Genomic Signature of the Standardized Uptake Value in 18F-Fluorodeoxyglucose Positron Emission Tomography in Breast Cancer

 , , ,
, , , 
Abstract
1. Introduction
2. Results
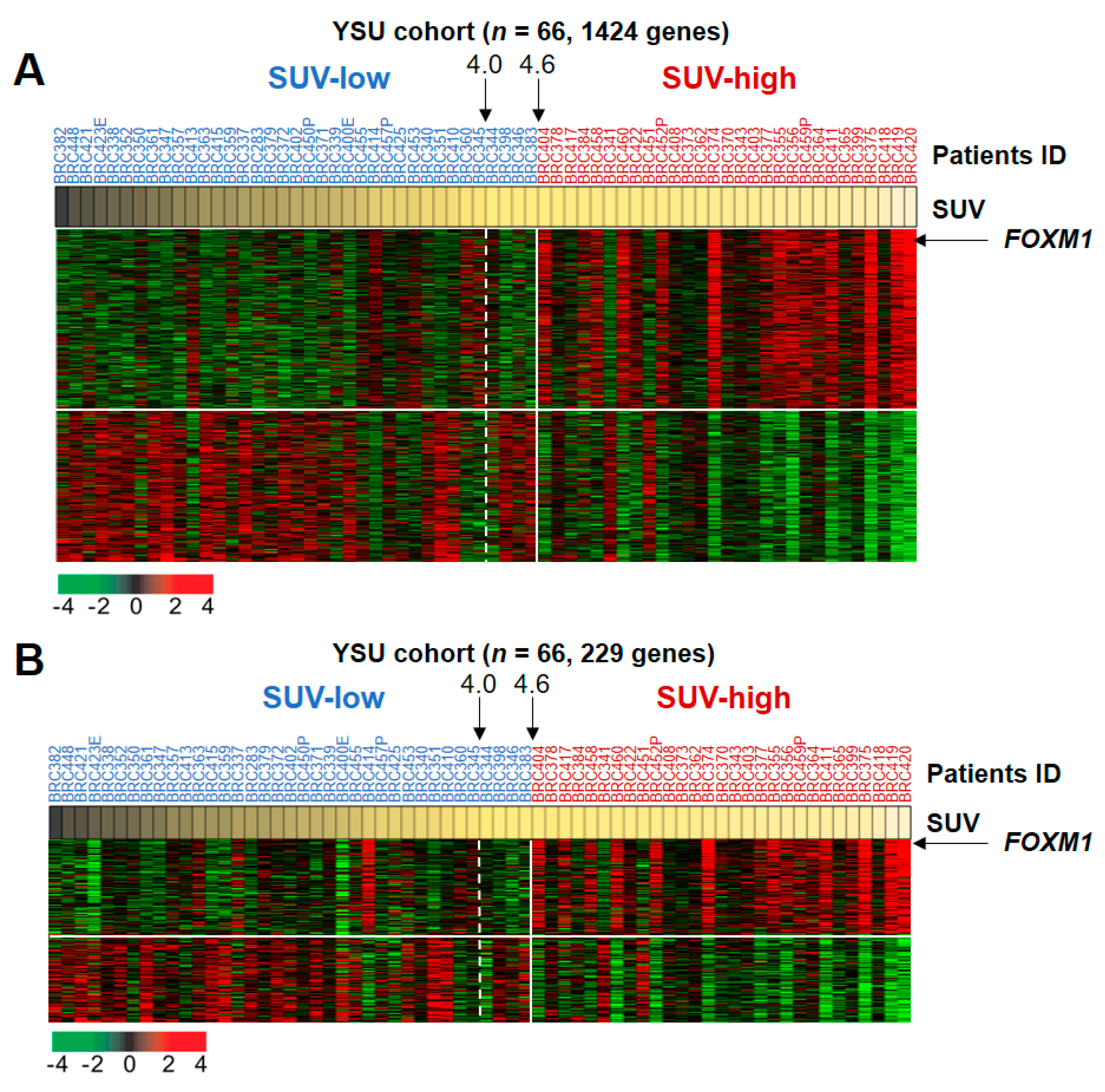
2.1. Transcriptomic Profiling Reflecting SUV Alterations in BRC
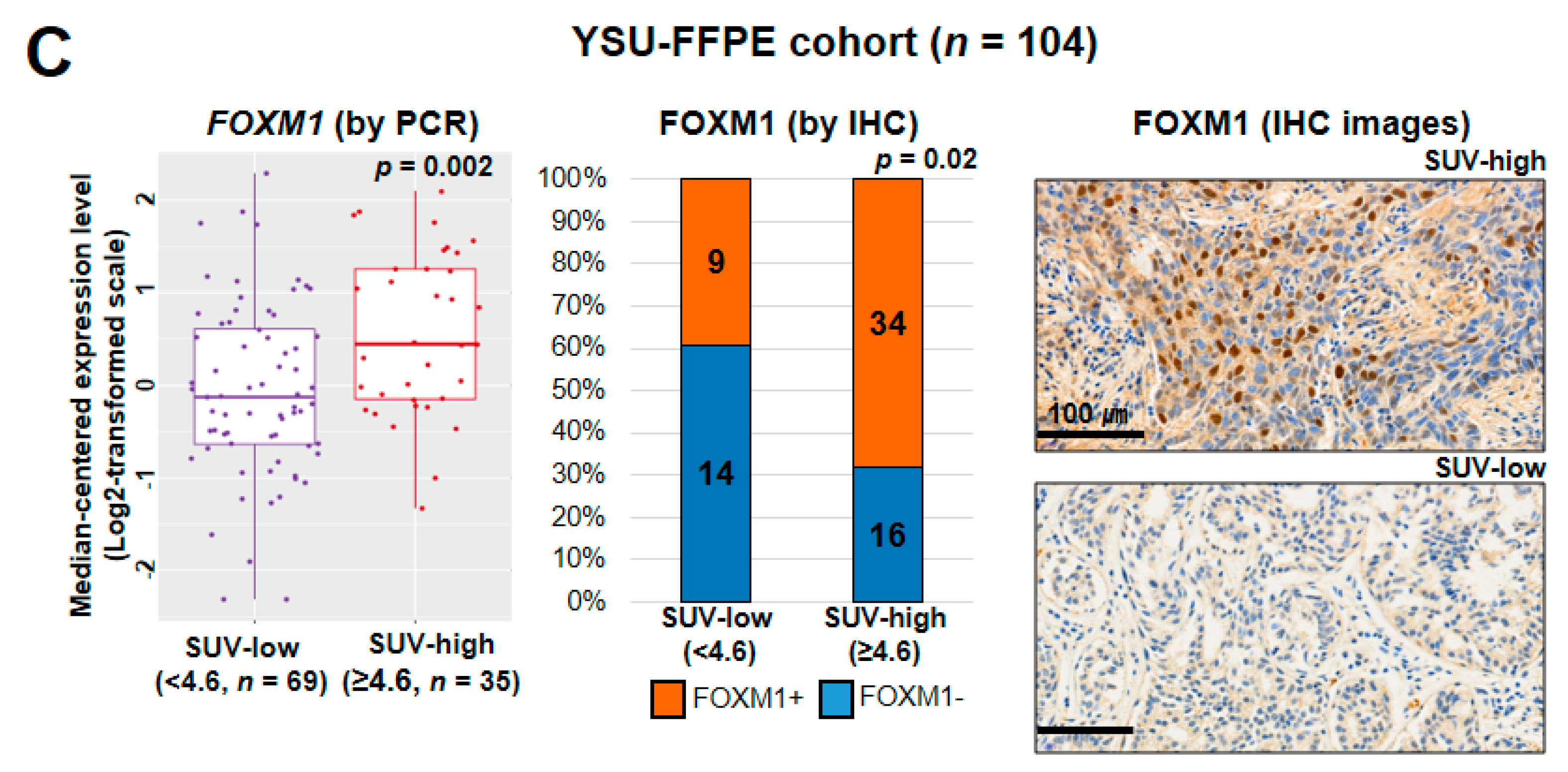
2.2. Biological Insight into the SUV-Associated Genes
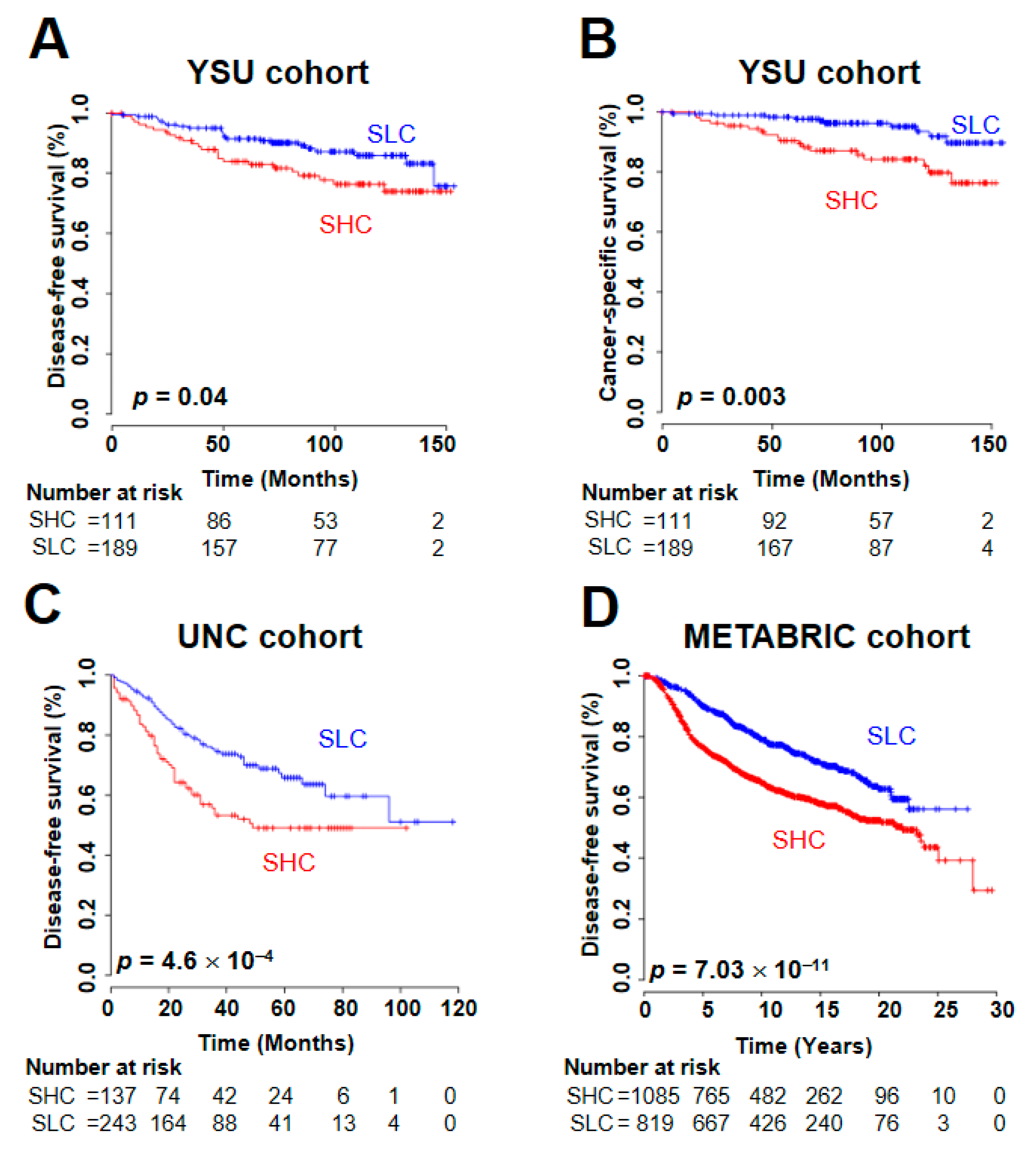
2.3. Prognostic Utility of the SUV Signature
2.4. Mutational Landscape of SUV Genomic Subtypes in BRC
2.5. Association Between the SUV Signature and the Response to Immune Checkpoint Inhibitors (ICIs)
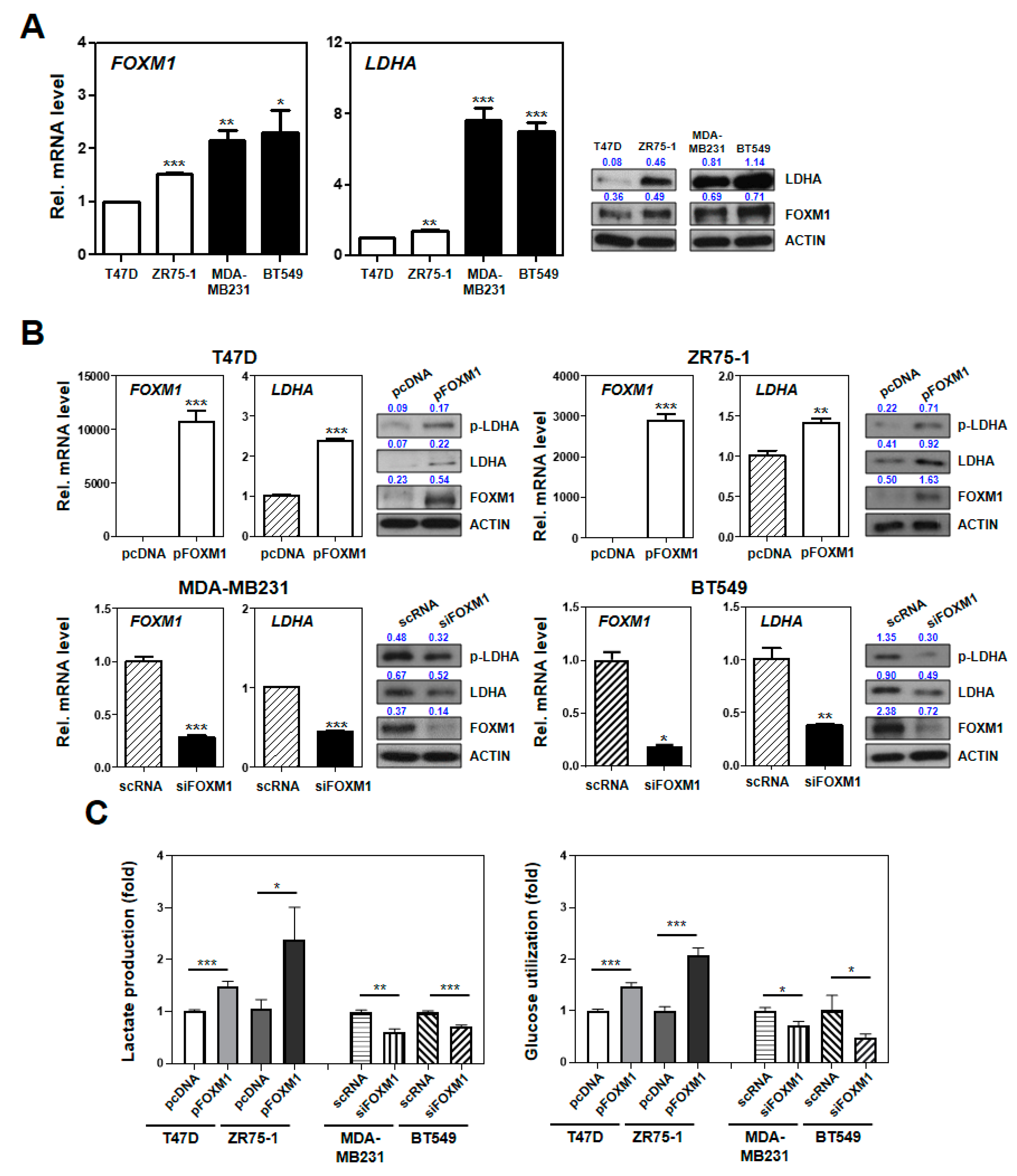
2.6. Experimental Characterization of the SUV Signature Mediated by FOXM1–LDHA Signaling
3. Discussion
4. Materials and Methods
4.1. Patients and Tissue Samples
4.2. 18F-Fluorodexoyglucose Positron Emission Tomography (FDG-PET)
4.3. RNA Extraction, Microarray Experiments, and Data Processing
4.4. Public Datasets of BRC Patients
4.5. Statistical Analysis
4.6. Cell Culture
4.7. Transfection, Plasmids, and Small Interfering RNA (siRNA)
4.8. Quantitative Real-Time (qRT) PCR
4.9. Western Blotting and Antibodies
4.10. Lactate Production and Glucose Utilization Assay
4.11. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ahn, S.G.; Park, J.T.; Lee, H.M.; Lee, H.W.; Jeon, T.J.; Han, K.; Lee, S.A.; Dong, S.M.; Ryu, Y.H.; Son, E.J.; et al. Standardized uptake value of (1)(8)F-fluorodeoxyglucose positron emission tomography for prediction of tumor recurrence in breast cancer beyond tumor burden. Breast Cancer Res. 2014, 16, 502. [Google Scholar] [CrossRef]
