Comprehensive Functional Characterization and Clinical Interpretation of 20 Splice-Site Variants of the RAD51C Gene

 ,
,  , , ,
, , ,  , , and
, , and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Bioinformatics Analysis
2.2. Functional Analysis
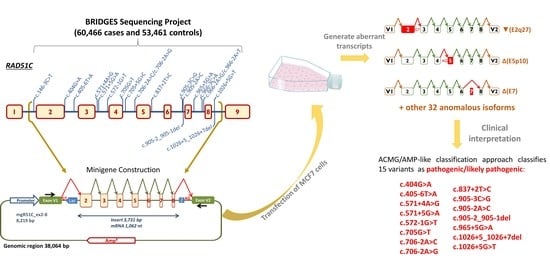
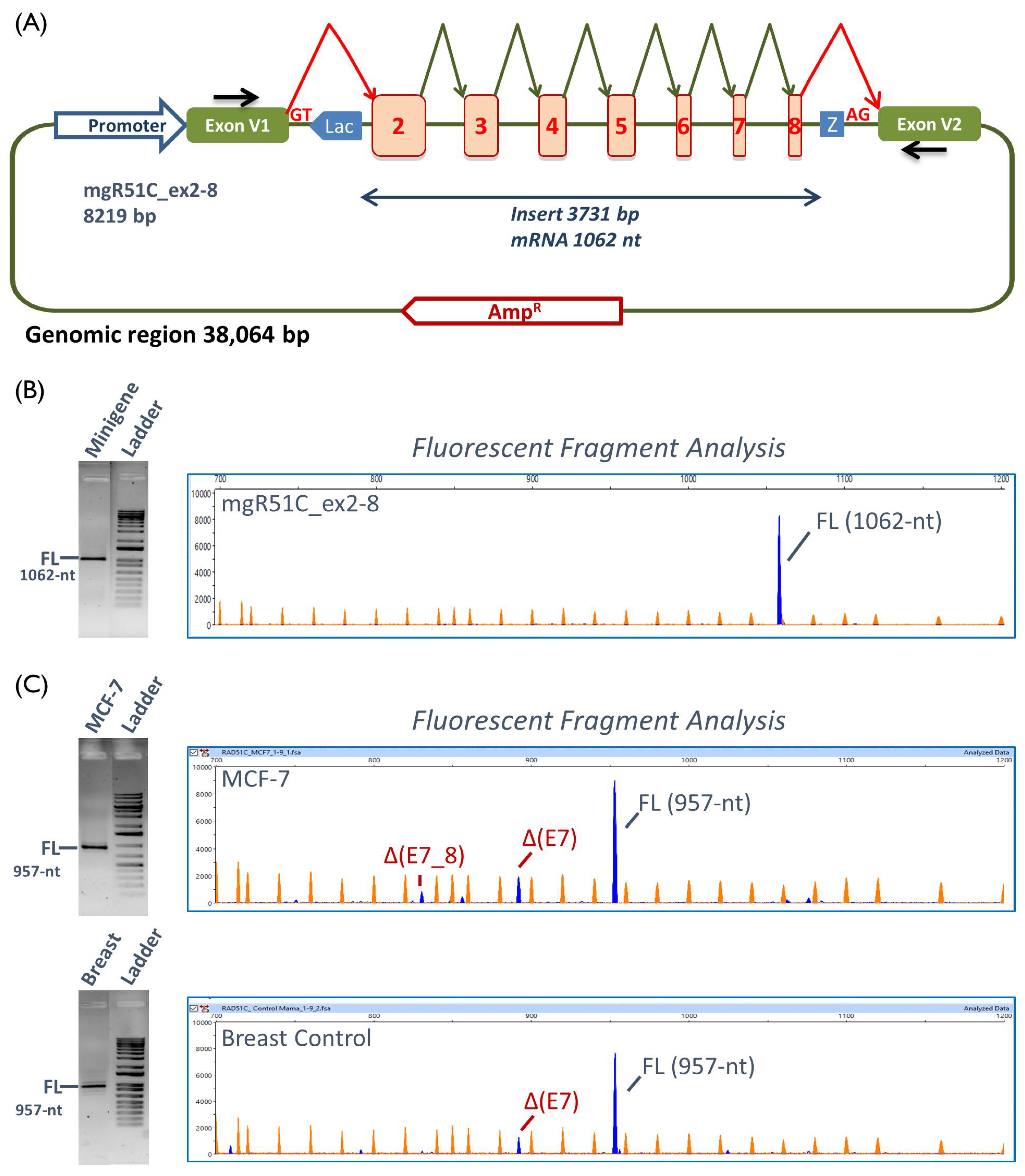
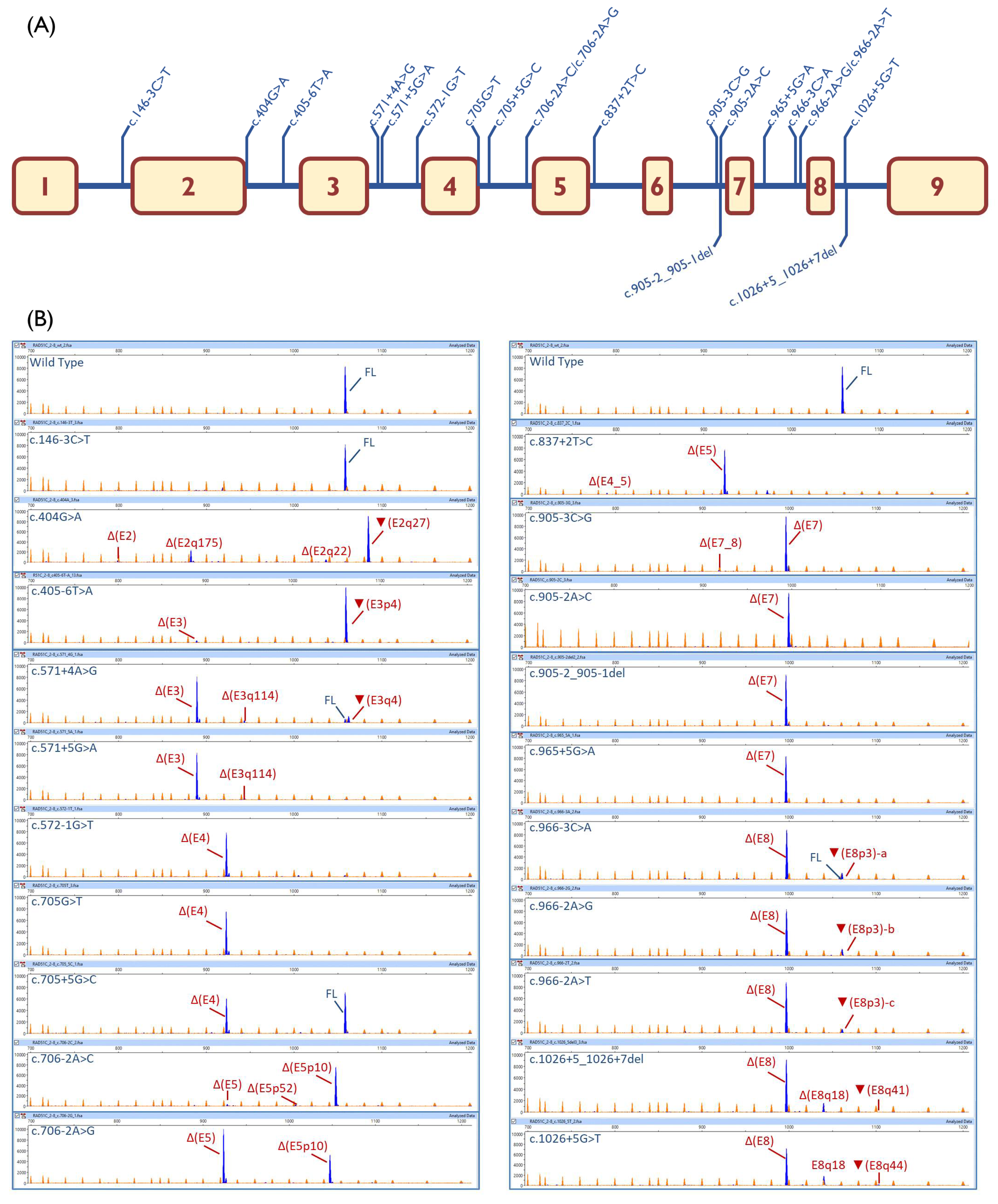
2.3. Transcript Analysis
2.4. ACMG/AMP-Like Classification of RAD51C Variants Based on PS3/BS3 Functional Evidence
3. Discussion
Clinical Interpretation of Variants
4. Materials and Methods
4.1. Ethics Approval
4.2. Variant and Transcript Annotations
4.3. Bioinformatics Analysis
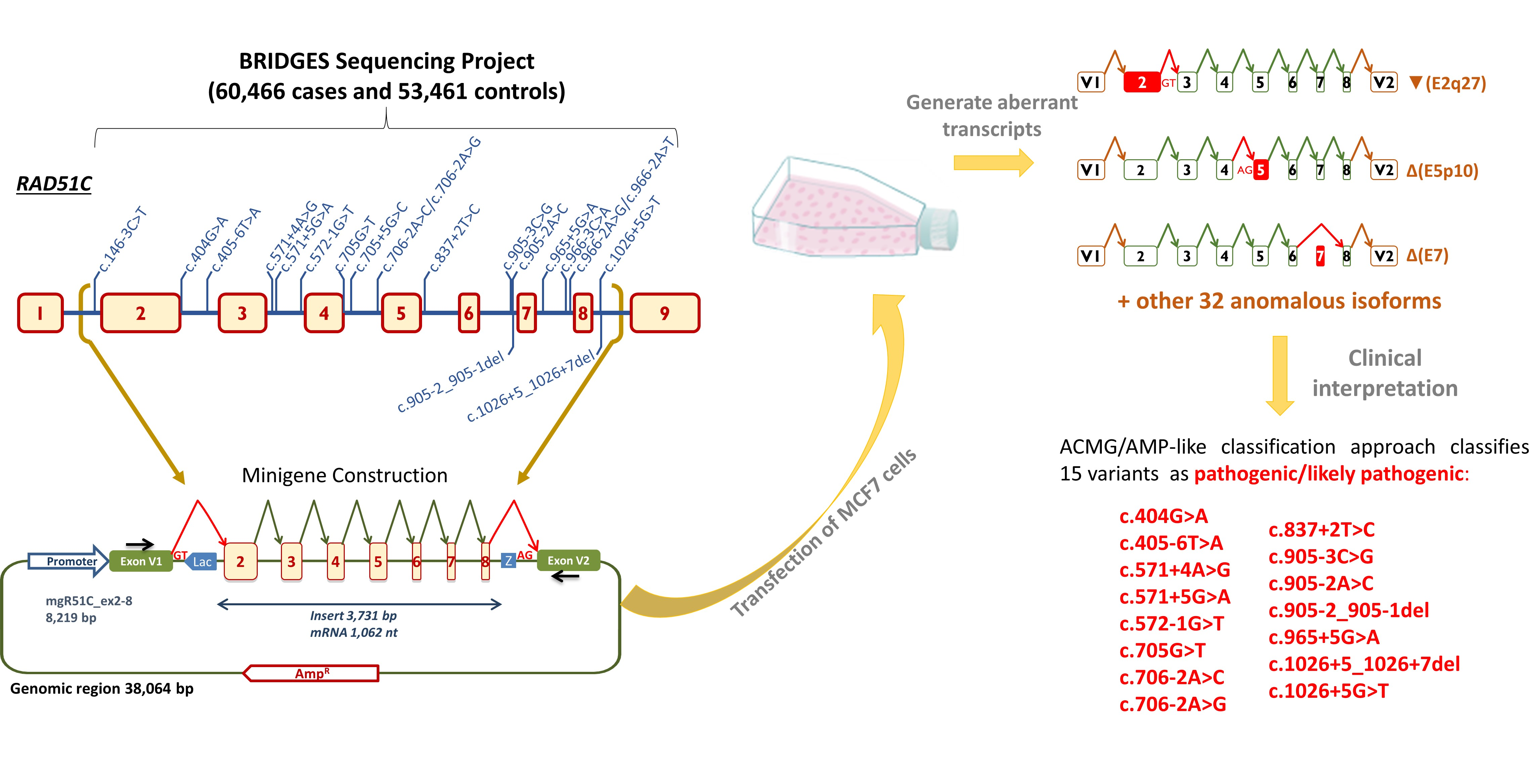
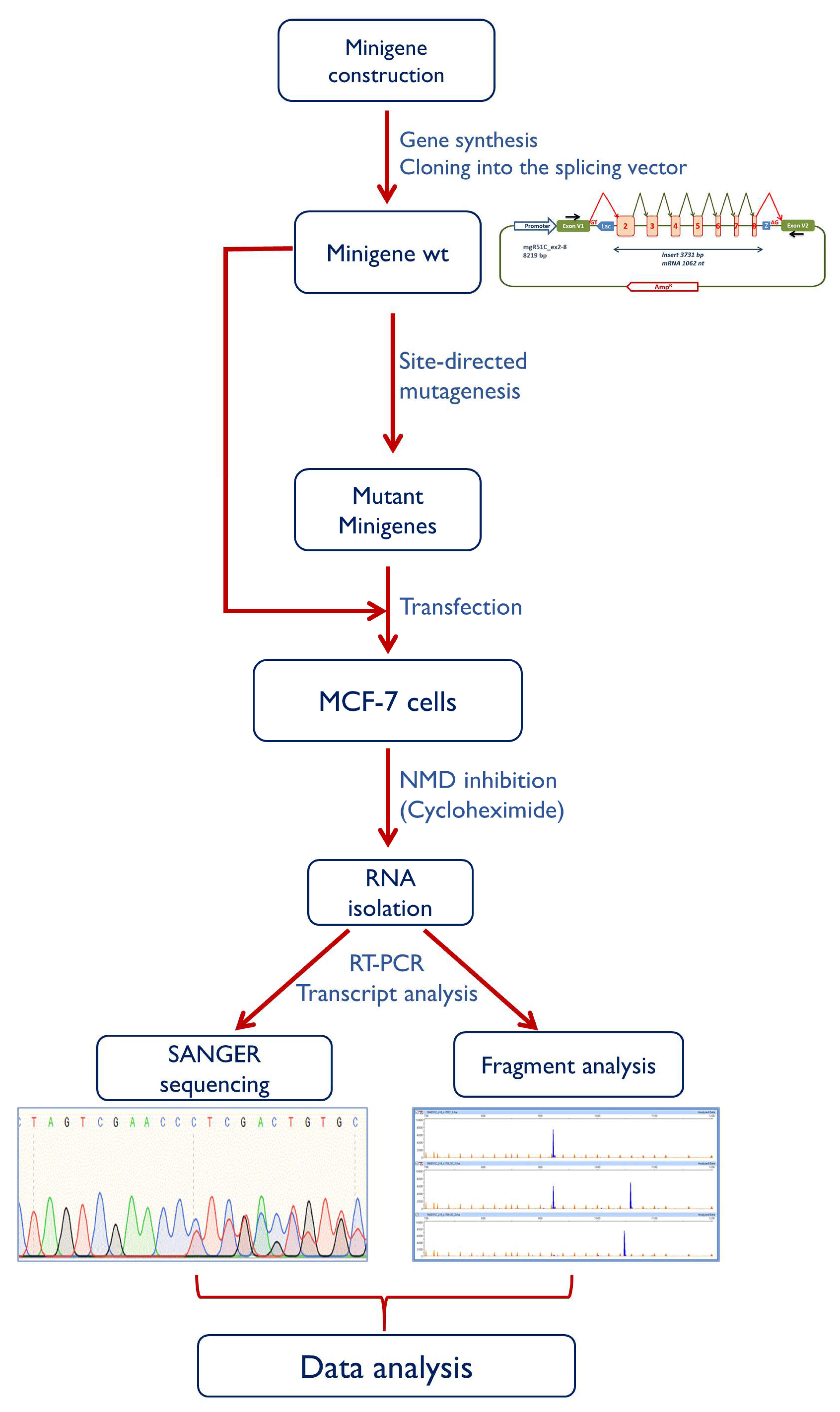
4.4. Minigene Construction and Mutagenesis
4.5. Transfection of Eukaryotic Cells
4.6. Reverse Transcription Polymerase Chain Reaction and Fragment Analysis
4.7. ACMG/AMP-Like Classification of 20 RAD51C Variants Based on PS3/BS3 Functional Evidence
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nielsen, F.C.; van Overeem Hansen, T.; Sørensen, C.S. Hereditary breast and ovarian cancer: New genes in confined pathways. Nat. Rev. Cancer 2016, 16, 599–612. [Google Scholar] [CrossRef]
- Schubert, S.; Luttikhuizen, J.L.; Auber, B.; Schmidt, G.; Hofmann, W.; Penkert, J.; Davenport, C.F.; Hille-Betz, U.; Wendeburg, L.; Bublitz, J.; et al. The identification of pathogenic variants in BRCA1/2 negative, high risk, hereditary breast and/or ovarian cancer patients: High frequency of FANCM pathogenic variants. Int. J. Cancer 2019, 144, 2683–2694. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef]
