Mechanisms of the Antitumor Activity of Low Molecular Weight Heparins in Pancreatic Adenocarcinomas


 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
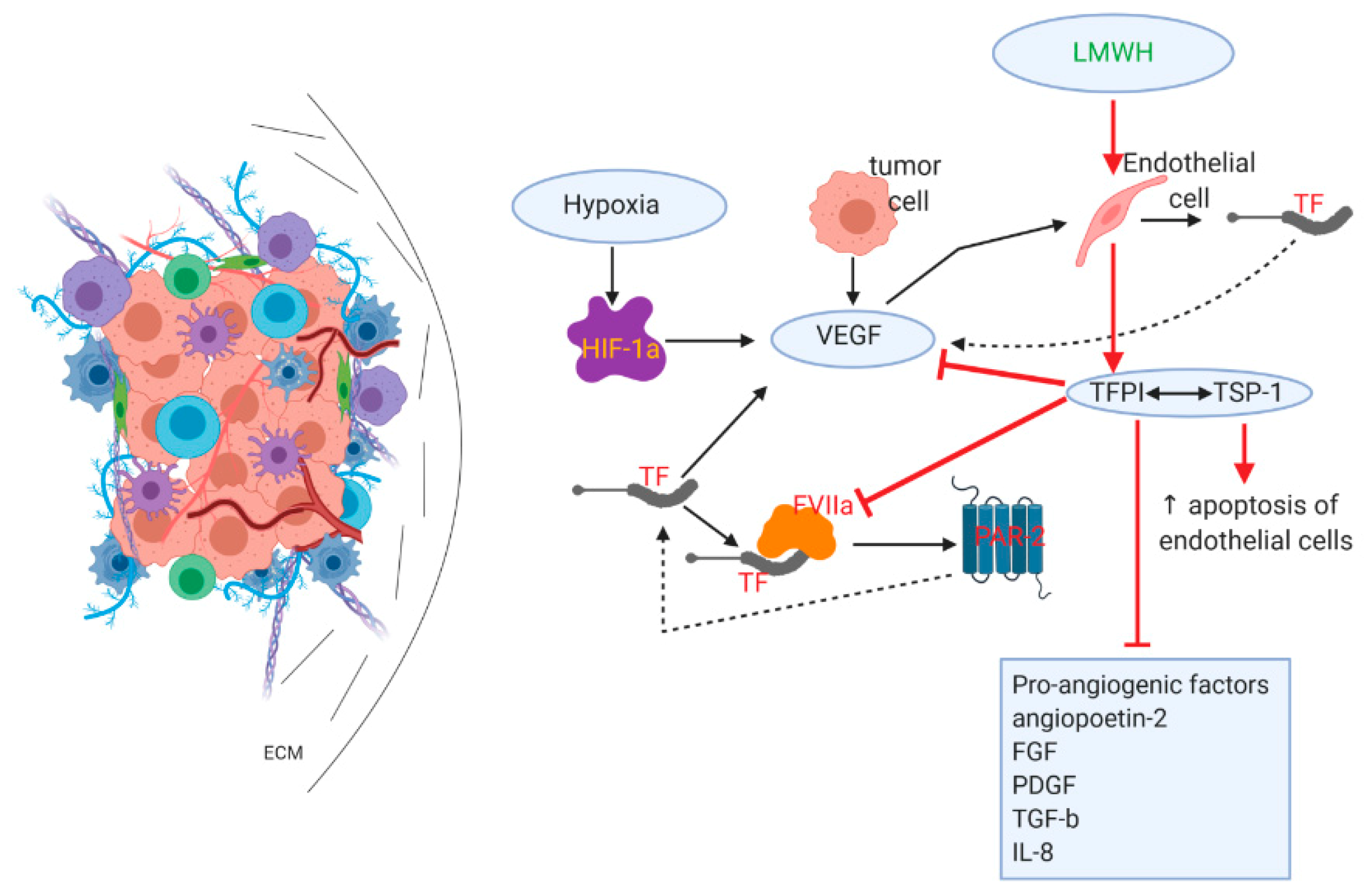{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Pancreatic Ductal Adenocarcinoma Microenvironment
3. The Role of LMWHs in Pancreatic Cancer
3.1. Effect on Heparan Sulfate Proteoglycans/Heparanase
3.2. Effect on Metastasis Formation
3.3. Effects on Angiogenesis/Tumor Vasculature
3.4. Effects on Immune-Suppressive/Therapy-Resistant TME
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Oiseth, S.J.; Aziz, M.S. Cancer immunotherapy: A brief review of the history, possibilities, and challenges ahead. J. Cancer Metastasis Treat. 2017. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Hilmi, M.; Bartholin, L.; Neuzillet, C. Immune therapies in pancreatic ductal adenocarcinoma: Where are we now? World J. Gastroenterol. 2018, 24, 2137–2151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wolfgang, C.L.; Zheng, L. Precision Immuno-Oncology: Prospects of Individualized Immunotherapy for Pancreatic Cancer. Cancers 2018, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A. Cancer and coagulation. Am. J. Hematol. 2012, 87 (Suppl. 1), S82–S87. [Google Scholar] [CrossRef] [PubMed]
- Farge, D.; Frere, C.; Connors, J.M.; Ay, C.; Khorana, A.A.; Munoz, A.; Brenner, B.; Kakkar, A.; Rafii, H.; Solymoss, S.; et al. International Initiative on Thrombosis and Cancer (ITAC) advisory panel 2019 international clinical practice guidelines for the treatment and prophylaxis of venous thromboembolism in patients with cancer. Lancet Oncol. 2019, 20, e566–e581. [Google Scholar] [CrossRef]
- Maraveyas, A.; Waters, J.; Roy, R.; Fyfe, D.; Propper, D.; Lofts, F.; Sgouros, J.; Gardiner, E.; Wedgwood, K.; Ettelaie, C.; et al. Gemcitabine versus gemcitabine plus dalteparin thromboprophylaxis in pancreatic cancer. Eur. J. Cancer 2012, 48, 1283–1292. [Google Scholar] [CrossRef]
- Pelzer, U.; Opitz, B.; Deutschinoff, G.; Stauch, M.; Reitzig, P.C.; Hahnfeld, S.; Müller, L.; Grunewald, M.; Stieler, J.M.; Sinn, M.; et al. Efficacy of prophylactic low-molecular weight heparin for ambulatory patients with advanced pancreatic cancer: Outcomes from the CONKO-004 trial. J. Clin. Oncol. 2015, 33, 2028–2034. [Google Scholar] [CrossRef]
- Thomas, D.P.; Hull, R.D.; Raskob, G.E.; Pineo, G.F. Low-Molecular-Weight Heparin. N. Engl. J. Med. 1992, 327, 817–818. [Google Scholar]
- Johnston, A.; Hsieh, S.C.; Carrier, M.; Kelly, S.E.; Bai, Z.; Skidmore, B.; Wells, G.A. A systematic review of clinical practice guidelines on the use of low molecular weight heparin and fondaparinux for the treatment and prevention of venous thromboembolism: Implications for research and policy decision-making. PLoS ONE 2018, 13, e0207410. [Google Scholar] [CrossRef] [PubMed]