- Seo, S.; Hatano, E.; Higashi, T.; Hara, T.; Tada, M.; Tamaki, N.; Iwaisako, K.; Ikai, I.; Uemoto, S. Fluorine-18 fluorodeoxyglucose positron emission tomography predicts tumor differentiation, P-glycoprotein expression, and outcome after resection in hepatocellular carcinoma. Clin. Cancer Res. 2007, 13, 427–433. [Google Scholar] [CrossRef]
- Cerfolio, R.J.; Bryant, A.S.; Ohja, B.; Bartolucci, A.A. The maximum standardized uptake values on positron emission tomography of a non-small cell lung cancer predict stage, recurrence, and survival. J. Thorac. Cardiovasc. Surg. 2005, 130, 151–159. [Google Scholar] [CrossRef]
- Ahn, S.G.; Lee, J.H.; Lee, H.W.; Jeon, T.J.; Ryu, Y.H.; Kim, K.M.; Sohn, J.; Yun, M.; Lee, S.A.; Jeong, J.; et al. Comparison of standardized uptake value of 18F-FDG-PET-CT with 21-gene recurrence score in estrogen receptor-positive, HER2-negative breast cancer. PLoS ONE 2017, 12, e0175048. [Google Scholar] [CrossRef]
- Palaskas, N.; Larson, S.M.; Schultz, N.; Komisopoulou, E.; Wong, J.; Rohle, D.; Campos, C.; Yannuzzi, N.; Osborne, J.R.; Linkov, I.; et al. 18F-fluorodeoxy-glucose positron emission tomography marks MYC-overexpressing human basal-like breast cancers. Cancer Res. 2011, 71, 5164–5174. [Google Scholar] [CrossRef]
- Basu, S.; Chen, W.; Tchou, J.; Mavi, A.; Cermik, T.; Czerniecki, B.; Schnall, M.; Alavi, A. Comparison of triple-negative and estrogen receptor-positive/progesterone receptor-positive/HER2-negative breast carcinoma using quantitative fluorine-18 fluorodeoxyglucose/positron emission tomography imaging parameters: A potentially useful method for disease characterization. Cancer 2008, 112, 995–1000. [Google Scholar] [CrossRef]
- Shimoda, W.; Hayashi, M.; Murakami, K.; Oyama, T.; Sunagawa, M. The relationship between FDG uptake in PET scans and biological behavior in breast cancer. Breast Cancer 2007, 14, 260–268. [Google Scholar] [CrossRef]