- Buys, S.S.; Sandbach, J.F.; Gammon, A.; Patel, G.; Kidd, J.; Brown, K.L.; Sharma, L.; Saam, J.; Lancaster, J.; Daly, M.B. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 2017, 123, 1721–1730. [Google Scholar] [CrossRef]
- Castéra, L.; Harter, V.; Muller, E.; Krieger, S.; Goardon, N.; Ricou, A.; Rousselin, A.; Paimparay, G.; Legros, A.; Bruet, O.; et al. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet. Med. 2018, 20, 1677–1686. [Google Scholar] [CrossRef]
- Taylor, M.R.G.; Špírek, M.; Chaurasiya, K.R.; Ward, J.D.; Carzaniga, R.; Yu, X.; Egelman, E.H.; Collinson, L.M.; Rueda, D.; Krejci, L.; et al. Rad51 Paralogs Remodel Pre-synaptic Rad51 Filaments to Stimulate Homologous Recombination. Cell 2015, 162, 271–286. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Bernstein, K.A. RAD-ical New Insights into RAD51 Regulation. Genes 2018, 9, 629. [Google Scholar] [CrossRef] [PubMed]
- Le Calvez-Kelm, F.; Oliver, J.; Damiola, F.; Forey, N.; Robinot, N.; Durand, G.; Voegele, C.; Vallée, M.P.; Byrnes, G.; Breast Cancer Family Registry; et al. RAD51 and Breast Cancer Susceptibility: No Evidence for Rare Variant Association in the Breast Cancer Family Registry Study. PLoS ONE 2012, 7, e52374. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Song, H.; Leslie, G.; Engel, C.; Hahnen, E.; Auber, B.; Horváth, J.; Kast, K.; Niederacher, D.; Turnbull, C.; et al. Ovarian and breast cancer risks associated with pathogenic variants in RAD51C and RAD51D. J. Natl. Cancer Inst. 2020, 37, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lhotova, K.; Stolarova, L.; Zemankova, P.; Vocka, M.; Janatova, M.; Borecka, M.; Cerna, M.; Jelinkova, S.; Kral, J.; Volkova, Z.; et al. Multigene panel germline testing of 1333 Czech patients with ovarian cancer. Cancers 2020, 12, 956. [Google Scholar] [CrossRef] [PubMed]