- Mousa, S.A.; Petersen, L.J. Anti-cancer properties of low-molecular-weight heparin: Preclinical evidence. Thromb. Haemost. 2009, 102, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Dimakakos, E.P.; Vathiotis, I.; Syrigos, K. The Role of Tinzaparin in Oncology. Clin. Appl. Thromb. 2018, 24, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.; Pfankuchen, D.B.; Wantoch von Rekowski, K.; Schlesinger, M.; Reipsch, F.; Bendas, G. The Impact of the Low Molecular Weight Heparin Tinzaparin on the Sensitization of Cisplatin-Resistant Ovarian Cancers-Preclinical In Vivo Evaluation in Xenograft Tumor Models. Molecules 2017, 22, 728. [Google Scholar] [CrossRef]
- Liu, Z.-L.; Wang, Q.; Wang, M.; Wang, B.; Huang, L.-N. Low molecular weight heparin in treating patients with lung cancer received chemotherapy: A meta-analysis. J. Cancer Res. Ther. 2018, 14, S437–S443. [Google Scholar] [CrossRef]
- Noble, S. PL-24 low-molecular-weight heparin and survival in lung cancer. Thromb. Res 2012, 129. [Google Scholar] [CrossRef]
- Seeholzer, N.; Thürlimann, B.; Köberle, D.; Hess, D.; Korte, W. Combining chemotherapy and low-molecular-weight heparin for the treatment of advanced breast cancer: Results on clinical response, transforming growth factor-beta 1 and fibrin monomer in a phase II study. Blood Coagul. Fibrinolysis 2007, 18, 415–423. [Google Scholar] [CrossRef]
- Rucki, A.A.; Foley, K.; Zhang, P.; Xiao, Q.; Kleponis, J.; Wu, A.A.; Sharma, R.; Mo, G.; Liu, A.; Van Eyk, J.; et al. Heterogeneous Stromal Signaling within the Tumor Microenvironment Controls the Metastasis of Pancreatic Cancer. Cancer Res. 2017, 77, 41–52. [Google Scholar] [CrossRef]
- Uzunparmak, B.; Sahin, I.H. Pancreatic cancer microenvironment: A current dilemma. Clin. Transl. Med. 2019, 8, 2. [Google Scholar] [CrossRef]
- Erkan, M.; Hausmann, S.; Michalski, C.W.; Fingerle, A.A.; Dobritz, M.; Kleeff, J.; Friess, H. The role of stroma in pancreatic cancer: Diagnostic and therapeutic implications. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 454–467. [Google Scholar] [CrossRef]
- Pu, N.; Zhao, G.; Lv, Y.; Wu, W. The Advancement of Tumor Microenvironment in Pancreatic Carcinoma. Cancer Cell Microenviron. 2016. [Google Scholar] [CrossRef]
- Oliver, A.J.; Lau, P.K.H.; Unsworth, A.S.; Loi, S.; Darcy, P.K.; Kershaw, M.H.; Slaney, C.Y. Tissue-dependent tumor microenvironments and their impact on immunotherapy responses. Front. Immunol. 2018, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef]
- Koustas, E.; Sarantis, P.; Kyriakopoulou, G.; Papavassiliou, A.G.; Karamouzis, M.V. The interplay of autophagy and tumor microenvironment in colorectal cancer—Ways of enhancing immunotherapy action. Cancers 2019, 11, 533. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed]
- Son, B.; Lee, S.; Youn, H.; Kim, E.; Kim, W.; Youn, B. The role of tumor microenvironment in therapeutic resistance. Oncotarget 2017, 8, 3933–3945. [Google Scholar] [CrossRef] [PubMed]
- Dougan, S.K. The Pancreatic Cancer Microenvironment. Cancer J. 2017, 23, 321–325. [Google Scholar] [CrossRef]