- Mavi, A.; Cermik, T.F.; Urhan, M.; Puskulcu, H.; Basu, S.; Yu, J.Q.; Zhuang, H.; Czerniecki, B.; Alavi, A. The effects of estrogen, progesterone, and C-erbB-2 receptor states on 18F-FDG uptake of primary breast cancer lesions. J. Nucl. Med. 2007, 48, 1266–1272. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Koo, H.R.; Park, J.S.; Kang, K.W.; Cho, N.; Chang, J.M.; Bae, M.S.; Kim, W.H.; Lee, S.H.; Kim, M.Y.; Kim, J.Y.; et al. 18F-FDG uptake in breast cancer correlates with immunohistochemically defined subtypes. Eur. Radiol. 2014, 24, 610–618. [Google Scholar] [CrossRef]
- Ueda, S.; Kondoh, N.; Tsuda, H.; Yamamoto, S.; Asakawa, H.; Fukatsu, K.; Kobayashi, T.; Yamamoto, J.; Tamura, K.; Ishida, J.; et al. Expression of centromere protein F (CENP-F) associated with higher FDG uptake on PET/CT, detected by cDNA microarray, predicts high-risk patients with primary breast cancer. BMC Cancer 2008, 8, 384. [Google Scholar] [CrossRef]
- Anders, C.K.; Fan, C.; Parker, J.S.; Carey, L.A.; Blackwell, K.L.; Klauber-DeMore, N.; Perou, C.M. Breast carcinomas arising at a young age: Unique biology or a surrogate for aggressive intrinsic subtypes? J. Clin. Oncol. 2011, 29, e18–e20. [Google Scholar] [CrossRef]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef]
- Kang, S.; Kim, E.H.; Hwang, J.E.; Shin, J.H.; Jeong, Y.S.; Yim, S.Y.; Joo, E.W.; Eun, Y.G.; Lee, D.J.; Sohn, B.H.; et al. Prognostic significance of high metabolic activity in breast cancer: PET signature in breast cancer. Biochem. Biophys. Res. Commun. 2019, 511, 185–191. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Teo, M.Y.; Seier, K.; Ostrovnaya, I.; Regazzi, A.M.; Kania, B.E.; Moran, M.M.; Cipolla, C.K.; Bluth, M.J.; Chaim, J.; Al-Ahmadie, H.; et al. Alterations in DNA Damage Response and Repair Genes as Potential Marker of Clinical Benefit From PD-1/PD-L1 Blockade in Advanced Urothelial Cancers. J. Clin. Oncol. 2018, 36, 1685–1694. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Bensch, F.; Van Der Veen, E.L.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Boellaard, R.; Kok, I.C.; Oosting, S.F.; Schroder, C.P.; Hiltermann, T.J.N.; Van Der Wekken, A.J.; et al. (89)Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med. 2018, 24, 1852–1858. [Google Scholar] [CrossRef]
- Lee, H.W.; Lee, H.M.; Choi, S.E.; Yoo, H.; Ahn, S.G.; Lee, M.K.; Jeong, J.; Jung, W.H. The Prognostic Impact of Early Change in 18F-FDG PET SUV After Neoadjuvant Chemotherapy in Patients with Locally Advanced Breast Cancer. J. Nucl. Med. 2016, 57, 1183–1188. [Google Scholar] [CrossRef][Green Version]
- Silwal-Pandit, L.; Langerod, A.; Borresen-Dale, A.L. TP53 Mutations in Breast and Ovarian Cancer. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef]
- Schon, K.; Tischkowitz, M. Clinical implications of germline mutations in breast cancer: TP53. Breast Cancer Res. Treat. 2018, 167, 417–423. [Google Scholar] [CrossRef]
- Anderson, G.R.; Wardell, S.E.; Cakir, M.; Crawford, L.; Leeds, J.C.; Nussbaum, D.P.; Shankar, P.S.; Soderquist, R.S.; Stein, E.M.; Tingley, J.P.; et al. PIK3CA mutations enable targeting of a breast tumor dependency through mTOR-mediated MCL-1 translation. Sci. Transl. Med. 2016, 8, 369ra175. [Google Scholar] [CrossRef]
- Van Keymeulen, A.; Lee, M.Y.; Ousset, M.; Brohee, S.; Rorive, S.; Giraddi, R.R.; Wuidart, A.; Bouvencourt, G.; Dubois, C.; Salmon, I.; et al. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 2015, 525, 119–123. [Google Scholar] [CrossRef]
