Ginsenoside Rp1, A Ginsenoside Derivative, Augments Anti-Cancer Effects of Actinomycin D via Downregulation of an AKT-SIRT1 Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
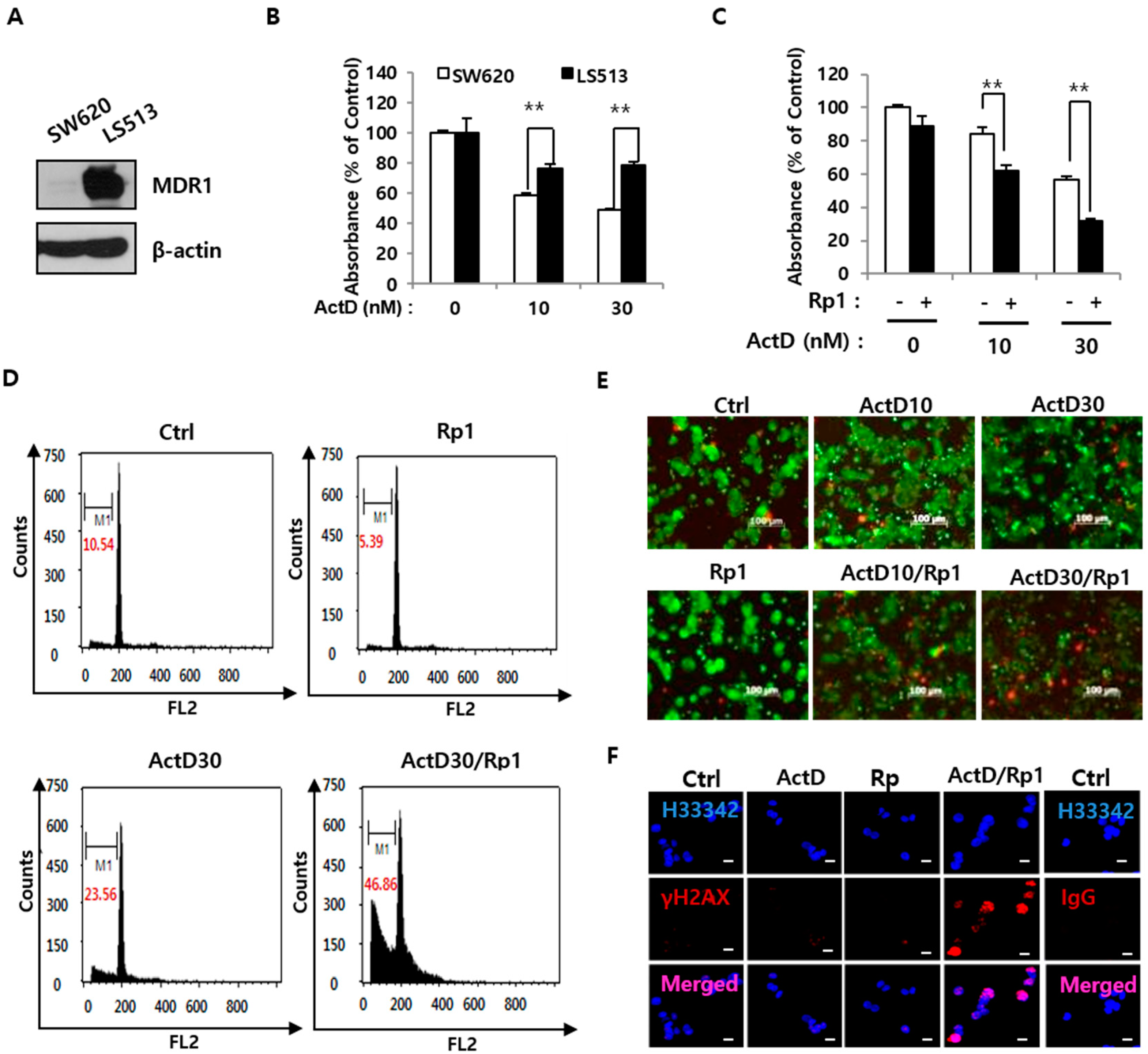
2.1. Rp1 Enhances ActD-Induced Cell Death in Multidrug-Resistant LS513 Cells
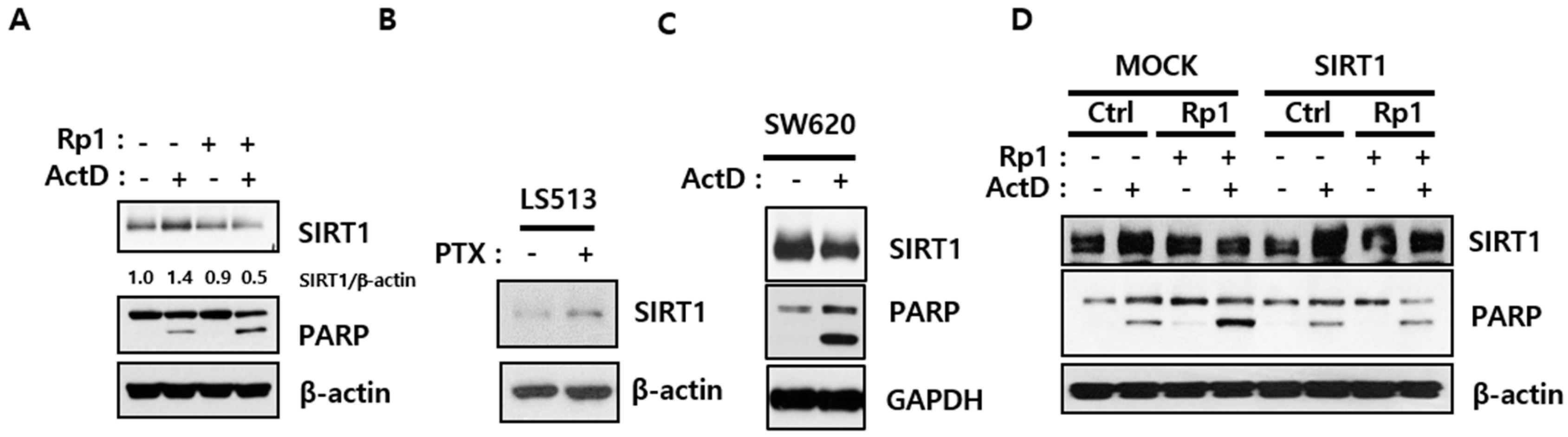
2.2. Rp1 Attenuates ActD-Induced SIRT1 Upregulation
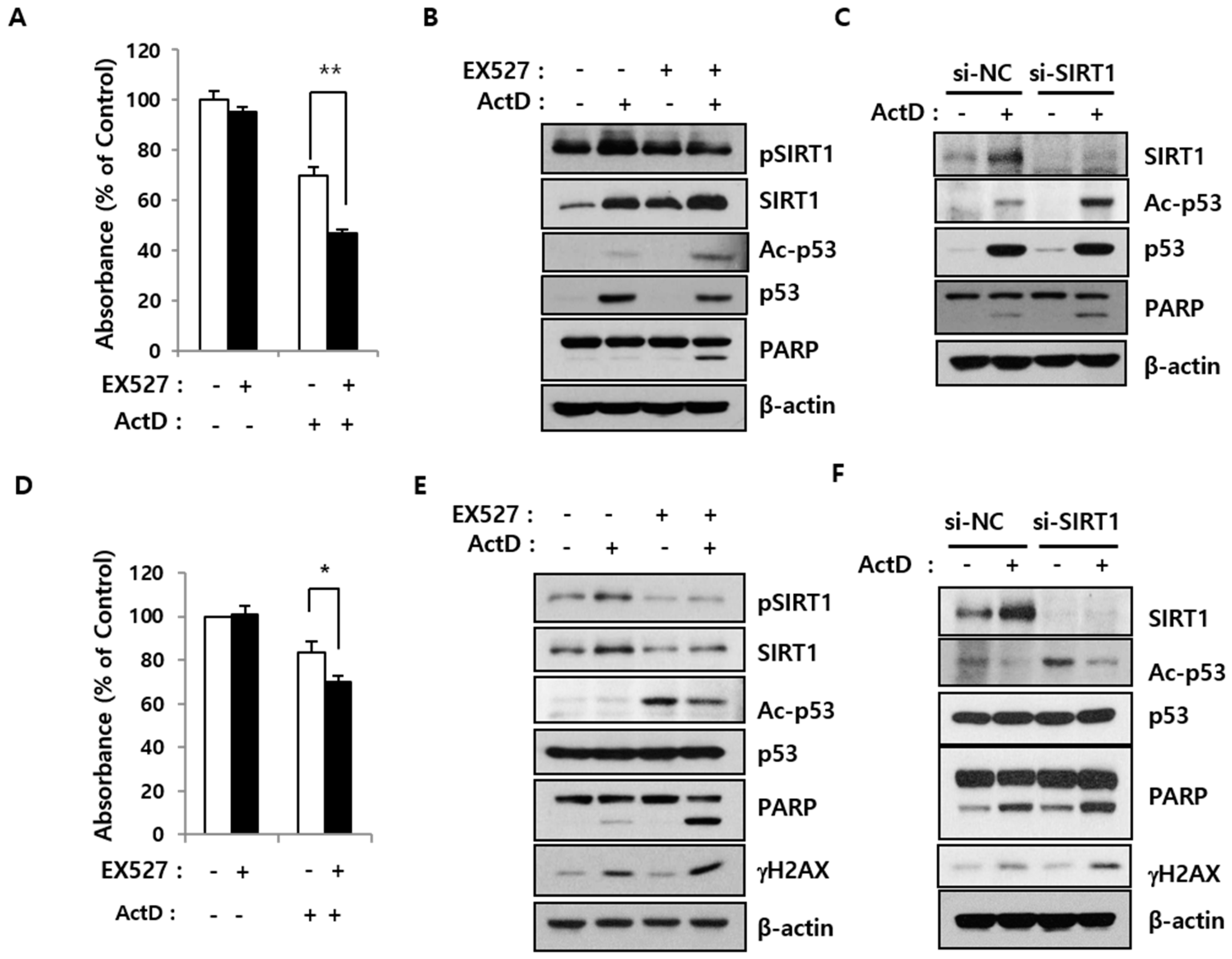
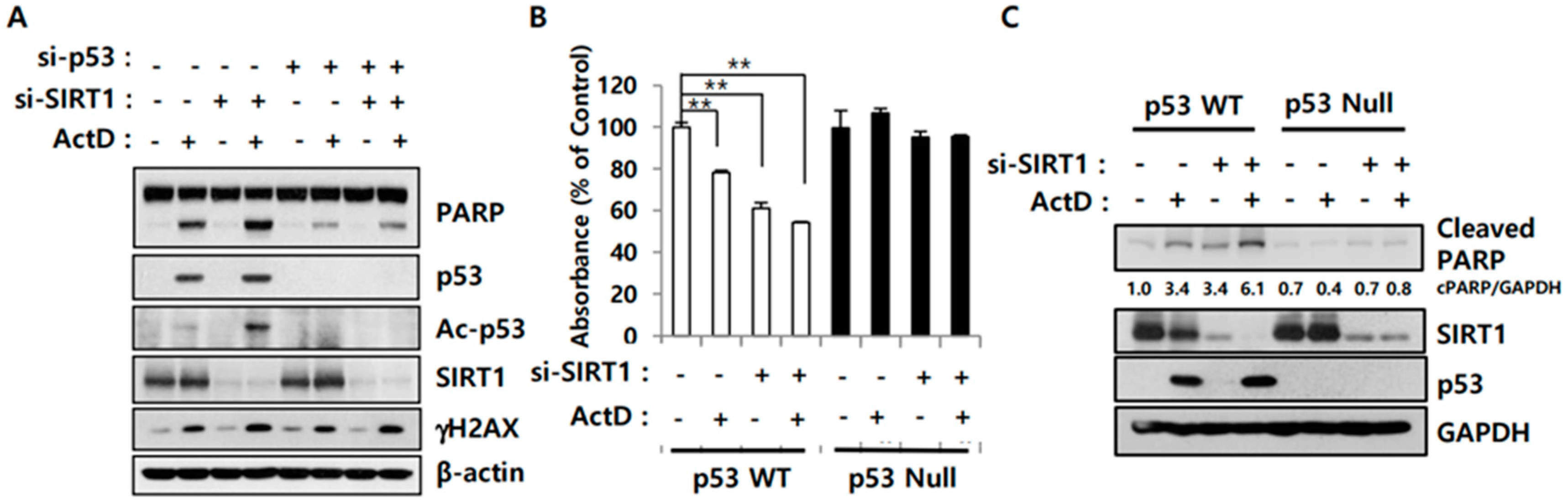
2.3. SIRT1 Inhibition Reverses Resistance to ActD through p53 Deacetylation
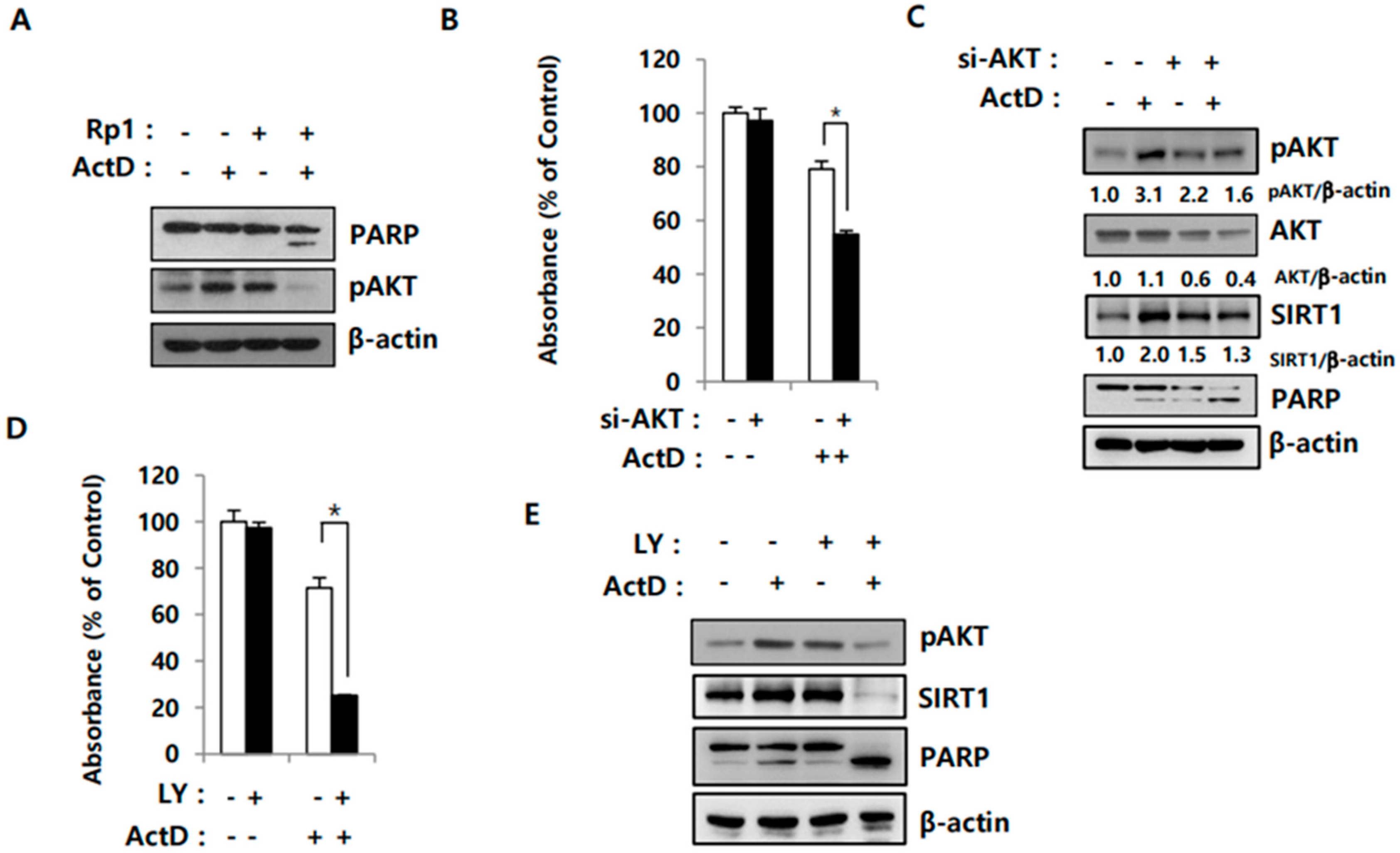
2.4. Activation of AKT Correlates with SIRT1 Upregulation in Drug-Resistant Cells
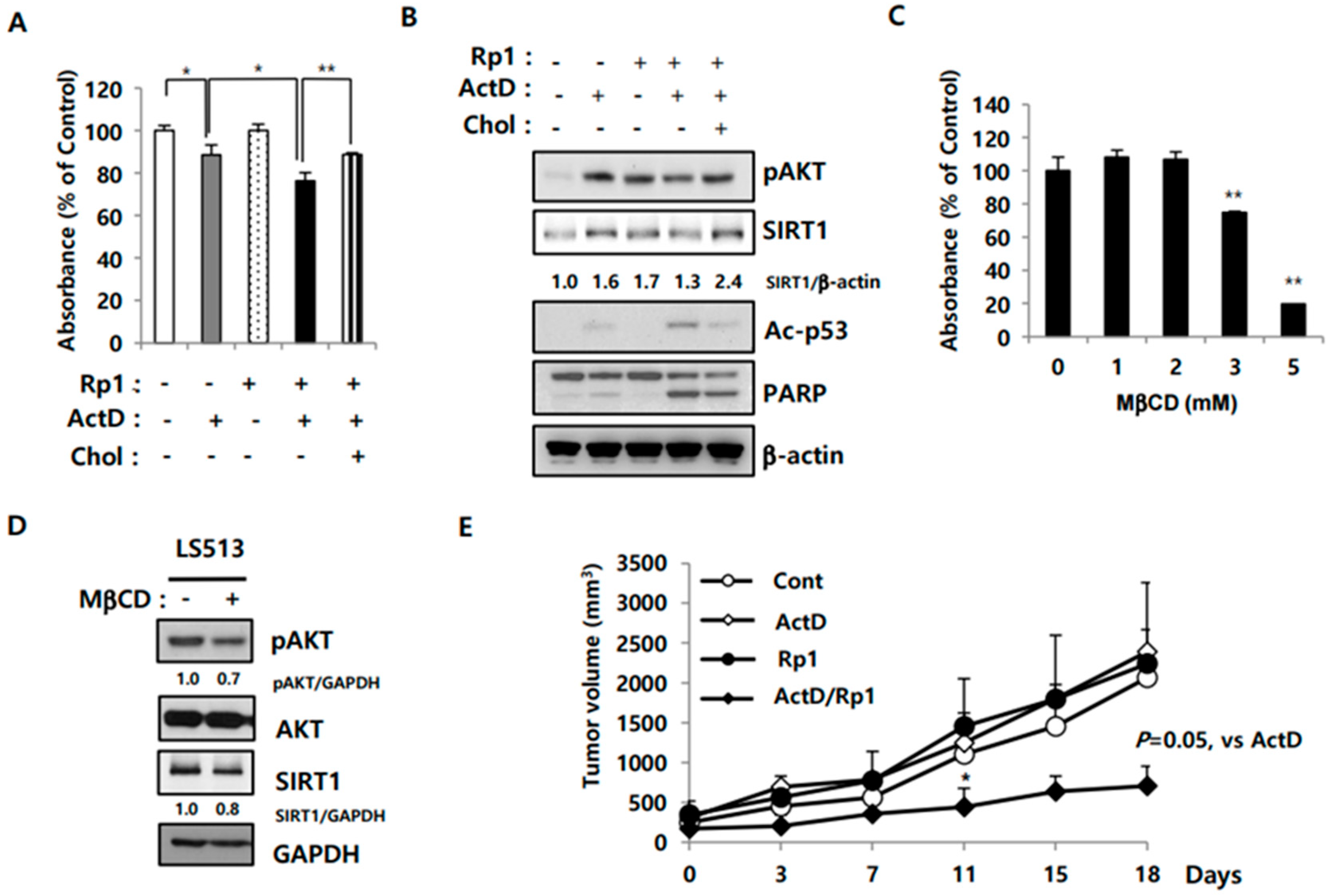
2.5. Rp1 Attenuates Resistance to ActD through the Cholesterol-Associated AKT/SIRT1 Pathway
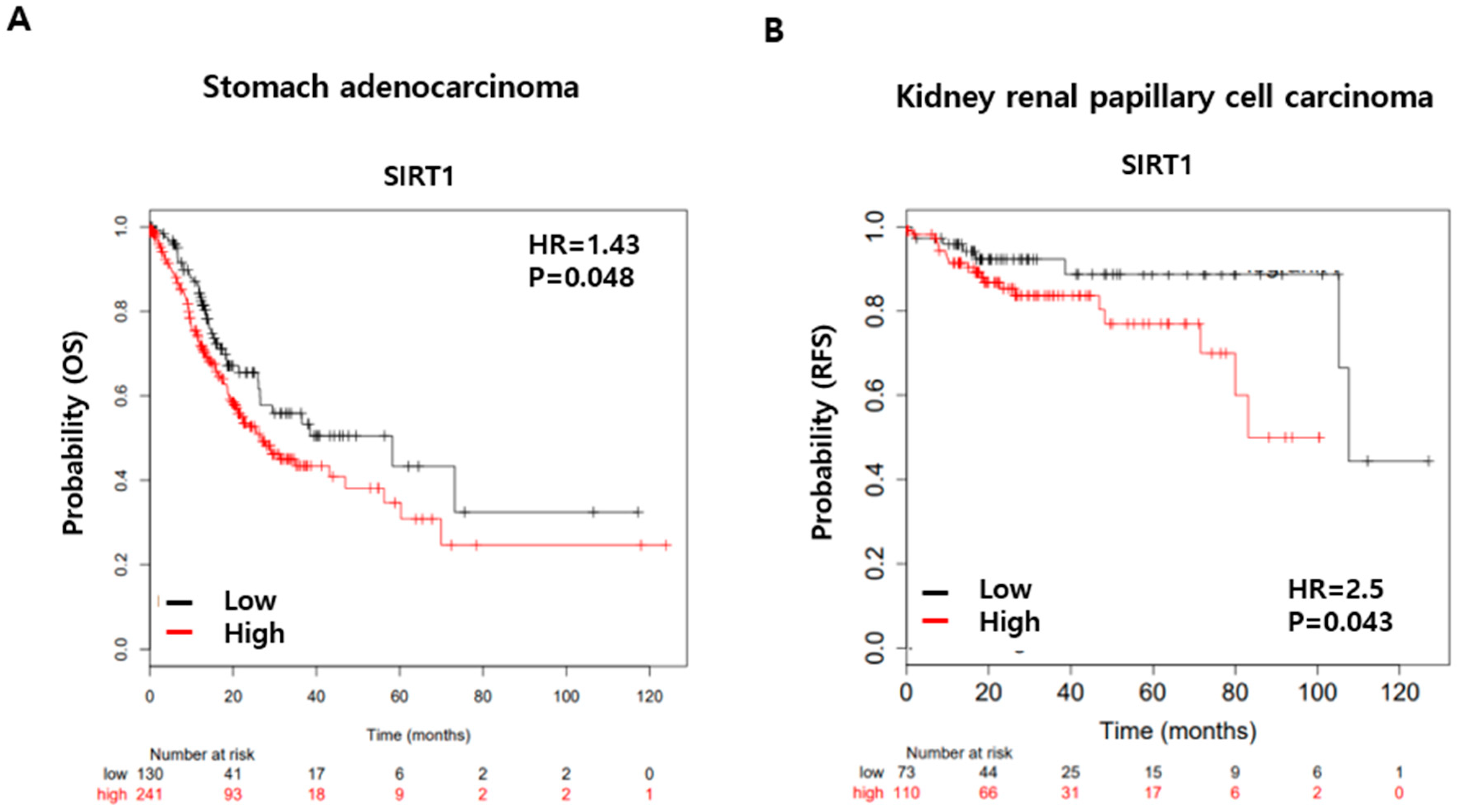
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Viability and Proliferation Assays
4.4. siRNA Transfection
4.5. Immunoblotting Analysis
4.6. Immunofluorescence and Confocal Microscopy Imaging
4.7. Flow Cytometric Analysis of Cell Death
4.8. Establishment of a Murine Xenograft Model
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Yun, U.J.; Lee, J.H.; Koo, K.H.; Ye, S.K.; Kim, S.Y.; Lee, C.H.; Kim, Y.N. Lipid raft modulation by Rp1 reverses multidrug resistance via inactivating MDR-1 and Src inhibition. Biochem. Pharmacol. 