- Wang, H.; Franco, F.; Ho, P.C. Metabolic Regulation of Tregs in Cancer: Opportunities for Immunotherapy. Trends Cancer 2017, 3, 583–592. [Google Scholar] [CrossRef]
- Vennin, C.; Murphy, K.J.; Morton, J.P.; Cox, T.R.; Pajic, M.; Timpson, P. Reshaping the Tumor Stroma for Treatment of Pancreatic Cancer. Gastroenterology 2018, 154, 820–838. [Google Scholar] [CrossRef]
- Evan, G.I.; Hah, N.; Littlewood, T.D.; Sodir, N.M.; Campos, T.; Downes, M.; Evans, R.M. Re-engineering the Pancreas Tumor Microenvironment: A “Regenerative Program” Hacked. Clin. Cancer Res. 2017, 23, 1647–1655. [Google Scholar] [CrossRef]
- Klimstra, D.S.; Longnecker, D.S.; Hruban, R.H.; DiGiuseppe, J.A.; Offerhaus, G.J.A. K-ras mutations in pancreatic ductal proliferative lesions. Am. J. Pathol. 1994, 145, 1547–1550. [Google Scholar] [PubMed]
- Rozenblum, E.; Schutte, M.; Goggins, M.; Hahn, S.A.; Panzer, S.; Zahurak, M.; Goodman, S.N.; Sohn, T.A.; Hruban, R.H.; Yeo, C.J.; et al. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res. 1997, 57, 1731–1734. [Google Scholar] [PubMed]
- Xie, D.; Xie, K. Pancreatic cancer stromal biology and therapy. Genes Dis. 2015, 2, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Gunturu, K.S.; Rossi, G.R.; Wasif, M.S. Immunotherapy updates in pancreatic cancer: Are we there yet? Ther. Adv. Med. Oncol. 2013, 5, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Yarchoan, M.; Lee, V.; Laheru, D.A.; Jaffee, E.M. Strategies for increasing pancreatic tumor immunogenicity. Clin. Cancer Res. 2017, 23, 1656–1669. [Google Scholar] [CrossRef]
- Bobek, V.; Kovarík, J. Antitumor and antimetastatic effect of warfarin and heparins. Biomed. Pharmacother. 2004, 58, 213–219. [Google Scholar] [CrossRef]
- Borsig, L. Heparin as an inhibitor of cancer progression. In Progress in Molecular Biology and Translational Science; Academic Press: Cambridge, MA, USA, 2010; Volume 93, pp. 335–349. [Google Scholar]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef]
- Arvatz, G.; Weissmann, M.; Ilan, N.; Vlodavsky, I. Heparanase and cancer progression: New directions, new promises. Hum. Vaccin. Immunother. 2016, 12, 2253–2256. [Google Scholar] [CrossRef]
- Koliopanos, A.; Friess, H.; Kleeff, J.; Shi, X.; Liao, Q.; Pecker, I.; Vlodavsky, I.; Zimmermann, A.; Büchler, M.W. Heparanase expression in primary and metastatic pancreatic cancer. Cancer Res. 2001, 61, 4655–4659. [Google Scholar]
- Casu, B.; Vlodavsky, I.; Sanderson, R.D. Non-anticoagulant heparins and inhibition of cancer. Pathophysiol. Haemost. Thromb. 2008, 36, 195–203. [Google Scholar] [CrossRef]
- Nadir, Y.; Brenner, B. Novel strategies of coagulation inhibition for reducing tumor growth and angiogenesis. Thromb. Res. 2018, 164, S153–S156. [Google Scholar] [CrossRef] [PubMed]
- Page, C. Heparin and related drugs: Beyond anticoagulant activity. ISRN Pharmacol. 2013, 2013, 910743. [Google Scholar] [CrossRef] [PubMed]