- Vaz, F.; Hanenberg, H.; Schuster, B.; Barker, K.; Wiek, C.; Erven, V.; Neveling, K.; Endt, D.; Kesterton, I.; Autore, F.; et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat. Genet. 2010, 42, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, H.; Sarai, N.; Kagawa, W.; Enomoto, R.; Shibata, T.; Kurumizaka, H.; Yokoyama, S. Preferential binding to branched DNA strands and strand-annealing activity of the human Rad51B, Rad51C, Rad51D and Xrcc2 protein complex. Nucleic Acids Res. 2004, 32, 2556–2565. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Masson, J.Y.; Shah, R.; O’Regan, P.; West, S.C. RAD51C Is Required for Holliday Junction Processing in Mammalian Cells. Science 2004, 303, 243–246. [Google Scholar] [CrossRef]
- Somyajit, K.; Subramanya, S.; Nagaraju, G. RAD51C: A novel cancer susceptibility gene is linked to Fanconi anemia and breast cancer. Carcinogenesis 2010, 31, 2031–2038. [Google Scholar] [CrossRef]
- Suwaki, N.; Klare, K.; Tarsounas, M. RAD51 paralogs: Roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin. Cell Dev. Biol. 2011, 22, 898–905. [Google Scholar] [CrossRef]
- Park, J.Y.; Zhang, F.; Andreassen, P.R. PALB2: The hub of a network of tumor suppressors involved in DNA damage responses. Biochim. Biophys. Acta Rev. Cancer 2014, 1846, 263–275. [Google Scholar] [CrossRef]
- Badie, S.; Liao, C.; Thanasoula, M.; Barber, P.; Hill, M.A.; Tarsounas, M. RAD51C facilitates checkpoint signaling by promoting CHK2 phosphorylation. J. Cell Biol. 2009, 185, 587–600. [Google Scholar] [CrossRef]
- Zannini, L.; Delia, D.; Buscemi, G. CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 2014, 6, 442–457. [Google Scholar] [CrossRef]