2013, 85, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Wang, Z.; Yuan, H.; Roth, M.; Stark, J.M.; Bhatia, R.; Chen, W.Y. SIRT1 deacetylase promotes acquisition of genetic mutations for drug resistance in CML cells. Oncogene 2013, 32, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Mao, B.; Hu, F.; Cheng, J.; Wang, P.; Xu, M.; Yuan, F.; Meng, S.; Wang, Y.; Yuan, Z.; Bi, W. SIRT1 regulates YAP2-mediated cell proliferation and chemoresistance in hepatocellular carcinoma. Oncogene 2014, 33, 1468–1474. [Google Scholar] [CrossRef]
- Alves-Fernandes, D.K.; Jasiulionis, M.G. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. Int. J. Mol. Sci. 2019, 20, 3153. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Jeng, Y.M.; Yuan, R.H.; Hsu, H.C.; Chen, Y.L. SIRT1 promotes tumorigenesis and resistance to chemotherapy in hepatocellular carcinoma and its expression predicts poor prognosis. Ann. Surg. Oncol. 2012, 19, 2011–2019. [Google Scholar] [CrossRef]
- Liang, X.J.; Finkel, T.; Shen, D.W.; Yin, J.J.; Aszalos, A.; Gottesman, M.M. SIRT1 contributes in part to cisplatin resistance in cancer cells by altering mitochondrial metabolism. Mol. Cancer Res. 2008, 6, 1499–1506. [Google Scholar] [CrossRef]
- Kojima, K.; Ohhashi, R.; Fujita, Y.; Hamada, N.; Akao, Y.; Nozawa, Y.; Deguchi, T.; Ito, M. A role for SIRT1 in cell growth and chemoresistance in prostate cancer PC3 and DU145 cells. Biochem. Biophys. Res. Commun. 2008, 373, 423–428. [Google Scholar] [CrossRef]