- Bendas, G.; Borsig, L. Cancer cell adhesion and metastasis: Selectins, integrins, and the inhibitory potential of heparins. Int. J. Cell Biol. 2012, 2012, 676731. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Jin, H.; Aiyer, A.; Su, J.; Borgstrom, P.; Stupack, D.; Friedlander, M.; Varner, J. A homing mechanism for bone marrow-derived progenitor cell recruitment to the neovasculature. J. Clin. Investig. 2006, 116, 652–662. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, C.; Mo, X.; Shi, H.; Li, S. Mechanisms by which CXCR4/CXCL12 cause metastatic behavior in pancreatic cancer. Oncol. Lett. 2018, 15, 1771–1776. [Google Scholar] [CrossRef]
- Cui, K.; Zhao, W.; Wang, C.; Wang, A.; Zhang, B.; Zhou, W.; Yu, J.; Sun, Z.; Li, S. The CXCR4-CXCL12 pathway facilitates the progression of pancreatic cancer via induction of angiogenesis and lymphangiogenesis. J. Surg. Res. 2011, 171, 143–150. [Google Scholar] [CrossRef]
- Borsig, L.; Wong, R.; Hynes, R.O.; Varki, N.M.; Varki, A. Synergistic effects of L- and P-selectin in facilitating tumor metastasis can involve non-mucin ligands and implicate leukocytes as enhancers of metastasis. Proc. Natl. Acad. Sci. USA 2002, 99, 2193–2198. [Google Scholar] [CrossRef]
- Schlesinger, M.; Roblek, M.; Ortmann, K.; Naggi, A.; Torri, G.; Borsig, L.; Bendas, G. The role of VLA-4 binding for experimental melanoma metastasis and its inhibition by heparin. Thromb. Res. 2014, 133, 855–862. [Google Scholar] [CrossRef]
- Harvey, J.R.; Mellor, P.; Eldaly, H.; Lennard, T.W.J.; Kirby, J.A.; Ali, S. Inhibition of CXCR4-mediated breast cancer metastasis: A potential role for heparinoids? Clin. Cancer Res. 2007, 13, 1562–1570. [Google Scholar] [CrossRef]
- Chen, I.X.; Chauhan, V.P.; Posada, J.; Ng, M.R.; Wu, M.W.; Adstamongkonkul, P.; Huang, P.; Lindeman, N.; Langer, R.; Jain, R.K. Blocking CXCR4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 4558–4566. [Google Scholar] [CrossRef]
- Manji, G.A.; Olive, K.P.; Saenger, Y.M.; Oberstein, P. Current and Emerging Therapies in Metastatic Pancreatic Cancer. Clin. Cancer Res. 2017, 23, 1670–1678. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- Abdollahi, A.; Folkman, J. Evading tumor evasion: Current concepts and perspectives of anti-angiogenic cancer therapy. Drug Resist. Updates 2010, 13, 16–28. [Google Scholar] [CrossRef]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 83. [Google Scholar] [CrossRef]
- Jun, J.C.; Rathore, A.; Younas, H.; Gilkes, D.; Polotsky, V.Y. Hypoxia-Inducible Factors and Cancer. Curr. Sleep Med. Rep. 2017, 3, 1–10. [Google Scholar] [CrossRef]
- Mizukami, Y.; Kohgo, Y.; Chung, D.C. Hypoxia inducible factor-1-independent pathways in tumor angiogenesis. Clin. Cancer Res. 2007, 13, 5670–5674. [Google Scholar] [CrossRef]
- Khorana, A.A.; Ahrendt, S.A.; Ryan, C.K.; Francis, C.W.; Hruban, R.H.; Hu, Y.C.; Hostetter, G.; Harvey, J.; Taubman, M.B. Tissue factor expression, angiogenesis, and thrombosis in pancreatic cancer. Clin. Cancer Res. 2007, 13, 2870–2875. [Google Scholar] [CrossRef]
