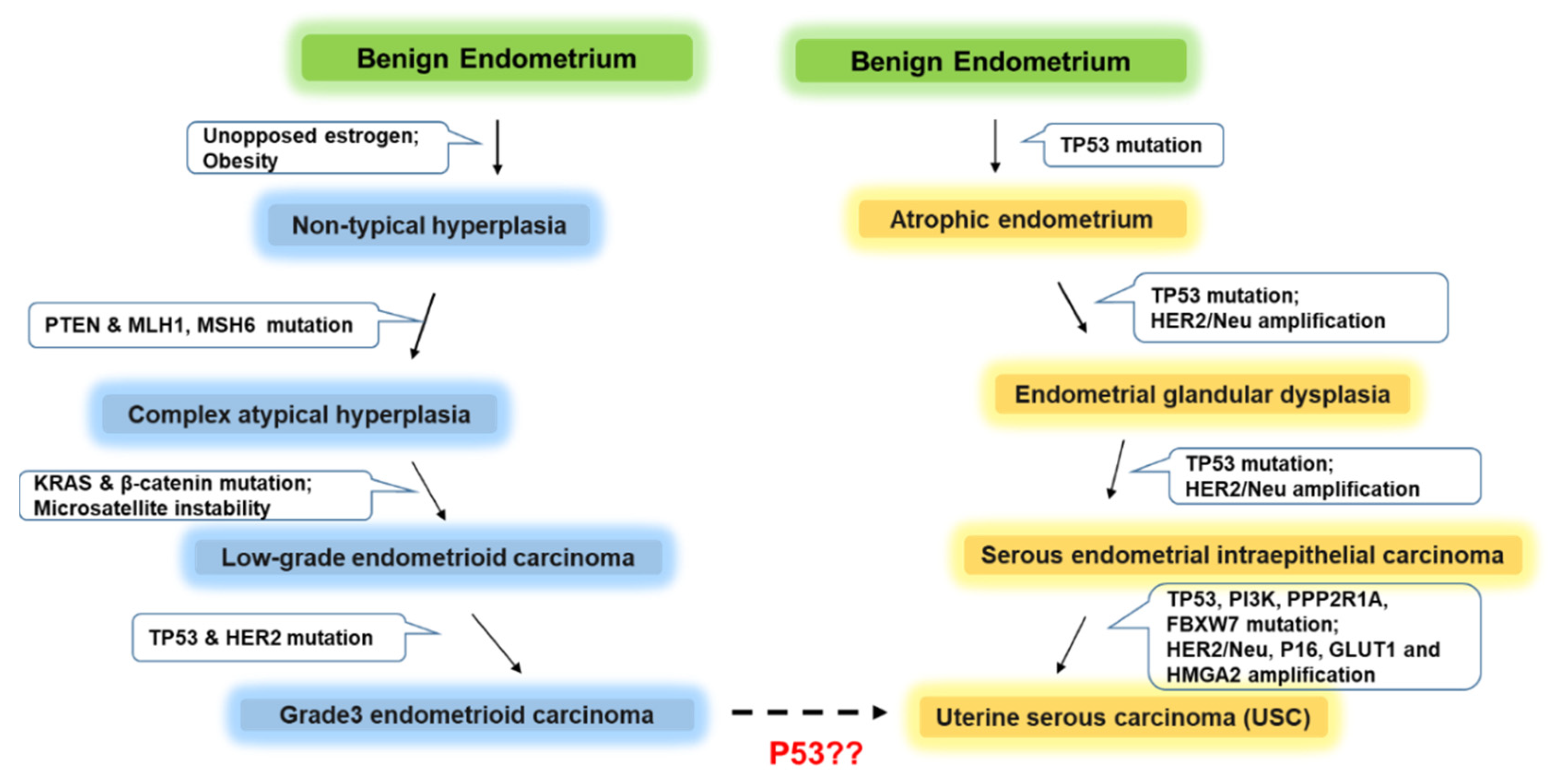
2. Histopathology of USC
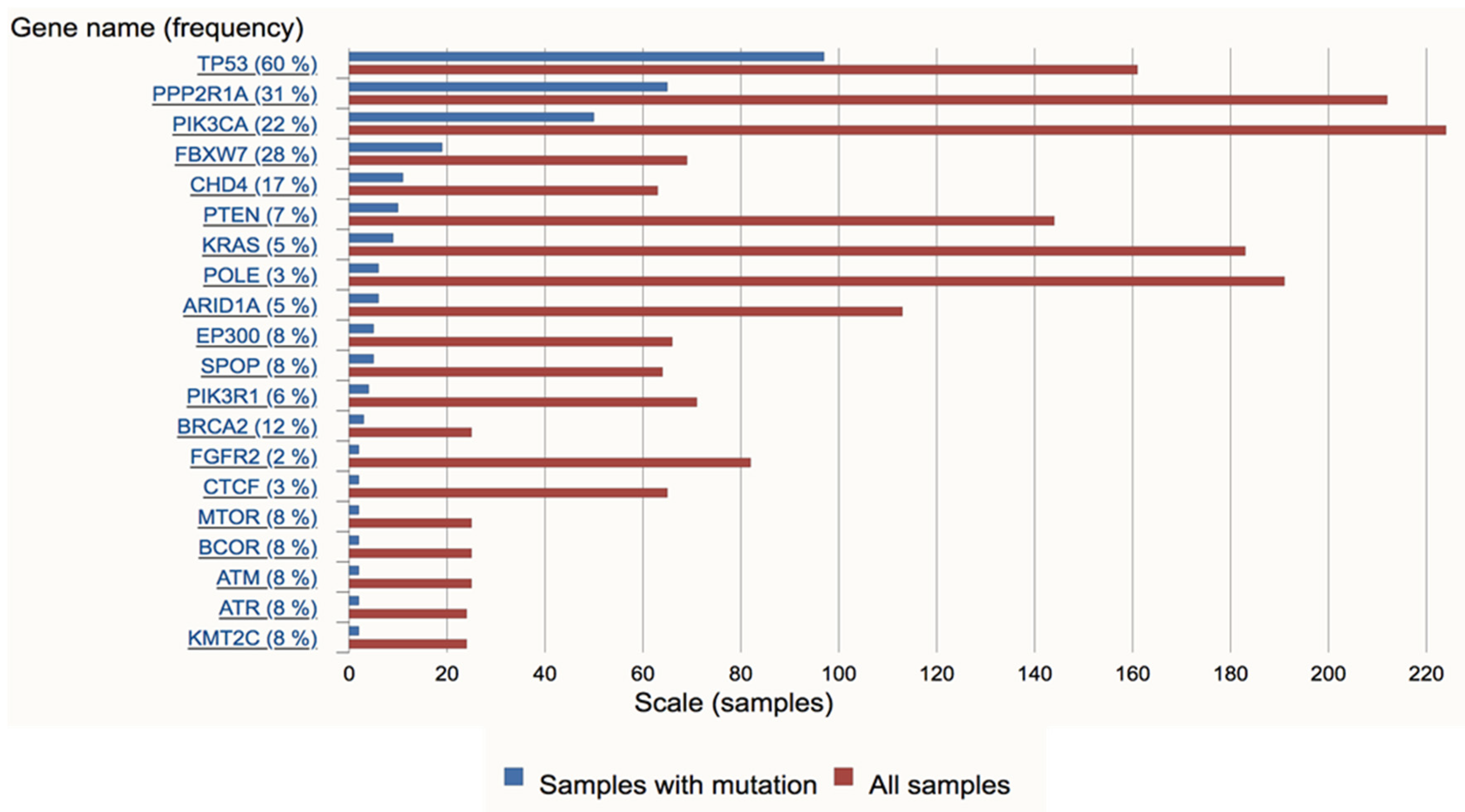
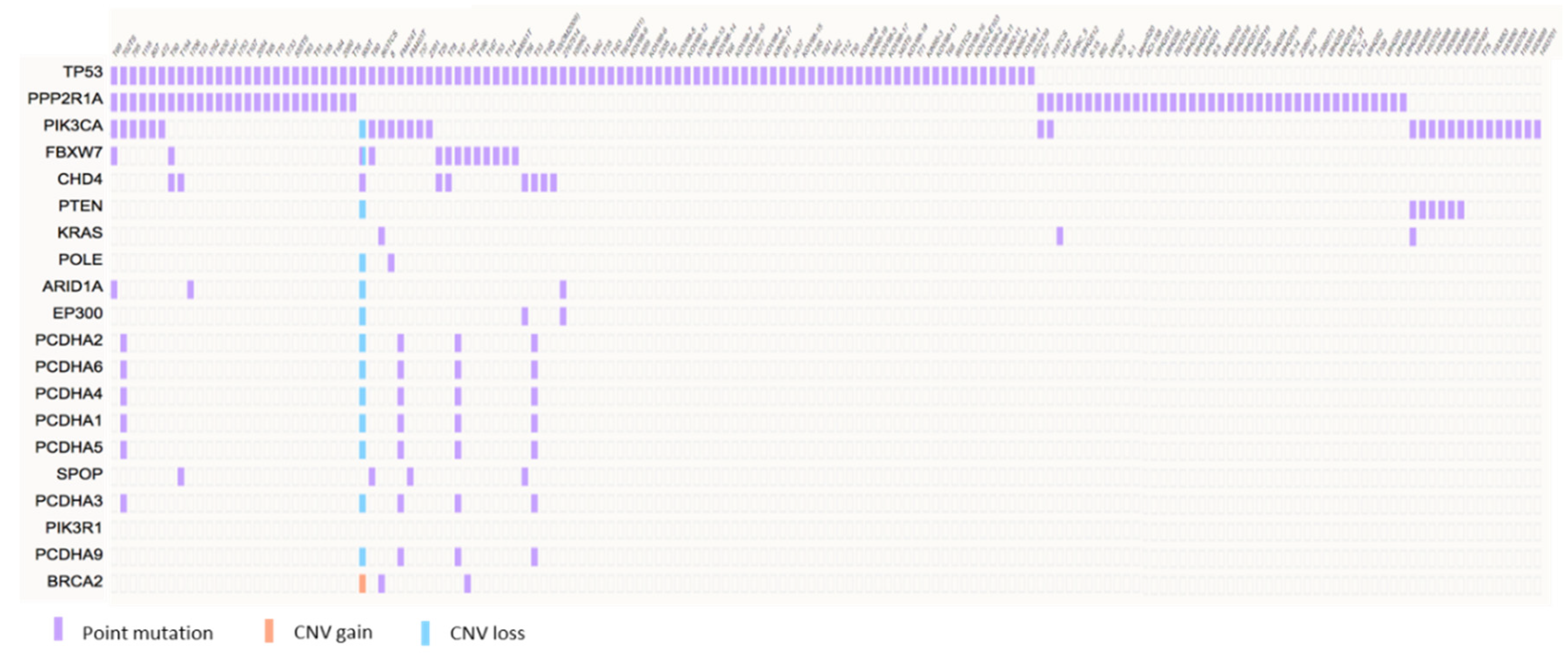
3. Molecular Pathogenesis of USC
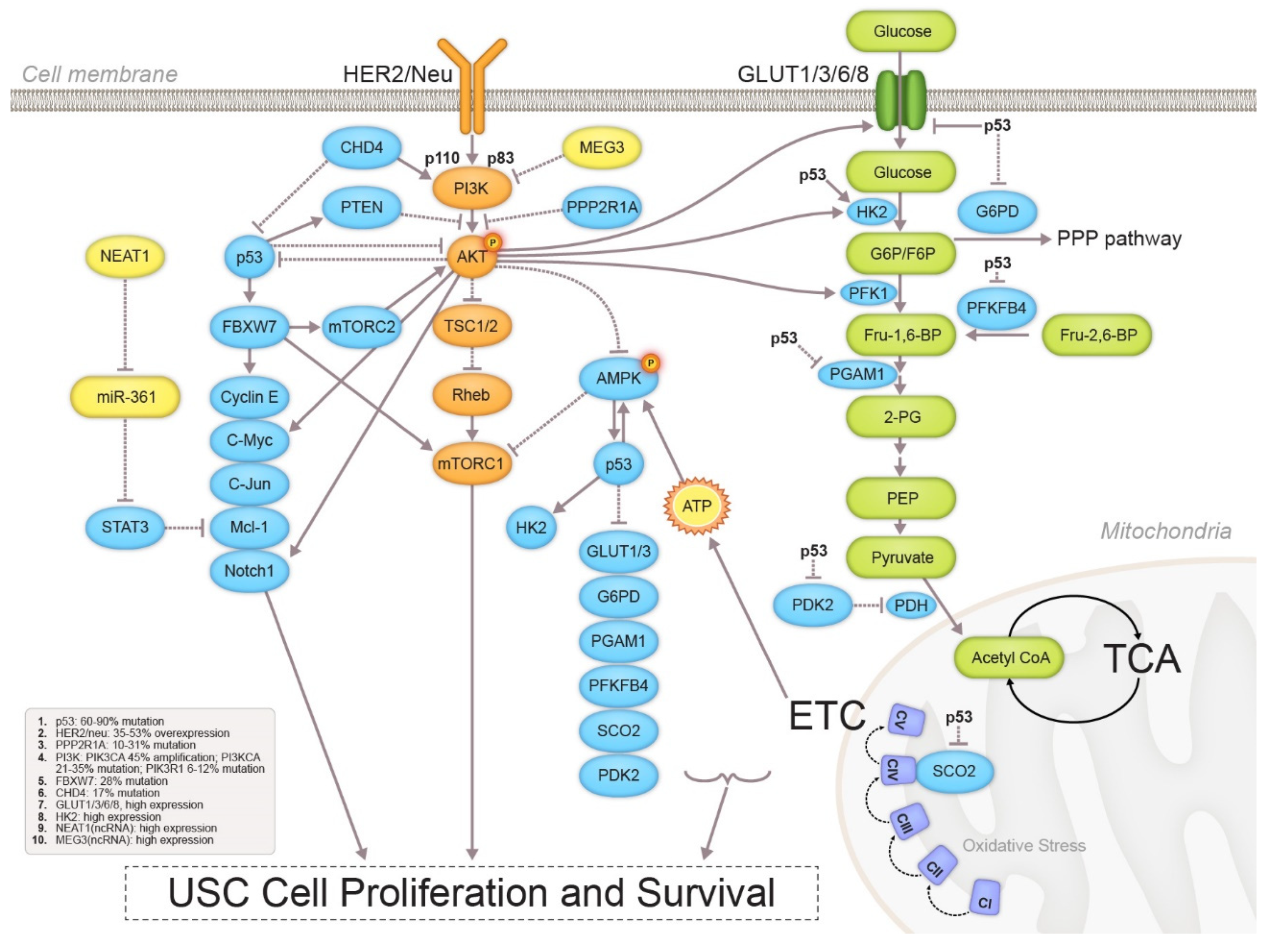
3.1. Metabolic Profile Alterations
3.1.1. Glycolysis
3.1.2. Mitochondrial Function
3.2. Epigenetic Alterations
3.3. Signaling Pathway Crosstalk in USC
4. Clinical Management of USC
4.1. Diagnosis
4.2. Treatment Approaches
4.2.1. Surgical Staging
4.2.2. Chemotherapy and Radiotherapy
4.2.3. Targeted Therapies
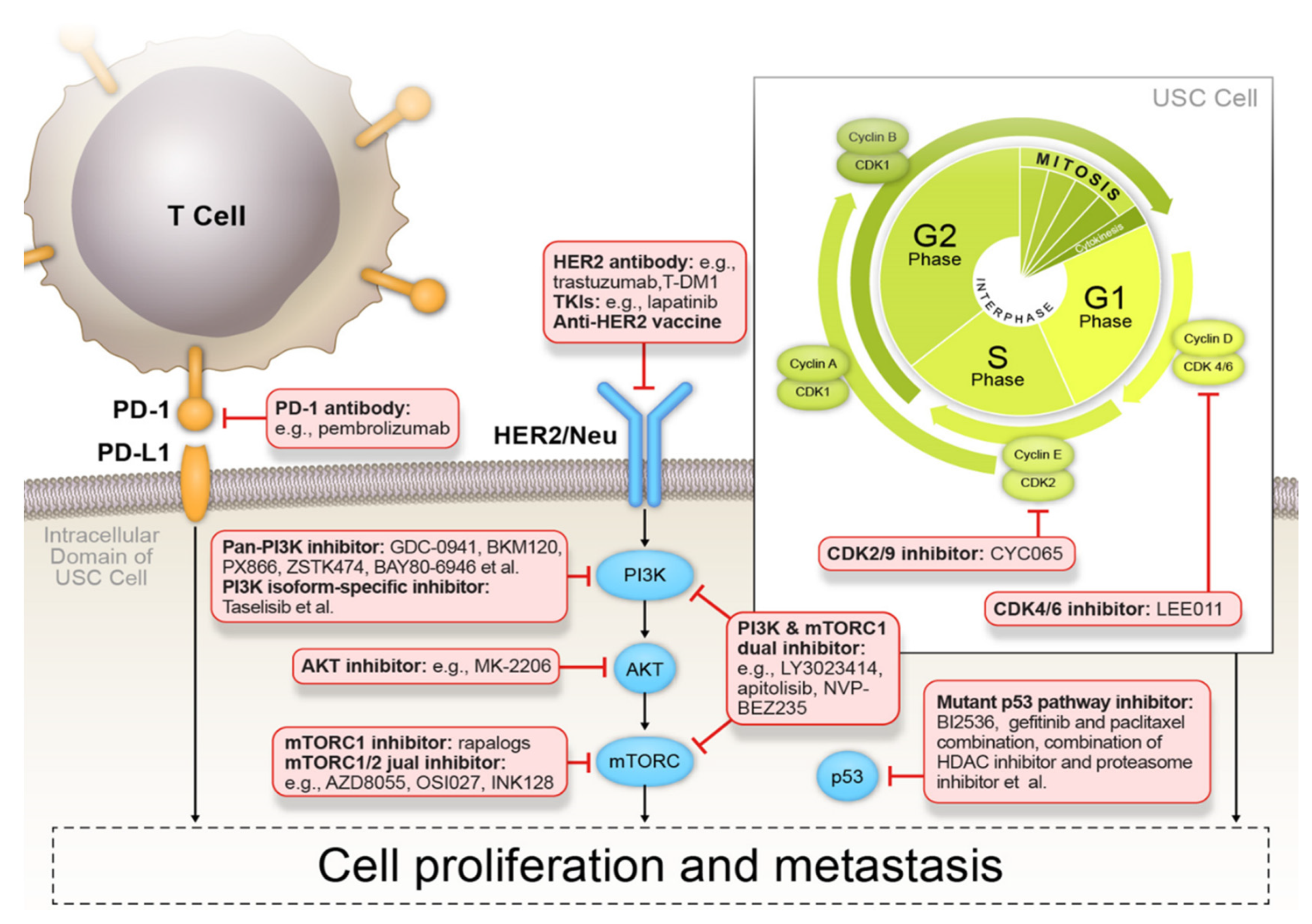
p53 Signaling Pathway Inhibitors
HER2/Neu Inhibitors
PI3K/AKT/mTOR Signaling Pathway Inhibitors
4.2.4. Cyclin-Dependent Kinase Inhibitors
4.3. Immune Profiling and Immunotherapy
| Drug | Function | Treatment Regimen | Phase | Status | Patient Cohort (EC Including USC) | ClinicalTrials.Gov Identifier | References |
|---|---|---|---|---|---|---|---|
| Trastuzumab | Anti-HER2/neu antibody | IV over 30–90 min on days 1, 8, 15, 22. Courses repeat every 28 days. | II | Completed | Stage III, IV, or recurrent EC with HER2/neu amplification (n = 34) | NCT00006089 | N/A |
| Trastuzumab-IL-12 | Trastuzumab: anti-HER2/neu antibody IL-I2: cytotoxic lymphocyte maturation factor | Trastuzumab: IV on day1, with maintenance dose on day 1 of each subsequent week. IL-I2: IV on days 2, 5 from week 3. | I | Completed | Recurrent cancers with high HER2/neu (n = 100) | NCT00004047 | [117] |
| Trastuzumab-paciliatxel-IL-I2 | Trastuzumab: anti-HER2/neu antibody IL-I2: cytotoxic lymphocyte maturation factor | Course 1: Trastuzumab IV on days 1, 8, 15; paclitaxel IV on day 1. Course 2: course 1 plus IL-12 SQ on days 2, 5, 9, 12, 16,19. Courses to be repeated q21 days. | I | Completed | Recurrent solid tumors (n = 18) | NCT00028535 | N/A |
| Trastuzumab-carboplatin-paclitaxel | Anti-HER2/neu antibody | Paclitaxel: 175 mg/m2 for 21 days for 6 cycles. Carboplatin: AUC 5 for 21 days for 6 cycles. Trastuzumab: 6 mg/kg for 21 days for 6 cycles from day 21. | II | Active, not recruiting | Stage III-IV or recurrent USC with HER2/neu amplification (n = 61) | NCT01367002 | [69] |
| Lapatinib | Dual tyrosine kinase inhibitor of HER2/neu and EGFR | PO once daily on days 1–28. Courses repeat every 28 days. | II | Completed; Has Result | Recurrent EC (n = 31) | NCT00096447 | [118] |
| Lapatinib-ixabepilone | Lapatinib: inhibitor of HER2/neu and EGFR; ixabepilone: antimicrotubule agent | Lapatinib: 500–1250 mg PO once daily. Cycle every 21 days for 6 cycles+ ixabepilone 32 mg/m2 every week. | I | Unknown | Recurrent EC with high HER2/neu | NCT01454479 | [26] |
| RAD001 | mTOR inhibitor | 10 mg PO daily. | II | Completed; Has Result | Progressive or recurrent EC (n = 35) | NCT00087685 | [119] |
| Temsirolimus | mTOR inhibitor | Temsirolimus: IV over 30 min on days 1, 8, 15, 22. Courses repeat every 28 days. | II | Completed; Has Result | Metastatic or locally advanced recurrent EC (n = 62) | NCT00072176 | [120] |
| Temsirolimus-RO4929097 | Temsirolimus: mTOR inhibitor; RO4929097: γ-secretase/Notch signaling pathway inhibitor | Temsirolimus: IV over 30 min on day1- 6 (course 1 only). Temsirolimus IV or PO on days 1, 8, 15 and RO4929097 PO once daily on days 1–3, 8–10, and 15–17. Courses repeat every 21 days. | I | Completed | Advanced solid tumors (n = 18) | NCT01198184 | [121] |
| Everolimus-letrozole | Everolimus: derivative of rapamycin, mTOR inhibitor; letrozole: aromatase inhibitor | Letrozole: 2.5 mg daily every 30 days. Everolimus: 10 mg daily every 28 days. | II/III | Active, not recruiting | Recurrent USC with PIK3CA gene mutation (n = 1) | NCT03285802 | N/A |
| MLN0128-bevacizumab | TORC1/2 inhibitor | INK128: PO daily on days 1–28 and bevacizumab IV on days 1 and 15. Courses repeat every 28 days. | I | Active, not recruiting | Recurrent glioblastoma and other solid tumors (n = 58) | NCT02142803 | [122] |
| MLN0128-MLN1117-paclitaxel | MLN0128: dual TORC1/2 inhibitor; MLN1117: PI3Kα inhibitor | Paclitaxel: 80 mg/m2 IV, weekly on days 1, 8, and 15 of a 28-day cycle. Paclitaxel 80 mg/m2 IV, weekly on days 1, 8, and 15 of a 28-day cycle along with MLN0128 4 mg capsule PO on days 2–4, 9–11, 16–18, and 23–25 of a 28-day cycle. MLN0128 30 mg capsule PO once weekly on days 1, 8, 15, and 22 of a 28-day cycle. MLN0128 4 mg capsule PO MLN1117 200 mg capsule PO on days 1–3, 8–10, 15–17, and 22–24 of a 28-day cycle. | II | Active, not recruiting | Advanced, recurrent or persistent EC (n = 245) | NCT02725268 | [94,123] |