- Yousafzai, N.A.; Zhou, Q.; Xu, W.; Shi, Q.; Xu, J.; Feng, L.; Chen, H.; Shin, V.Y.; Jin, H.; Wang, X. SIRT1 deacetylated and stabilized XRCC1 to promote chemoresistance in lung cancer. Cell Death Dis. 2019, 10, 363. [Google Scholar] [CrossRef]
- Shin, H.R.; Kim, J.Y.; Yun, T.K.; Morgan, G.; Vainio, H. The cancer-preventive potential of Panax ginseng: A review of human and experimental evidence. Cancer Causes Control 2000, 11, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.Y.; Mak, N.K.; Cheng, Y.K.; Leung, K.W.; Ng, T.B.; Fan, D.T.; Yeung, H.W.; Wong, R.N. Pharmacogenomics and the Yin/Yang actions of ginseng: Anti-tumor, angiomodulating and steroid-like activities of ginsenosides. Chin. Med. 2007, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.Y.; Kim, E.H.; Kim, S.W.; Kim, S.N.; Park, J.D.; Rhee, D.K. Selective toxicity of ginsenoside Rg3 on multidrug resistant cells by membrane fluidity modulation. Arch. Pharm. Res. 2008, 31, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, F.; Wu, X.; Zhang, X.; Chen, Y.; Zha, B.S.; Niu, F.; Lu, M.; Hao, G.; Sun, Y.; et al. Cellular pharmacokinetic mechanisms of adriamycin resistance and its modulation by 20(S)-ginsenoside Rh2 in MCF-7/Adr cells. Br. J. Pharmacol. 2012, 165, 120–134. [Google Scholar] [CrossRef]
- Perry, R.P.; Kelley, D.E. Inhibition of RNA synthesis by actinomycin D: Characteristic dose-response of different RNA species. J. Cell. Physiol. 1970, 76, 127–139. [Google Scholar] [CrossRef]