- Haas, S.L.; Jesnowski, R.; Steiner, M.; Hummel, F.; Ringel, J.; Burstein, C.; Nizze, H.; Liebe, S.; Löhr, J.M. Expression of tissue factor in pancreatic adenocarcinoma is associated with activation of coagulation. World J. Gastroenterol. 2006, 12, 4843–4849. [Google Scholar] [CrossRef]
- Dahlbäck, B. Blood coagulation. Lancet 2000, 355, 1627–1632. [Google Scholar] [CrossRef]
- Bluff, J.E.; Brown, N.J.; Reed, M.W.R.; Staton, C.A. Tissue factor, angiogenesis and tumour progression. Breast Cancer Res. 2008, 10, 204. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Yasuda, M.; Yamamoto, T. Angiogenesis induced by tissue factor in vitro and in vivo. Thromb. Res. 1999, 96, 183–189. [Google Scholar] [CrossRef]
- Zucker, S.; Mirza, H.; Conner, C.E.; Lorenz, A.F.; Drews, M.H.; Bahou, W.F.; Jesty, J. Vascular endothelial growth factor induces tissue factor and matrix metalloproteinase production in endothelial cells: Conversion of prothrombin to thrombin results in progelatinase A activation and cell proliferation. Int. J. Cancer 1998, 75, 780–786. [Google Scholar] [CrossRef]
- Belting, M.; Dorrell, M.I.; Sandgren, S.; Aguilar, E.; Ahamed, J.; Dorfleutner, A.; Carmeliet, P.; Mueller, B.M.; Friedlander, M.; Ruf, W. Regulation of angiogenesis by tissue factor cytoplasmic domain signaling. Nat. Med. 2004, 10, 502–509. [Google Scholar] [CrossRef]
- Ahamed, J.; Ruf, W. Protease-activated receptor 2-dependent phosphorylation of the tissue factor cytoplasmic domain. J. Biol. Chem. 2004, 279, 23038–23044. [Google Scholar] [CrossRef]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Mousa, S.A.; Bozarth, J.; Barrett, J.S. Pharmacodynamic properties of the low molecular weight heparin, tinzaparin: Effect of molecular weight distribution on plasma tissue factor pathway inhibitor in healthy human subjects. J. Clin. Pharmacol. 2003, 43, 727–734. [Google Scholar] [CrossRef]
- Sandset, P.M.; Abildgaard, U.; Larsen, M.L. Heparin induces release of extrinsic: Coagulation pathway inhibitor (EPI). Thromb. Res. 1988, 50, 803–813. [Google Scholar] [CrossRef]
- Mousa, S.A.; Mohamed, S. Inhibition of endothelial cell tube formation by the low molecular weight heparin, tinzaparin, is mediated by tissue factor pathway inhibitor. Thromb. Haemost. 2004, 92, 627–633. [Google Scholar] [CrossRef]
- Mousa, S.A.; Mohamed, S. Anti-angiogenic mechanisms and efficacy of the low molecular weight heparin, tinzaparin: Anti-cancer efficacy. Oncol. Rep. 2004, 12, 683–688. [Google Scholar] [CrossRef]
- Mast, A.E.; Stadanlick, J.E.; Lockett, J.M.; Dietzen, D.J.; Hasty, K.A.; Hall, C.L. Tissue factor pathway inhibitor binds to platelet thrombospondin-1. J. Biol. Chem. 2000, 275, 31715–31721. [Google Scholar] [CrossRef]
- Good, D.J.; Polverini, P.J.; Rastinejad, F.; Le Beau, M.M.; Lemons, R.S.; Frazier, W.A.; Bouck, N.P. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc. Natl. Acad. Sci. USA 1990, 87, 6624–6628. [Google Scholar] [CrossRef]