| BKM120 | Pan-PI3K inhibitor | 100 mg/day PO as a second-line therapy. | II | Completed | Advanced EC (n = 70) | NCT01289041 | [124] |
| LY3023414 | mTOR and PI3K dual inhibitor | RP2D of 200 mg PO twice daily. | II | Active, not recruiting | Recurrent or persistent EC (n = 31) | NCT02549989 | N/A |
| GDC-0980 | mTOR and PI3K dual inhibitor | PO daily. | II | Completed | Recurrent or persistent EC (n = 56) | NCT01455493 | [125] |
| NVP-BEZ235 | mTOR and PI3K dual inhibitor | BEZ235: dose escalation PO once daily. | I | Completed | Adult Japanese patients with advanced solid tumors (n = 35) | NCT01195376 | [126] |
| MK2206 | AKT inhibitor | II | Completed | Recurrent or persistent EC (n = 37) | NCT01312753 | [127] | |
| Ribociclib (LEE011)-everolimus-letrozole | Ribociclib: CDK4/6 inhibitor; everolimus: mTOR inhibitor; letrozole: aromatase inhibitor | Ribociclib: 250 mg PO daily for a 28 day cycle. Everolimus: 2.5 mg PO daily for a 28-day cycle. Letrozole: 2.5 mg PO daily for a 28-day cycle. | II | Recruiting | Malignant neoplasms of female genital organs; endometrial carcinoma (n = 76) | NCT03008408 | [128] |
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Charo, L.M.; Plaxe, S.C. Recent advances in endometrial cancer: A review of key clinical trials from 2015 to 2019. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Creasman, W.T.; Eddy, G.L. Recent advances in endometrial cancer. Semin. Surg. Oncol. 1990, 6, 339–342. [Google Scholar] [CrossRef]
- Saso, S.; Chatterjee, J.; Georgiou, E.; Ditri, A.M.; Smith, J.R.; Ghaem-Maghami, S. Endometrial cancer. BMJ 2011, 343, d3954. [Google Scholar] [CrossRef]
- Amant, F.; Moerman, P.; Neven, P.; Timmerman, D.; Van Limbergen, E.; Vergote, I. Endometrial cancer. Lancet 2005, 366, 491–505. [Google Scholar] [CrossRef]
- Soslow, R.A.; Tornos, C.; Park, K.J.; Malpica, A.; Matias-Guiu, X.; Oliva, E.; Parkash, V.; Carlson, J.; McCluggage, W.G.; Gilks, C.B. Endometrial Carcinoma Diagnosis: Use of FIGO Grading and Genomic Subcategories in Clinical Practice: Recommendations of the International Society of Gynecological Pathologists. Int. J. Gynecol. Pathol. 2019, 38 (Suppl. 1), S64–S74. [Google Scholar] [CrossRef]
- Goebel, E.A.; Vidal, A.; Matias-Guiu, X.; Blake Gilks, C. The evolution of endometrial carcinoma classification through application of immunohistochemistry and molecular diagnostics: Past, present and future. Virchows Arch. 2018, 472, 885–896. [Google Scholar] [CrossRef]
- Group, S.G.O.C.P.E.C.W.; Burke, W.M.; Orr, J.; Leitao, M.; Salom, E.; Gehrig, P.; Olawaiye, A.B.; Brewer, M.; Boruta, D.; Herzog, T.J.; et al. Endometrial cancer: A review and current management strategies: Part II. Gynecol. Oncol. 2014, 134, 393–402. [Google Scholar] [CrossRef]
- Moore, K.N.; Fader, A.N. Uterine papillary serous carcinoma. Clin. Obstet. Gynecol. 2011, 54, 278–291. [Google Scholar] [CrossRef]
- Hamilton, C.A.; Cheung, M.K.; Osann, K.; Chen, L.; Teng, N.N.; Longacre, T.A.; Powell, M.A.; Hendrickson, M.R.; Kapp, D.S.; Chan, J.K. Uterine papillary serous and clear cell carcinomas predict for poorer survival compared to grade 3 endometrioid corpus cancers. Br. J. Cancer 2006, 94, 642–646. [Google Scholar] [CrossRef]
- Group, S.G.O.C.P.E.C.W.; Burke, W.M.; Orr, J.; Leitao, M.; Salom, E.; Gehrig, P.; Olawaiye, A.B.; Brewer, M.; Boruta, D.; Villella, J.; et al. Endometrial cancer: A review and current management strategies: Part I. Gynecol. Oncol. 2014, 134, 385–392. [Google Scholar] [CrossRef]
- Yeramian, A.; Moreno-Bueno, G.; Dolcet, X.; Catasus, L.; Abal, M.; Colas, E.; Reventos, J.; Palacios, J.; Prat, J.; Matias-Guiu, X. Endometrial carcinoma: Molecular alterations involved in tumor development and progression. Oncogene 2013, 32, 403–413. [Google Scholar] [CrossRef]
- Bell, D.W.; Ellenson, L.H. Molecular Genetics of Endometrial Carcinoma. Annu. Rev. Pathol. 2019, 14, 339–367. [Google Scholar] [CrossRef]
- Zheng, W.; Chambers, S.K. Histopathology of endometrial hyperplasia and endometrial carcinoma. An update by Dr LC Horn et al in Annals of Diagnostic Pathology 2007;11:297–311. Ann. Diagn. Pathol. 2008, 12, 231–232. [Google Scholar] [CrossRef]
- Nakayama, K.; Nakayama, N.; Ishikawa, M.; Miyazaki, K. Endometrial serous carcinoma: Its molecular characteristics and histology-specific treatment strategies. Cancers 2012, 4, 799–807. [Google Scholar] [CrossRef]
- Ambros, R.A.; Sherman, M.E.; Zahn, C.M.; Bitterman, P.; Kurman, R.J. Endometrial intraepithelial carcinoma: A distinctive lesion specifically associated with tumors displaying serous differentiation. Hum. Pathol. 1995, 26, 1260–1267. [Google Scholar] [CrossRef]