- Pandit, B.; Halasi, M.; Gartel, A.L. p53 negatively regulates expression of FoxM1. Cell Cycle 2009, 8, 3425–3427. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, N.; Jia, Z.; Le, X.; Dai, B.; Wei, D.; Huang, S.; Tan, D.; Xie, K. Critical role and regulation of transcription factor FoxM1 in human gastric cancer angiogenesis and progression. Cancer Res. 2009, 69, 3501–3509. [Google Scholar] [CrossRef]
- Qian, J.; Luo, Y.; Gu, X.; Zhan, W.; Wang, X. Twist1 promotes gastric cancer cell proliferation through up-regulation of FoxM1. PLoS ONE 2013, 8, e77625. [Google Scholar] [CrossRef]
- Cui, J.; Shi, M.; Xie, D.; Wei, D.; Jia, Z.; Zheng, S.; Gao, Y.; Huang, S.; Xie, K. FOXM1 promotes the warburg effect and pancreatic cancer progression via transactivation of LDHA expression. Clin. Cancer Res. 2014, 20, 2595–2606. [Google Scholar] [CrossRef]
- Jiang, W.; Zhou, F.; Li, N.; Li, Q.; Wang, L. FOXM1-LDHA signaling promoted gastric cancer glycolytic phenotype and progression. Int. J. Clin. Exp. Pathol. 2015, 8, 6756–6763. [Google Scholar]
- Roh, Y.G.; Mun, M.H.; Jeong, M.S.; Kim, W.T.; Lee, S.R.; Chung, J.W.; Kim, S.I.; Kim, T.N.; Nam, J.K.; Leem, S.H. Drug resistance of bladder cancer cells through activation of ABCG2 by FOXM1. BMB Rep. 2018, 51, 98–103. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | YSU Cohort | YSU-TNBC Cohort | YSU-FFPE Cohort | UNC Cohort | TCGA Cohort | METABRIC Cohort |
|---|---|---|---|---|---|---|
| Patients (n) | 301 | 84 | 104 | 500 | 817 | 1904 |
| Age (years) | ||||||
| Median | 47 | 49 | 48 | 55 | 59 | 62 |
| Range | 22–78 | 34–77 | 28–72 | 24–93 | 26–90 | 22–91 |
| Histology (%) | ||||||
| Infiltrating Ductal Carcinoma | 260 (86.38) | 74 (88.1) | 84 (80.8) | 599 (73.3) | 1454 (76.37) | |