- Huong, P.T.; Nguyen, L.T.; Nguyen, X.B.; Lee, S.K.; Bach, D.H. The role of platelets in the tumor-microenvironment and the drug resistance of cancer cells. Cancers 2019, 11, 240. [Google Scholar] [CrossRef]
- Kazerounian, S.; Yee, K.O.; Lawler, J. Thrombospondins: From structure to therapeutics—Thrombospondins in cancer. Cell. Mol. Life Sci. 2008, 65, 700–712. [Google Scholar] [CrossRef]
- Huang, T.; Sun, L.; Yuan, X.; Qiu, H. Thrombospondin-1 is a multifaceted player in tumor progression. Oncotarget 2017, 8, 84546–84558. [Google Scholar] [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.-I.; Cheng, P.; Cho, H.-I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Nagaraj, S.; Gabrilovich, D.I. Tumor-Associated CD8 + T Cell Tolerance Induced by Bone Marrow-Derived Immature Myeloid Cells. J. Immunol. 2005, 175, 4583–4592. [Google Scholar] [CrossRef]
- Jain, R.K. Antiangiogenesis Strategies Revisited: From Starving Tumors to Alleviating Hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef]
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-Associated Fibroblasts Build and Secure the Tumor Microenvironment. Front. Cell Dev. Biol. 2019, 7, 60. [Google Scholar] [CrossRef]
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. 2010, 15, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Franco, O.E.; Shaw, A.K.; Strand, D.W.; Hayward, S.W. Cancer associated fibroblasts in cancer pathogenesis. Semin. Cell Dev. Biol. 2010, 21, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fan, X.; Houghton, J. Tumor microenvironment: The role of the tumor stroma in cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef]
- Bükki, J. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 2139–2141. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bokas, A.; Papakotoulas, P.; Sarantis, P.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Mechanisms of the Antitumor Activity of Low Molecular Weight Heparins in Pancreatic Adenocarcinomas. Cancers 2020, 12, 432. https://doi.org/10.3390/cancers12020432
Bokas A, Papakotoulas P, Sarantis P, Papadimitropoulou A, Papavassiliou AG, Karamouzis MV. Mechanisms of the Antitumor Activity of Low Molecular Weight Heparins in Pancreatic Adenocarcinomas. Cancers. 2020; 12(2):432. https://doi.org/10.3390/cancers12020432
Chicago/Turabian StyleBokas, Alexandros, Pavlos Papakotoulas, Panagiotis Sarantis, Adriana Papadimitropoulou, Athanasios G Papavassiliou, and Michalis V Karamouzis. 2020. "Mechanisms of the Antitumor Activity of Low Molecular Weight Heparins in Pancreatic Adenocarcinomas" Cancers 12, no. 2: 432. https://doi.org/10.3390/cancers12020432
APA StyleBokas, A., Papakotoulas, P., Sarantis, P., Papadimitropoulou, A., Papavassiliou, A. G., & Karamouzis, M. V. (2020). Mechanisms of the Antitumor Activity of Low Molecular Weight Heparins in Pancreatic Adenocarcinomas. Cancers, 12(2), 432. https://doi.org/10.3390/cancers12020432