- Zheng, W.; Liang, S.X.; Yu, H.; Rutherford, T.; Chambers, S.K.; Schwartz, P.E. Endometrial glandular dysplasia: A newly defined precursor lesion of uterine papillary serous carcinoma. Part I: Morphologic features. Int. J. Surg. Pathol. 2004, 12, 207–223. [Google Scholar] [CrossRef]
- Fadare, O.; Zheng, W. Endometrial Glandular Dysplasia (EmGD): Morphologically and biologically distinctive putative precursor lesions of Type II endometrial cancers. Diagn. Pathol. 2008, 3, 6. [Google Scholar] [CrossRef]
- Kajo, K.; Vallova, M.; Biro, C.; Bognar, G.; Machalekova, K.; Zavodna, K.; Galbavy, S.; Zubor, P. Molecular pathology of endometrial carcinoma-a review. Ceskoslovenska Patol. 2015, 51, 65–73. [Google Scholar]
- Matias-Guiu, X.; Prat, J. Molecular pathology of endometrial carcinoma. Histopathology 2013, 62, 111–123. [Google Scholar] [CrossRef]
- Llobet, D.; Pallares, J.; Yeramian, A.; Santacana, M.; Eritja, N.; Velasco, A.; Dolcet, X.; Matias-Guiu, X. Molecular pathology of endometrial carcinoma: Practical aspects from the diagnostic and therapeutic viewpoints. J. Clin. Pathol. 2009, 62, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.C.; Blanchard, Z.; Maurer, K.A.; Gertz, J. Estrogen Signaling in Endometrial Cancer: A Key Oncogenic Pathway with Several Open Questions. Horm. Cancer 2019, 10, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Li, J.; Cragun, J.; Hatch, K.; Chambers, S.K.; Zheng, W. Lynch syndrome related endometrial cancer: Clinical significance beyond the endometrium. J. Hematol. Oncol. 2013, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.J.; Bell, D.W. The genomics and genetics of endometrial cancer. Adv. Genom. Genet. 2012, 2012, 33–47. [Google Scholar] [CrossRef]
- Althubiti, M.A. Mutation Frequencies in Endometrial Cancer Patients of Different Ethnicities and Tumor Grades: An Analytical Study. Saudi J. Med. Med. Sci. 2019, 7, 16–21. [Google Scholar] [CrossRef]
- Le Gallo, M.; Bell, D.W. The emerging genomic landscape of endometrial cancer. Clin. Chem. 2014, 60, 98–110. [Google Scholar] [CrossRef]
- Alkushi, A.; Lim, P.; Coldman, A.; Huntsman, D.; Miller, D.; Gilks, C.B. Interpretation of p53 immunoreactivity in endometrial carcinoma: Establishing a clinically relevant cut-off level. Int. J. Gynecol. Pathol. 2004, 23, 129–137. [Google Scholar] [CrossRef]
- Kobel, M.; Ronnett, B.M.; Singh, N.; Soslow, R.A.; Gilks, C.B.; McCluggage, W.G. Interpretation of P53 Immunohistochemistry in Endometrial Carcinomas: Toward Increased Reproducibility. Int. J. Gynecol. Pathol. 2019, 38 (Suppl. 1), S123–S131. [Google Scholar] [CrossRef]
- Garcia-Dios, D.A.; Lambrechts, D.; Coenegrachts, L.; Vandenput, I.; Capoen, A.; Webb, P.M.; Ferguson, K.; Akslen, L.A.; Claes, B.; Vergote, I.; et al. High-throughput interrogation of PIK3CA, PTEN, KRAS, FBXW7 and TP53 mutations in primary endometrial carcinoma. Gynecol. Oncol. 2013, 128, 327–334. [Google Scholar] [CrossRef]
- Lokich, E.; Kole, M.; Raker, C.; Quddus, M.R.; Mathews, C. Molecular markers in uterine serous cancer: Correlation between endometrial biopsy and hysterectomy specimens. Gynecol. Oncol. Rep. 2019, 29, 98–101. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Clem, B.F.; O’Neal, J.; Klarer, A.C.; Telang, S.; Chesney, J. Clinical development of cancer therapeutics that target metabolism. QJM 2016, 109, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef]
- Mathupala, S.P. Metabolic targeting of malignant tumors: Small-molecule inhibitors of bioenergetic flux. Recent Pat. Anticancer Drug Discov. 2011, 6, 6–14. [Google Scholar] [CrossRef]
- Akins, N.S.; Nielson, T.C.; Le, H.V. Inhibition of Glycolysis and Glutaminolysis: An Emerging Drug Discovery Approach to Combat Cancer. Curr. Top. Med. Chem. 2018, 18, 494–504. [Google Scholar] [CrossRef]
- Aft, R.L.; Zhang, F.W.; Gius, D. Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: Mechanism of cell death. Br. J. Cancer 2002, 87, 805–812. [Google Scholar] [CrossRef]
- Benjamin, F.; Romney, S. Disturbed Carbohydrate Metabolism in Endometrial Carcinoma. Cancer 1964, 17, 386–390. [Google Scholar] [CrossRef]
- Byrne, F.L.; Poon, I.K.; Modesitt, S.C.; Tomsig, J.L.; Chow, J.D.; Healy, M.E.; Baker, W.D.; Atkins, K.A.; Lancaster, J.M.; Marchion, D.C.; et al. Metabolic vulnerabilities in endometrial cancer. Cancer Res. 2014, 74, 5832–5845. [Google Scholar] [CrossRef]
- Fan, T.; Sun, G.; Sun, X.; Zhao, L.; Zhong, R.; Peng, Y. Tumor Energy Metabolism and Potential of 3-Bromopyruvate as an Inhibitor of Aerobic Glycolysis: Implications in Tumor Treatment. Cancers 2019, 11, 317. [Google Scholar] [CrossRef]