- Lo, Y.S.; Tseng, W.H.; Chuang, C.Y.; Hou, M.H. The structural basis of actinomycin D-binding induces nucleotide flipping out, a sharp bend and a left-handed twist in CGG triplet repeats. Nucleic Acids Res. 2013, 41, 4284–4294. [Google Scholar] [CrossRef]
- Mischo, H.E.; Hemmerich, P.; Grosse, F.; Zhang, S. Actinomycin D induces histone gamma-H2AX foci and complex formation of gamma-H2AX with Ku70 and nuclear DNA helicase II. J. Biol. Chem. 2005, 280, 9586–9594. [Google Scholar] [CrossRef]
- Vellinga, T.T.; Borovski, T.; de Boer, V.C.; Fatrai, S.; van Schelven, S.; Trumpi, K.; Verheem, A.; Snoeren, N.; Emmink, B.L.; Koster, J.; et al. SIRT1/PGC1alpha-Dependent Increase in Oxidative Phosphorylation Supports Chemotherapy Resistance of Colon Cancer. Clin. Cancer Res. 2015, 21, 2870–2879. [Google Scholar] [CrossRef]
- Mvunta, D.H.; Miyamoto, T.; Asaka, R.; Yamada, Y.; Ando, H.; Higuchi, S.; Ida, K.; Kashima, H.; Shiozawa, T. SIRT1 Regulates the Chemoresistance and Invasiveness of Ovarian Carcinoma Cells. Transl. Oncol. 2017, 10, 621–631. [Google Scholar] [CrossRef]
- Lau, A.W.; Liu, P.; Inuzuka, H.; Gao, D. SIRT1 phosphorylation by AMP-activated protein kinase regulates p53 acetylation. Am. J. Cancer Res. 2014, 4, 245–255. [Google Scholar]
- Cao, C.; Lu, S.; Kivlin, R.; Wallin, B.; Card, E.; Bagdasarian, A.; Tamakloe, T.; Wang, W.J.; Song, X.; Chu, W.M.; et al. SIRT1 confers protection against UVB- and H2O2-induced cell death via modulation of p53 and JNK in cultured skin keratinocytes. J. Cell. Mol. Med. 2009, 13, 3632–3643. [Google