- Goldgar, D.E.; Easton, D.F.; Deffenbaugh, A.M.; Monteiro, A.N.A.; Tavtigian, S.V.; Couch, F.J.; Information, C.; Bic, C. Integrated evaluation of DNA sequence variants of unknown clinical significance: Application to BRCA1 and BRCA2. Am. J. Hum. Genet. 2004, 75, 535–544. [Google Scholar] [CrossRef]
- Sanz, D.J.; Acedo, A.; Infante, M.; Durán, M.; Pérez-Cabornero, L.; Esteban-Cardeñosa, E.; Lastra, E.; Pagani, F.; Miner, C.; Velasco, E.A. A high proportion of DNA variants of BRCA1 and BRCA2 is associated with aberrant splicing in breast/ovarian cancer patients. Clin. Cancer Res. 2010, 16, 1957–1967. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, D.J.; Agarwal, M.K.; Pankratz, V.S.; Deffenbaugh, A.M.; Pruss, D.; Frye, C.; Wadum, L.; Johnson, K.; Mentlick, J.; Tavtigian, S.V.; et al. Functional Assays for Classification of BRCA2 Variants of Uncertain Significance. Cancer Res. 2008, 68, 3523–3531. [Google Scholar] [CrossRef]
- Fraile-Bethencourt, E.; Valenzuela-Palomo, A.; Díez-Gómez, B.; Infante, M.; Durán, M.; Marcos, G.; Lastra, E.; Gómez-Barrero, S.; Velasco, E.A. Genetic dissection of the BRCA2 promoter and transcriptional impact of DNA variants. Breast Cancer Res. Treat. 2018, 171, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Brewster, B.L.; Rossiello, F.; French, J.D.; Edwards, S.L.; Wong, M.; Wronski, A.; Whiley, P.; Waddell, N.; Chen, X.; Bove, B.; et al. Identification of fifteen novel germline variants in the BRCA1 3’UTR reveals a variant in a breast cancer case that introduces a functional miR-103 target site. Hum. Mutat. 2012, 33, 1665–1675. [Google Scholar] [CrossRef]