- Krzeslak, A.; Wojcik-Krowiranda, K.; Forma, E.; Jozwiak, P.; Romanowicz, H.; Bienkiewicz, A.; Brys, M. Expression of GLUT1 and GLUT3 glucose transporters in endometrial and breast cancers. Pathol. Oncol. Res. 2012, 18, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.Y.; Kalir, T.; Sabo, E.; Sherman, D.E.; Cohen, C.; Burstein, D.E. Immunohistochemical staining of GLUT1 in benign, hyperplastic, and malignant endometrial epithelia. Cancer 2000, 88, 2774–2781. [Google Scholar] [CrossRef]
- Goldman, N.A.; Katz, E.B.; Glenn, A.S.; Weldon, R.H.; Jones, J.G.; Lynch, U.; Fezzari, M.J.; Runowicz, C.D.; Goldberg, G.L.; Charron, M.J. GLUT1 and GLUT8 in endometrium and endometrial adenocarcinoma. Mod. Pathol. 2006, 19, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Dysregulated TCA Cycle Pathway in Endometrial Cancer. Adv. Mod. Oncol. Res. 2018, 4, 10. [Google Scholar] [CrossRef]
- Lee, T.Y.; Martinez-Outschoorn, U.E.; Schilder, R.J.; Kim, C.H.; Richard, S.D.; Rosenblum, N.G.; Johnson, J.M. Metformin as a Therapeutic Target in Endometrial Cancers. Front. Oncol. 2018, 8, 341. [Google Scholar] [CrossRef]
- Qiang, P.; Shao, Y.; Sun, Y.P.; Zhang, J.; Chen, L.J. Metformin inhibits proliferation and migration of endometrial cancer cells through regulating PI3K/AKT/MDM2 pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1778–1785. [Google Scholar] [CrossRef]
- Cao, C.; Zhou, J.Y.; Xie, S.W.; Guo, X.J.; Li, G.T.; Gong, Y.J.; Yang, W.J.; Li, Z.; Zhong, R.H.; Shao, H.H.; et al. Metformin Enhances Nomegestrol Acetate Suppressing Growth of Endometrial Cancer Cells and May Correlate to Downregulating mTOR Activity In Vitro and In Vivo. Int. J. Mol. Sci. 2019, 20, 3308. [Google Scholar] [CrossRef]
- Sato, E.; Nakayama, K.; Nakamura, K.; Ishikawa, N.; Ishikawa, M.; Minamoto, T.; Ishibashi, T.; Kyo, S. Efficacy of metformin for advanced-stage endometrial cancer: A case report. Mol. Clin. Oncol. 2017, 6, 441–443. [Google Scholar] [CrossRef]
- Ko, E.M.; Walter, P.; Jackson, A.; Clark, L.; Franasiak, J.; Bolac, C.; Havrilesky, L.J.; Secord, A.A.; Moore, D.T.; Gehrig, P.A.; et al. Metformin is associated with improved survival in endometrial cancer. Gynecol. Oncol. 2014, 132, 438–442. [Google Scholar] [CrossRef]
- Musicco, C.; Cormio, G.; Pesce, V.; Loizzi, V.; Cicinelli, E.; Resta, L.; Ranieri, G.; Cormio, A. Mitochondrial Dysfunctions in Type I Endometrial Carcinoma: Exploring Their Role in Oncogenesis and Tumor Progression. Int. J. Mol. Sci. 2018, 19, 2076. [Google Scholar] [CrossRef]
- Mohammad, H.P.; Barbash, O.; Creasy, C.L. Targeting epigenetic modifications in cancer therapy: Erasing the roadmap to cancer. Nat. Med. 2019, 25, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Seeber, L.M.; Zweemer, R.P.; Marchionni, L.; Massuger, L.F.; Smit, V.T.; van Baal, W.M.; Verheijen, R.H.; van Diest, P.J. Methylation profiles of endometrioid and serous endometrial cancers. Endocr. Relat. Cancer 2010, 17, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Tao, M.H.; Freudenheim, J.L. DNA methylation in endometrial cancer. Epigenetics 2010, 5, 491–498. [Google Scholar] [CrossRef]
- Dong, P.; Xiong, Y.; Yue, J.; Hanley, S.; Kobayashi, N.; Todo, Y.; Watari, H. Exploring lncRNA-Mediated Regulatory Networks in Endometrial Cancer Cells and the Tumor Microenvironment: Advances and Challenges. Cancers 2019, 11, 234. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Xiong, Y.; Yue, J.; Xu, D.; Ihira, K.; Konno, Y.; Kobayashi, N.; Todo, Y.; Watari, H. Long noncoding RNA NEAT1 drives aggressive endometrial cancer progression via miR-361-regulated networks involving STAT3 and tumor microenvironment-related genes. J. Exp. Clin. Cancer Res. 2019, 38, 295. [Google Scholar] [CrossRef]
- Sun, K.X.; Wu, D.D.; Chen, S.; Zhao, Y.; Zong, Z.H. LncRNA MEG3 inhibit endometrial carcinoma tumorigenesis and progression through PI3K pathway. Apoptosis 2017, 22, 1543–1552. [Google Scholar] [CrossRef]
- Black, J.D.; English, D.P.; Roque, D.M.; Santin, A.D. Targeted therapy in uterine serous carcinoma: An aggressive variant of endometrial cancer. Womens Health 2014, 10, 45–57. [Google Scholar] [CrossRef]
- Tate, K.; Yoshida, H.; Ishikawa, M.; Uehara, T.; Ikeda, S.I.; Hiraoka, N.; Kato, T. Prognostic factors for patients with early-stage uterine serous carcinoma without adjuvant therapy. J. Gynecol. Oncol. 2018, 29, e34. [Google Scholar] [CrossRef]
- Abbasi, J. A Pap-Based Test to Detect Endometrial and Ovarian Cancers Early. JAMA 2018, 319, 1853. [Google Scholar] [CrossRef]