Scholar] [CrossRef] [PubMed]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.G.; Wei, C.S.; Zhang, S.; Zhang, Z.; Gao, H.M. Matrine reversed multidrug resistance of breast cancer MCF-7/ADR cells through PI3K/AKT signaling pathway. J. Cell. Biochem. 2018, 119, 3885–3891. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kang, G.J.; Kang, J.I.; Boo, H.J.; Hyun, J.W.; Koh, Y.S.; Chang, W.Y.; Kim, Y.R.; Kwon, J.M.; Maeng, Y.H.; et al. Over-activation of AKT signaling leading to 5-Fluorouracil resistance in SNU-C5/5-FU cells. Oncotarget 2018, 9, 19911–19928. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Pillai, V.B.; Wolfgeher, D.; Samant, S.; Vasudevan, P.; Parekh, V.; Raghuraman, H.; Cunningham, J.M.; Gupta, M.; Gupta, M.P. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci. Signal. 2011, 4, ra46. [Google Scholar] [CrossRef]
- Li, Y.C.; Park, M.J.; Ye, S.K.; Kim, C.W.; Kim, Y.N. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am. J. Pathol. 2006, 168, 1107–1118. [Google Scholar] [CrossRef]
- Zhuang, L.; Lin, J.; Lu, M.L.; Solomon, K.R.; Freeman, M.R. Cholesterol-rich lipid rafts mediate akt-regulated survival in prostate cancer cells. Cancer Res. 2002, 62, 2227–2231. [Google Scholar]
- Park, E.K.; Lee, E.J.; Lee, S.H.; Koo, K.H.; Sung, J.Y.; Hwang, E.H.; Park, J.H.; Kim, C.W.; Jeong, K.C.; Park, B.K.; et al. Induction of apoptosis by the ginsenoside Rh2 by internalization of lipid rafts and caveolae and inactivation of Akt. Br. J. Pharmacol. 2010, 160, 1212–1223. [Google Scholar] [CrossRef]