- Diederichs, S.; Bartsch, L.; Berkmann, J.C.; Fröse, K.; Heitmann, J.; Hoppe, C.; Iggena, D.; Jazmati, D.; Karschnia, P.; Linsenmeier, M.; et al. The dark matter of the cancer genome: Aberrations in regulatory elements, untranslated regions, splice sites, non-coding RNA and synonymous mutations. EMBO Mol. Med. 2016, 8, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Burke, L.J.; Sevcik, J.; Gambino, G.; Tudini, E.; Mucaki, E.J.; Shirley, B.C.; Whiley, P.; Parsons, M.T.; De Leeneer, K.; Gutiérrez-Enríquez, S.; et al. BRCA1 and BRCA2 5′ noncoding region variants identified in breast cancer patients alter promoter activity and protein binding. Hum. Mutat. 2018, 39, 2025–2039. [Google Scholar] [CrossRef] [PubMed]
- Gelli, E.; Colombo, M.; Pinto, A.; De Vecchi, G.; Foglia, C.; Amitrano, S.; Morbidoni, V.; Imperatore, V.; Manoukian, S.; Baldassarri, M.; et al. Usefulness and Limitations of Comprehensive Characterization of mRNA Splicing Profiles in the Definition of the Clinical Relevance of BRCA1/2 Variants of Uncertain Significance. Cancers 2019, 11, 295. [Google Scholar] [CrossRef]
- Yoshida, K.; Ogawa, S. Splicing factor mutations and cancer. Wiley Interdiscip. Rev. RNA 2014, 5, 445–459. [Google Scholar] [CrossRef]
- Canson, D.; Glubb, D.; Spurdle, A.B. Variant effect on splicing regulatory elements, branchpoint usage, and pseudoexonization: Strategies to enhance bioinformatic prediction using hereditary cancer genes as exemplars. Hum. Mutat. 2020, 41, 1705–1721. [Google Scholar] [CrossRef]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef]
- Fraile-Bethencourt, E.; Díez-Gómez, B.; Velásquez-Zapata, V.; Acedo, A.; Sanz, D.J.; Velasco, E.A. Functional classification of DNA variants by hybrid minigenes: Identification of 30 spliceogenic variants of BRCA2 exons 17 and 18. PLoS Genet. 2017, 13, e1006691. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E.A.; Stewart, C.; Abdel-Wahab, O. Altered RNA processing in cancer pathogenesis and therapy. Cancer Discov. 2019, 9, 1493–1510. [Google Scholar] [CrossRef] [PubMed]