- Lachance, J.A.; Darus, C.J.; Rice, L.W. Surgical management and postoperative treatment of endometrial carcinoma. Rev. Obstet. Gynecol. 2008, 1, 97–105. [Google Scholar]
- DeNardis, S.A.; Holloway, R.W.; Bigsby, G.E.; Pikaart, D.P.; Ahmad, S.; Finkler, N.J. Robotically assisted laparoscopic hysterectomy versus total abdominal hysterectomy and lymphadenectomy for endometrial cancer. Gynecol. Oncol. 2008, 111, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Rungruang, B.; Olawaiye, A.B. Comprehensive surgical staging for endometrial cancer. Rev. Obstet. Gynecol. 2012, 5, 28–34. [Google Scholar] [PubMed]
- Frederick, P.J.; Straughn, J.M., Jr. The role of comprehensive surgical staging in patients with endometrial cancer. Cancer Control 2009, 16, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Salehi, S.; Brandberg, Y.; Avall-Lundqvist, E.; Suzuki, C.; Johansson, H.; Legerstam, B.; Falconer, H. Long-term quality of life after comprehensive surgical staging of high-risk endometrial cancer-results from the RASHEC trial. Acta Oncol. 2018, 57, 1671–1676. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.; Filiaci, V.; Randall, M.E.; Mutch, D.; Steinhoff, M.M.; DiSilvestro, P.A.; Moxley, K.M.; Kim, Y.M.; Powell, M.A.; O’Malley, D.M.; et al. Adjuvant Chemotherapy plus Radiation for Locally Advanced Endometrial Cancer. N. Engl. J. Med. 2019, 380, 2317–2326. [Google Scholar] [CrossRef] [PubMed]
- Mandato, V.D.; Torricelli, F.; Palomba, S.; Uccella, S.; Pirillo, D.; Ciarlini, G.; De Iaco, P.; Lucia, E.; Giorda, G.; Ditto, A.; et al. Uterine Papillary Serous Carcinoma Arising in a Polyp: A Multicenter Retrospective Analysis on 75 Patients. Am. J. Clin. Oncol. 2019, 42, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.B.; Mariani, A.; Cliby, W.A.; Keeney, G.A.; Podratz, K.C.; Dowdy, S.C. Role of systematic lymphadenectomy and adjuvant therapy in stage I uterine papillary serous carcinoma. Gynecol. Oncol. 2007, 107, 186–189. [Google Scholar] [CrossRef]
- Bestvina, C.M.; Fleming, G.F. Chemotherapy for Endometrial Cancer in Adjuvant and Advanced Disease Settings. Oncologist 2016, 21, 1250–1259. [Google Scholar] [CrossRef]
- Fader, A.N.; Roque, D.M.; Siegel, E.; Buza, N.; Hui, P.; Abdelghany, O.; Chambers, S.K.; Secord, A.A.; Havrilesky, L.; O’Malley, D.M.; et al. Randomized Phase II Trial of Carboplatin-Paclitaxel Versus Carboplatin-Paclitaxel-Trastuzumab in Uterine Serous Carcinomas That Overexpress Human Epidermal Growth Factor Receptor 2/neu. J. Clin. Oncol. 2018, 36, 2044–2051. [Google Scholar] [CrossRef]
- Viswanathan, A.N.; Macklin, E.A.; Berkowitz, R.; Matulonis, U. The importance of chemotherapy and radiation in uterine papillary serous carcinoma. Gynecol. Oncol. 2011, 123, 542–547. [Google Scholar] [CrossRef]
- Shaeffer, D.T.; Randall, M.E. Adjuvant radiotherapy in endometrial carcinoma. Oncologist 2005, 10, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Roelofsen, T.; van Ham, M.A.; de Hullu, J.A.; Massuger, L.F. Clinical management of uterine papillary serous carcinoma. Expert Rev. Anticancer Ther. 2011, 11, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.L.; Santin, A.D. Targeted therapy in the treatment of uterine serous carcinoma. Pharmacogenomics 2015, 16, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.L.; Yen, M.S.; Chao, K.C.; Yuan, C.C.; Ng, H.T.; Chao, H.T.; Lee, F.K.; Wang, P.H. Hormone therapy for patients with advanced or recurrent endometrial cancer. J. Chin. Med. Assoc. 2014, 77, 221–226. [Google Scholar] [CrossRef]
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 2018, 37, 30. [Google Scholar] [CrossRef]
- Melo Dos Santos, N.; de Oliveira, G.A.P.; Ramos Rocha, M.; Pedrote, M.M.; Diniz da Silva Ferretti, G.; Pereira Rangel, L.; Morgado-Diaz, J.A.; Silva, J.L.; Rodrigues Pereira Gimba, E. Loss of the p53 transactivation domain results in high amyloid aggregation of the Delta40p53 isoform in endometrial carcinoma cells. J. Biol. Chem. 2019, 294, 9430–9439. [Google Scholar] [CrossRef]
- Meng, X.; Laidler, L.L.; Kosmacek, E.A.; Yang, S.; Xiong, Z.; Zhu, D.; Wang, X.; Dai, D.; Zhang, Y.; Wang, X.; et al. Induction of mitotic cell death by overriding G2/M checkpoint in endometrial cancer cells with non-functional p53. Gynecol. Oncol. 2013, 128, 461–469. [Google Scholar] [CrossRef]
- Meng, X.; Yang, S.; Li, Y.; Li, Y.; Devor, E.J.; Bi, J.; Wang, X.; Umesalma, S.; Quelle, D.E.; Thiel, W.H.; et al. Combination of Proteasome and Histone Deacetylase Inhibitors Overcomes the Impact of Gain-of-Function p53 Mutations. Dis. Markers 2018, 2018, 3810108. [Google Scholar] [CrossRef]