- Nag, S.A.; Qin, J.J.; Wang, W.; Wang, M.H.; Wang, H.; Zhang, R. Ginsenosides as Anticancer Agents: In vitro and in vivo Activities, Structure-Activity Relationships, and Molecular Mechanisms of Action. Front. Pharmacol. 2012, 3, 25. [Google Scholar] [CrossRef]
- Lu, J.M.; Yao, Q.; Chen, C. Ginseng compounds: An update on their molecular mechanisms and medical applications. Curr. Vasc. Pharmacol. 2009, 7, 293–302. [Google Scholar] [CrossRef]
- Sun, M.; Ye, Y.; Xiao, L.; Duan, X.; Zhang, Y.; Zhang, H. Anticancer effects of ginsenoside Rg3 (Review). Int. J. Mol. Med. 2017, 39, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.Y.; Hwang, S.H.; Kwon, K.S.; Kim, K.R.; Choi, H.N.; Lee, N.R.; Kwak, J.Y.; Park, B.H.; Park, H.S.; Chung, M.J.; et al. SIRT1 expression is associated with poor prognosis of diffuse large B-cell lymphoma. Am. J. Surg. Pathol. 2008, 32, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, A.; Kikuchi, K.; Zheng, H.; Takahashi, H.; Miyagi, Y.; Aoki, I.; Takano, Y. SIRT1 expression is associated with a poor prognosis, whereas DBC1 is associated with favorable outcomes in gastric cancer. Cancer Med. 2014, 3, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, L.; Zheng, L.; Zhan, X.; Xu, B.; Jiang, J.; Wu, C. SIRT1 expression is associated with poor prognosis of lung adenocarcinoma. Onco Targets Ther. 2015, 8, 977–984. [Google Scholar] [CrossRef]