- Baralle, D.; Buratti, E. RNA splicing in human disease and in the clinic. Clin. Sci. 2017, 131, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Matlin, A.J.; Clark, F.; Smith, C.W.J. Understanding alternative splicing: Towards a cellular code. Nat. Rev. Mol. Cell Biol. 2005, 6, 386–398. [Google Scholar] [CrossRef]
- Moles-Fernández, A.; Duran-Lozano, L.; Montalban, G.; Bonache, S.; López-Perolio, I.; Menéndez, M.; Santamariña, M.; Behar, R.; Blanco, A.; Carrasco, E.; et al. Computational Tools for Splicing Defect Prediction in Breast/Ovarian Cancer Genes: How Efficient Are They at Predicting RNA Alterations? Front. Genet. 2018, 9, 366. [Google Scholar] [CrossRef]
- Tosi, M.; Stamm, S.; Baralle, D. RNA splicing meets genetic testing: Detection and interpretation of splicing defects in genetic diseases. Eur. J. Hum. Genet. 2010, 18, 737–738. [Google Scholar] [CrossRef]
- Whiley, P.J.; De La Hoya, M.; Thomassen, M.; Becker, A.; Brandão, R.; Pedersen, I.S.; Montagna, M.; Menéndez, M.; Quiles, F.; Gutiérrez-Enríquez, S.; et al. Comparison of mRNA splicing assay protocols across multiple laboratories: Recommendations for best practice in standardized clinical testing. Clin. Chem. 2014, 60, 341–352. [Google Scholar] [CrossRef]
- Baralle, D.; Lucassen, A.; Buratti, E. Missed threads. The impact of pre-mRNA splicing defects on clinical practice. EMBO Rep. 2009, 10, 810–816. [Google Scholar] [CrossRef]
- Gaildrat, P.; Killian, A.; Martins, A.; Tournier, I.; Frébourg, T.; Tosi, M. Use of splicing reporter minigene assay to evaluate the effect on splicing of unclassified genetic variants. Methods Mol. Biol. 2010, 653, 249–257. [Google Scholar]