| Infiltrating Lobular Carcinoma | 7 (2.33) | 1 (1.2) | 12 (11.5) | 143 (17.5) | 142 (7.46) | |
| Mucinous Carcinoma | 12 (3.99) | 1 (1.2) | 2 (1.9) | 14 (1.7) | 22 (1.16) | |
| Medullary Carcinoma | 5 (1.66) | 2 (2.4) | 0 (0) | 5 (0.6) | 25 (1.31) | |
| Metaplastic Carcinoma | 2 (0.66) | 1 (1.2) | 0 (0) | 3 (0.4) | 1 (0.05) | |
| Others | 15 (4.98) | 4 (4.8) | 6 (5.8) | 54 (6.6) | 245 (12.87) | |
| NA | 1 (1.2) | 500 (100) | 15 (0.79) | |||
| T classification | ||||||
| T1 | 108 (35.88) | 35 (41.7) | 77 (74.0) | 219 (26.8) | 475 (24.95) | |
| T2 | 166 (55.15) | 46 (54.8) | 26 (25.0) | 459 (56.2) | 800 (42.02) | |
| T3 | 22 (7.31) | 3 (3.6) | 1 (1.0) | 105 (12.9) | 115 (6.04) | |
| T4 | 0 (0) | 0 (0) | 0 (0) | 34 (4.2) | 9 (0.47) | |
| NA | 5 (1.66) | 500 (100) | 505 (26.52) | |||
| N classification | ||||||
| N0 | 161 (53.49) | 58 (69.0) | 88 (84.6) | 182 (36.4) | 382 (46.8) | 993 (52.15) |
| N1 | 83 (27.57) | 21 (25.0) | 15 (14.4) | 189 (37.8) | 278 (34) | 604 (31.72) |
| N2 | 35 (11.63) | 4 (4.8) | 26 (5.2) | 85 (10.4) | 204 (10.71) | |
| N3 | 22 (7.31) | 1 (1.2) | 0 (0) | 58 (7.1) | 103 (5.41) | |
| NA | 1 (1.0) | 103 (20.6) | 14 (1.7) | |||
| AJCC stage | ||||||
| I | 77 (25.58) | 29 (34.5) | 70 (67.3) | 140 (17.1) | 470 (24.68) | |
| II | 157 (52.16) | 50 (59.5) | 33 (31.7) | 461 (56.4) | 681 (35.77) | |
| III | 62 (20.6) | 5 (6.0) | 1 (1.0) | 184 (22.5) | 280 (14.71) | |
| IV | 0 (0) | 0 (0) | 0 (0) | 13 (1.6) | 0 (0) | |
| NA | 5 (1.66) | 500 (100) | 19 (2.3) | 473 (24.84) | ||
| ER | ||||||
| Positive | 188 (62.46) | 102 (98.0) | 243 (48.6) | 593 (72.6) | 1459 (76.63) | |
| Negative | 113 (37.54) | 84 (100) | 1 (1.0) | 243 (48.6) | 174 (21.3) | 445 (23.37) |
| NA | 1 (1.0) | 14 (2.8) | 50 (6.1) | |||