- Koskas, M.; Depreeuw, J.; Moens, S.; Annibali, D.; Cuppens, T.; Moerman, P.; Lambrechts, D.; Amant, F. Genomic Characterisation and Response to Trastuzumab and Paclitaxel in Advanced or Recurrent HER2-positive Endometrial Carcinoma. Anticancer Res. 2016, 36, 5381–5384. [Google Scholar] [CrossRef]
- English, D.P.; Bellone, S.; Schwab, C.L.; Bortolomai, I.; Bonazzoli, E.; Cocco, E.; Buza, N.; Hui, P.; Lopez, S.; Ratner, E.; et al. T-DM1, a novel antibody-drug conjugate, is highly effective against primary HER2 overexpressing uterine serous carcinoma in vitro and in vivo. Cancer Med. 2014, 3, 1256–1265. [Google Scholar] [CrossRef]
- Menderes, G.; Bonazzoli, E.; Bellone, S.; Black, J.; Predolini, F.; Pettinella, F.; Masserdotti, A.; Zammataro, L.; Altwerger, G.; Buza, N.; et al. SYD985, a Novel Duocarmycin-Based HER2-Targeting Antibody-Drug Conjugate, Shows Antitumor Activity in Uterine and Ovarian Carcinosarcoma with HER2/Neu Expression. Clin. Cancer Res. 2017, 23, 5836–5845. [Google Scholar] [CrossRef] [PubMed]
- Remmerie, M.; Janssens, V. Targeted Therapies in Type II Endometrial Cancers: Too Little, but Not Too Late. Int. J. Mol. Sci. 2018, 19, 2380. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, J.W.; Hernandez, S.F.; Byron, V.F.; DiGloria, C.M.; Lopez, H.; Scialabba, V.; Kim, M.; Zhang, L.; Borger, D.R.; Tambouret, R.; et al. Dual HER2 targeting impedes growth of HER2 gene-amplified uterine serous carcinoma xenografts. Clin. Cancer Res. 2014, 20, 6517–6528. [Google Scholar] [CrossRef] [PubMed]
- Leslie, K.K.; Sill, M.W.; Lankes, H.A.; Fischer, E.G.; Godwin, A.K.; Gray, H.; Schilder, R.J.; Walker, J.L.; Tewari, K.; Hanjani, P.; et al. Lapatinib and potential prognostic value of EGFR mutations in a Gynecologic Oncology Group phase II trial of persistent or recurrent endometrial cancer. Gynecol. Oncol. 2012, 127, 345–350. [Google Scholar] [CrossRef]
- Jackson, D.O.; Trappey, F.A.; Clifton, G.T.; Vreeland, T.J.; Peace, K.M.; Hale, D.F.; Litton, J.K.; Murray, J.L.; Perez, S.A.; Papamichail, M.; et al. Effects of HLA status and HER2 status on outcomes in breast cancer patients at risk for recurrence-Implications for vaccine trial design. Clin. Immunol. 2018, 195, 28–35. [Google Scholar] [CrossRef]
- Schneble, E.J.; Berry, J.S.; Trappey, F.A.; Clifton, G.T.; Ponniah, S.; Mittendorf, E.; Peoples, G.E. The HER2 peptide nelipepimut-S (E75) vaccine (NeuVax) in breast cancer patients at risk for recurrence: Correlation of immunologic data with clinical response. Immunotherapy 2014, 6, 519–531. [Google Scholar] [CrossRef]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- Ballou, L.M.; Lin, R.Z. Rapamycin and mTOR kinase inhibitors. J. Chem. Biol. 2008, 1, 27–36. [Google Scholar] [CrossRef]
- Royce, M.E.; Osman, D. Everolimus in the Treatment of Metastatic Breast Cancer. Breast Cancer 2015, 9, 73–79. [Google Scholar] [CrossRef]
- Houghton, P.J. Everolimus. Clin. Cancer Res. 2010, 16, 1368–1372. [Google Scholar] [CrossRef]
- Zanardi, E.; Verzoni, E.; Grassi, P.; Necchi, A.; Giannatempo, P.; Raggi, D.; De Braud, F.; Procopio, G. Clinical experience with temsirolimus in the treatment of advanced renal cell carcinoma. Ther. Adv. Urol. 2015, 7, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Y. mTOR kinase inhibitors as potential cancer therapeutic drugs. Cancer Lett. 2013, 340, 1–8. [Google Scholar] [CrossRef] [PubMed]
- English, D.P.; Roque, D.M.; Carrara, L.; Lopez, S.; Bellone, S.; Cocco, E.; Bortolomai, I.; Schwartz, P.E.; Rutherford, T.; Santin, A.D. HER2/neu gene amplification determines the sensitivity of uterine serous carcinoma cell lines to AZD8055, a novel dual mTORC1/2 inhibitor. Gynecol. Oncol. 2013, 131, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Brana, I.; Siu, L.L.; De Jonge, M.J.; Homji, N.; Mills, D.; Di Tomaso, E.; Sarr, C.; Trandafir, L.; Massacesi, C.; et al. Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2014, 32, 670–681. [Google Scholar] [CrossRef]
- Patnaik, A.; Appleman, L.J.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Weiss, G.J.; Sachdev, J.C.; Chadha, M.; Fulk, M.; et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann. Oncol. 2016, 27, 1928–1940. [Google Scholar] [CrossRef]
- Juric, D.; de Bono, J.S.; LoRusso, P.M.; Nemunaitis, J.; Heath, E.I.; Kwak, E.L.; Macarulla Mercade, T.; Geuna, E.; Jose de Miguel-Luken, M.; Patel, C.; et al. A First-in-Human, Phase I, Dose-Escalation Study of TAK-117, a Selective PI3Kalpha Isoform Inhibitor, in Patients with Advanced Solid Malignancies. Clin. Cancer Res. 2017, 23, 5015–5023. [Google Scholar] [CrossRef]