- Chu, F.; Chou, P.M.; Zheng, X.; Mirkin, B.L.; Rebbaa, A. Control of multidrug resistance gene mdr1 and cancer resistance to chemotherapy by the longevity gene sirt1. Cancer Res. 2005, 65, 10183–10187. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, W. Emerging Roles of SIRT1 in Cancer Drug Resistance. Genes Cancer 2013, 4, 82–90. [Google Scholar] [CrossRef]
- Cheng, H.L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; Li, L.; Wang, Z.; Ho, Y.; McDonald, T.; Holyoake, T.L.; Chen, W.; Bhatia, R. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell 2012, 21, 266–281. [Google Scholar] [CrossRef]
- Kume, S.; Haneda, M.; Kanasaki, K.; Sugimoto, T.; Araki, S.; Isono, M.; Isshiki, K.; Uzu, T.; Kashiwagi, A.; Koya, D. Silent information regulator 2 (SIRT1) attenuates oxidative stress-induced mesangial cell apoptosis via p53 deacetylation. Free Radic. Biol. Med. 2006, 40, 2175–2182. [Google Scholar] [CrossRef]
- Martelli, A.M.; Tazzari, P.L.; Tabellini, G.; Bortul, R.; Billi, A.M.; Manzoli, L.; Ruggeri, A.; Conte, R.; Cocco, L. A new selective AKT pharmacological inhibitor reduces resistance to chemotherapeutic drugs, TRAIL, all-trans-retinoic acid, and ionizing radiation of human leukemia cells. Leukemia 2003, 17, 1794–1805. [Google Scholar] [CrossRef]
- West, K.A.; Castillo, S.S.; Dennis, P.A. Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resist. Updates 2002, 5, 234–248. [Google Scholar] [CrossRef]