| PR | ||||||
| Positive | 184 (61.13) | 85 (81.7) | 522 (63.9) | 1009 (52.99) | ||
| Negative | 117 (38.87) | 84 (100) | 18 (17.3) | 251 (30.7) | 895 (47.01) | |
| NA | 1 (1.0) | 500 (100) | 44 (5.4) | |||
| HER2 | ||||||
| Positive | 44 (14.62) | 1 (1.0) | 78 (15.6) | 121 (14.8) | 236 (12.39) | |
| Negative | 216 (71.76) | 84 (100) | 101 (97.1) | 369 (73.8) | 417 (51) | 1668 (87.61) |
| NA | 41 (13.62) | 2 (1.9) | 53 (10.6) | 279 (34.1) | ||
| SUV data availability | ||||||
| Yes | 66 (21.9) | 84 (100) | 104 (100) | |||
| No | 235 (78.1) | 500 (100) | 817 (100) | 1904 (100) | ||
| Adjuvant chemotherapy | ||||||
| Yes | 270 (89.7) | 83 (98.8) | 27 (26.0) | 40 (4.9) | 396 (20.8) | |
| No | 18 (5.98) | 1 (1.2) | 76 (73.0) | 12 (1.5) | 1508 (79.2) | |
| NA | 13 (4.32) | 1 (1.0) | 500 (100) | 765 (93.6) | ||
| Recurrence, n | 47 | 16 | 1 | 117 | 85 | 623 |
| Death, n | 35 | 5 | 1 | 91 | 120 | 1103 |
| Median follow-up (month) | 93.5 | 67.5 | 45.5 | 30.5 | 28.9 | 115.6 |
| Variable | Univariate | Multivariate | |||||
|---|---|---|---|---|---|---|---|
| n | HR (95% CI) | p-Value | n | HR (95% CI) | p-Value | ||
| Combined with YSU and UNC cohorts | Age (>35 years or not) | 681 | 0.61 (0.38–0.98) | 0.04 | 681 | 0.74 (0.45–1.25) | 0.26 |
| Node status (Positive or Negative) | 681 | 1.02 (0.65–1.6) | 0.93 | 0.94 (0.58–1.5) | 0.79 | ||
| ER status (Positive or Negative) | 681 | 0.55 (0.4–0.75) | 1.84 × 10−4 | 0.66 (0.47–0.94) | 0.02 | ||
| HER2 status (Positive or Negative) | 681 | 1.36 (0.95–1.94) | 0.09 | 1.43 (0.99–2.08) | 0.06 | ||
| SUV-signature (SHC vs. SLC *) | 681 | 1.78 (1.31–2.42) | 2.1 × 10−4 | 1.48 (1.05–2.09) | 0.02 | ||
| METABRIC cohort | Age (>35 years or not) | 1904 | 0.62 (0.4–0.97) | 0.04 | 1904 | 1.26 (0.81–1.95) | 0.31 |