- Lopez, S.; Schwab, C.L.; Cocco, E.; Bellone, S.; Bonazzoli, E.; English, D.P.; Schwartz, P.E.; Rutherford, T.; Angioli, R.; Santin, A.D. Taselisib, a selective inhibitor of PIK3CA, is highly effective on PIK3CA-mutated and HER2/neu amplified uterine serous carcinoma in vitro and in vivo. Gynecol. Oncol. 2014, 135, 312–317. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, K.N.; Li, R.; Shao, R.; Chen, C. Activation of PI3K/Akt/mTOR pathway and dual inhibitors of PI3K and mTOR in endometrial cancer. Curr. Med. Chem. 2014, 21, 3070–3080. [Google Scholar] [CrossRef]
- Bendell, J.C.; Varghese, A.M.; Hyman, D.M.; Bauer, T.M.; Pant, S.; Callies, S.; Lin, J.; Martinez, R.; Wickremsinhe, E.; Fink, A.; et al. A First-in-Human Phase 1 Study of LY3023414, an Oral PI3K/mTOR Dual Inhibitor, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 3253–3262. [Google Scholar] [CrossRef]
- Hyman, D.M.; Smyth, L.M.; Donoghue, M.T.A.; Westin, S.N.; Bedard, P.L.; Dean, E.J.; Bando, H.; El-Khoueiry, A.B.; Perez-Fidalgo, J.A.; Mita, A.; et al. AKT Inhibition in Solid Tumors with AKT1 Mutations. J. Clin. Oncol. 2017, 35, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- de Melo, A.C.; Paulino, E.; Garces, A.H. A Review of mTOR Pathway Inhibitors in Gynecologic Cancer. Oxid. Med. Cell. Longev. 2017, 2017, 4809751. [Google Scholar] [CrossRef] [PubMed]
- Winder, A.; Unno, K.; Yu, Y.; Lurain, J.; Kim, J.J. The allosteric AKT inhibitor, MK2206, decreases tumor growth and invasion in patient derived xenografts of endometrial cancer. Cancer Biol. Ther. 2017, 18, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.; Li, Y.; Abramson, V.; Litton, J.; et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019, 21, 78. [Google Scholar] [CrossRef]
- Ching, C.B.; Hansel, D.E. Expanding therapeutic targets in bladder cancer: The PI3K/Akt/mTOR pathway. Lab. Investig. 2010, 90, 1406–1414. [Google Scholar] [CrossRef]
- Shimizu, T.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Smith, L.S.; Gunn, S.; Smetzer, L.; Mays, T.A.; Kaiser, B.; et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 2316–2325. [Google Scholar] [CrossRef]
- Lopez, S.; Cocco, E.; Black, J.; Bellone, S.; Bonazzoli, E.; Predolini, F.; Ferrari, F.; Schwab, C.L.; English, D.P.; Ratner, E.; et al. Dual HER2/PIK3CA Targeting Overcomes Single-Agent Acquired Resistance in HER2-Amplified Uterine Serous Carcinoma Cell Lines In Vitro and In Vivo. Mol. Cancer Ther. 2015, 14, 2519–2526. [Google Scholar] [CrossRef]
- Tripathy, D.; Bardia, A.; Sellers, W.R. Ribociclib (LEE011): Mechanism of Action and Clinical Impact of This Selective Cyclin-Dependent Kinase 4/6 Inhibitor in Various Solid Tumors. Clin. Cancer Res. 2017, 23, 3251–3262. [Google Scholar] [CrossRef]
- Cocco, E.; Lopez, S.; Black, J.; Bellone, S.; Bonazzoli, E.; Predolini, F.; Ferrari, F.; Schwab, C.L.; Menderes, G.; Zammataro, L.; et al. Dual CCNE1/PIK3CA targeting is synergistic in CCNE1-amplified/PIK3CA-mutated uterine serous carcinomas in vitro and in vivo. Br. J. Cancer 2016, 115, 303–311. [Google Scholar] [CrossRef]
- Di Tucci, C.; Capone, C.; Galati, G.; Iacobelli, V.; Schiavi, M.C.; Di Donato, V.; Muzii, L.; Panici, P.B. Immunotherapy in endometrial cancer: New scenarios on the horizon. J. Gynecol. Oncol. 2019, 30, e46. [Google Scholar] [CrossRef]
- Sungu, N.; Yildirim, M.; Desdicioglu, R.; Basaran Aydogdu, O.; Kilicarslan, A.; Tatli Dogan, H.; Kilic Yazgan, A.; Akyol, M.; Erdogan, F. Expression of Immunomodulatory Molecules PD-1, PD-L1, and PD-L2, and their Relationship With Clinicopathologic Characteristics in Endometrial Cancer. Int. J. Gynecol. Pathol. 2019, 38, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, O. PD-L1 expression and outcome of advanced melanoma patients treated with anti-PD-1/PD-L1 agents: A meta-analysis. Immunotherapy 2016, 8, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Liu, J.; Zhang, Q.; Chen, Z.; Mei, J.; Liu, L.; Yang, S.; Li, H.; Zhou, L.; You, Z. Expression of PD-1, PD-L1 and PD-L2 is associated with differentiation status and histological type of endometrial cancer. Oncol. Lett. 2016, 12, 944–950. [Google Scholar] [CrossRef]
- Makker, V.; Rasco, D.; Vogelzang, N.J.; Brose, M.S.; Cohn, A.L.; Mier, J.; Di Simone, C.; Hyman, D.M.; Stepan, D.E.; Dutcus, C.E.; et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: An interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 711–718. [Google Scholar] [CrossRef]