- Wang, L.; Lin, N.; Li, Y. The PI3K/AKT signaling pathway regulates ABCG2 expression and confers resistance to chemotherapy in human multiple myeloma. Oncol. Rep. 2019, 41, 1678–1690. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.Y.; Yoo, E.S.; Baik, K.U.; Park, M.H.; Han, B.H. In vitro inhibitory effect of protopanaxadiol ginsenosides on tumor necrosis factor (TNF)-alpha production and its modulation by known TNF-alpha antagonists. Planta Med. 2001, 67, 213–218. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, U.-J.; Lee, I.H.; Lee, J.-S.; Shim, J.; Kim, Y.-N. Ginsenoside Rp1, A Ginsenoside Derivative, Augments Anti-Cancer Effects of Actinomycin D via Downregulation of an AKT-SIRT1 Pathway. Cancers 2020, 12, 605. https://doi.org/10.3390/cancers12030605
Yun U-J, Lee IH, Lee J-S, Shim J, Kim Y-N. Ginsenoside Rp1, A Ginsenoside Derivative, Augments Anti-Cancer Effects of Actinomycin D via Downregulation of an AKT-SIRT1 Pathway. Cancers. 2020; 12(3):605. https://doi.org/10.3390/cancers12030605
Chicago/Turabian StyleYun, Un-Jung, In Hye Lee, Jae-Seon Lee, Jaegal Shim, and Yong-Nyun Kim. 2020. "Ginsenoside Rp1, A Ginsenoside Derivative, Augments Anti-Cancer Effects of Actinomycin D via Downregulation of an AKT-SIRT1 Pathway" Cancers 12, no. 3: 605. https://doi.org/10.3390/cancers12030605
APA StyleYun, U.-J., Lee, I. H., Lee, J.-S., Shim, J., & Kim, Y.-N. (2020). Ginsenoside Rp1, A Ginsenoside Derivative, Augments Anti-Cancer Effects of Actinomycin D via Downregulation of an AKT-SIRT1 Pathway. Cancers, 12(3), 605. https://doi.org/10.3390/cancers12030605