| Node status (Positive or Negative) | 1904 | 2.26 (1.92–2.66) | 7.07 × 10−23 | 1.68 (1.46–1.86) | 2.88 × 10−16 | ||
| ER status (Positive or Negative) | 1904 | 0.58 (0.49–0.69) | 7.32 × 10−10 | 1.04 (0.89–1.21) | 0.66 | ||
| HER2 status (Positive or Negative) | 1904 | 2.05 (1.67–2.51) | 3.57 × 10−12 | 1.37 (1.14–1.64) | 8.27 × 10−4 | ||
| SUV-signature (SHC vs. SLC *) | 1904 | 1.74 (1.47–2.05) | 1.21 × 10−10 | 1.27 (1.12–1.45) | 2.23 × 10−4 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-K.; Ahn, S.G.; Mun, J.-Y.; Jeong, M.-S.; Bae, S.J.; Lee, J.-S.; Jeong, J.; Leem, S.-H.; Chu, I.-S. Genomic Signature of the Standardized Uptake Value in 18F-Fluorodeoxyglucose Positron Emission Tomography in Breast Cancer. Cancers 2020, 12, 497. https://doi.org/10.3390/cancers12020497
Kim S-K, Ahn SG, Mun J-Y, Jeong M-S, Bae SJ, Lee J-S, Jeong J, Leem S-H, Chu I-S. Genomic Signature of the Standardized Uptake Value in 18F-Fluorodeoxyglucose Positron Emission Tomography in Breast Cancer. Cancers. 2020; 12(2):497. https://doi.org/10.3390/cancers12020497
Chicago/Turabian StyleKim, Seon-Kyu, Sung Gwe Ahn, Jeong-Yeon Mun, Mi-So Jeong, Soong June Bae, Ju-Seog Lee, Joon Jeong, Sun-Hee Leem, and In-Sun Chu. 2020. "Genomic Signature of the Standardized Uptake Value in 18F-Fluorodeoxyglucose Positron Emission Tomography in Breast Cancer" Cancers 12, no. 2: 497. https://doi.org/10.3390/cancers12020497
APA StyleKim, S.-K., Ahn, S. G., Mun, J.-Y., Jeong, M.-S., Bae, S. J., Lee, J.-S., Jeong, J., Leem, S.-H., & Chu, I.-S. (2020). Genomic Signature of the Standardized Uptake Value in 18F-Fluorodeoxyglucose Positron Emission Tomography in Breast Cancer. Cancers, 12(2), 497. https://doi.org/10.3390/cancers12020497