- Barrington, D.A.; Dilley, S.E.; Smith, H.J.; Straughn, J.M., Jr. Pembrolizumab in advanced recurrent endometrial cancer: A cost-effectiveness analysis. Gynecol. Oncol. 2019, 153, 381–384. [Google Scholar] [CrossRef]
- Giles, F.J.; Cortes, J.E.; Thomas, D.A.; Garcia-Manero, G.; Faderl, S.; Jeha, S.; De Jager, R.L.; Kantarjian, H.M. Phase I and pharmacokinetic study of DX-8951f (exatecan mesylate), a hexacyclic camptothecin, on a daily-times-five schedule in patients with advanced leukemia. Clin. Cancer Res. 2002, 8, 2134–2141. [Google Scholar]
- Iglesias, D.A.; Bodurka, D.C. Personalized care in uterine cancer. Clin. Adv. Hematol. Oncol. 2012, 10, 797–805. [Google Scholar]
- Myers, A.P. New strategies in endometrial cancer: Targeting the PI3K/mTOR pathway--the devil is in the details. Clin. Cancer Res. 2013, 19, 5264–5274. [Google Scholar] [CrossRef]
- Oza, A.M.; Elit, L.; Tsao, M.S.; Kamel-Reid, S.; Biagi, J.; Provencher, D.M.; Gotlieb, W.H.; Hoskins, P.J.; Ghatage, P.; Tonkin, K.S.; et al. Phase II study of temsirolimus in women with recurrent or metastatic endometrial cancer: A trial of the NCIC Clinical Trials Group. J. Clin. Oncol. 2011, 29, 3278–3285. [Google Scholar] [CrossRef]
- Diaz-Padilla, I.; Hirte, H.; Oza, A.M.; Clarke, B.A.; Cohen, B.; Reedjik, M.; Zhang, T.; Kamel-Reid, S.; Ivy, S.P.; Hotte, S.J.; et al. A phase Ib combination study of RO4929097, a gamma-secretase inhibitor, and temsirolimus in patients with advanced solid tumors. Investig. New Drugs 2013, 31, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Skaga, E.; Kulesskiy, E.; Fayzullin, A.; Sandberg, C.J.; Potdar, S.; Kyttala, A.; Langmoen, I.A.; Laakso, A.; Gaal-Paavola, E.; Perola, M.; et al. Intertumoral heterogeneity in patient-specific drug sensitivities in treatment-naive glioblastoma. BMC Cancer 2019, 19, 628. [Google Scholar] [CrossRef] [PubMed]
- Bottino, D.C.; Patel, M.; Kadakia, E.; Zhou, J.; Patel, C.; Neuwirth, R.; Iartchouk, N.; Brake, R.; Venkatakrishnan, K.; Chakravarty, A. Dose Optimization for Anticancer Drug Combinations: Maximizing Therapeutic Index via Clinical Exposure-Toxicity/Preclinical Exposure-Efficacy Modeling. Clin. Cancer Res. 2019, 25, 6633–6643. [Google Scholar] [CrossRef] [PubMed]
- Heudel, P.E.; Fabbro, M.; Roemer-Becuwe, C.; Kaminsky, M.C.; Arnaud, A.; Joly, F.; Roche-Forestier, S.; Meunier, J.; Foa, C.; You, B.; et al. Phase II study of the PI3K inhibitor BKM120 in patients with advanced or recurrent endometrial carcinoma: A stratified type I-type II study from the GINECO group. Br. J. Cancer 2017, 116, 303–309. [Google Scholar] [CrossRef]
- Makker, V.; Recio, F.O.; Ma, L.; Matutonis, U.; Lauchle, J.O.; Parmar, H.; Gilbert, H.; Wang, Y.L.; Koeppen, H.; Spoerke, J.M.; et al. Phase II trial of GDC-0980 (dual PI3K/mTOR inhibitor) in patients with advanced endometrial carcinoma: Final study results. J. Clin. Oncol. 2014, 32, 5513. [Google Scholar] [CrossRef]
- Toyoda, M.; Watanabe, K.; Amagasaki, T.; Natsume, K.; Takeuchi, H.; Quadt, C.; Shirao, K.; Minami, H. A phase I study of single-agent BEZ235 special delivery system sachet in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2019, 83, 289–299. [Google Scholar] [CrossRef]
- Myers, A.P.; Konstantinopoulos, P.A.; Barry, W.T.; Luo, W.; Broaddus, R.R.; Makker, V.; Drapkin, R.; Liu, J.; Doyle, A.; Horowitz, N.S.; et al. Phase II, 2-stage, 2-arm, PIK3CA mutation stratified trial of MK-2206 in recurrent endometrial cancer. Int. J. Cancer 2019. [Google Scholar] [CrossRef]
- Giannone, G.; Tuninetti, V.; Ghisoni, E.; Genta, S.; Scotto, G.; Mittica, G.; Valabrega, G. Role of Cyclin-Dependent Kinase Inhibitors in Endometrial Cancer. Int. J. Mol. Sci. 2019, 20, 2353. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Kwan, S.Y.; Wong, K.K.; Soliman, P.T.; Lu, K.H.; Mok, S.C. Pathogenesis and Clinical Management of Uterine Serous Carcinoma. Cancers 2020, 12, 686. https://doi.org/10.3390/cancers12030686
Zhang L, Kwan SY, Wong KK, Soliman PT, Lu KH, Mok SC. Pathogenesis and Clinical Management of Uterine Serous Carcinoma. Cancers. 2020; 12(3):686. https://doi.org/10.3390/cancers12030686
Chicago/Turabian StyleZhang, Li, Suet Ying Kwan, Kwong Kwok Wong, Pamela T. Soliman, Karen H. Lu, and Samuel C. Mok. 2020. "Pathogenesis and Clinical Management of Uterine Serous Carcinoma" Cancers 12, no. 3: 686. https://doi.org/10.3390/cancers12030686
APA StyleZhang, L., Kwan, S. Y., Wong, K. K., Soliman, P. T., Lu, K. H., & Mok, S. C. (2020). Pathogenesis and Clinical Management of Uterine Serous Carcinoma. Cancers, 12(3), 686. https://doi.org/10.3390/cancers12030686