- Cooper, T.A. Use of minigene systems to dissect alternative splicing elements. Methods 2005, 37, 331–340. [Google Scholar] [CrossRef]
- Acedo, A.; Hernández-Moro, C.; Curiel-García, Á.; Díez-Gómez, B.; Velasco, E.A. Functional classification of BRCA2 DNA variants by splicing assays in a large minigene with 9 exons. Hum. Mutat. 2015, 36, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Fraile-Bethencourt, E.; Valenzuela-Palomo, A.; Díez-Gómez, B.; Acedo, A.; Velasco, E.A. Identification of Eight Spliceogenic Variants in BRCA2 Exon 16 by Minigene Assays. Front. Genet. 2018, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Baralle, D.; Baralle, M. Splicing in action: Assessing disease causing sequence changes. J. Med. Genet. 2005, 42, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Luccarini, C.; Wahlström, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; Wang, Q.; et al. Breast cancer risk genes: Association analysis in more than 113,000 women. N. Engl. J. Med. 2020, in press. [Google Scholar]
- Miller, K.A.; Sawicka, D.; Barsky, D.; Albala, J.S. Domain mapping of the Rad51 paralog protein complexes. Nucleic Acids Res. 2004, 32, 169–178. [Google Scholar] [CrossRef]
- Jacquinet, A.; Brown, L.; Sawkins, J.; Liu, P.; Pugash, D.; Van Allen, M.I.; Patel, M.S. Expanding the FANCO/RAD51C associated phenotype: Cleft lip and palate and lobar holoprosencephaly, two rare findings in Fanconi anemia. Eur. J. Med. Genet. 2018, 61, 257–261. [Google Scholar] [CrossRef]
- Suszynska, M.; Ratajska, M.; Kozlowski, P. BRIP1, RAD51C, and RAD51D mutations are associated with high susceptibility to ovarian cancer: Mutation prevalence and precise risk estimates based on a pooled analysis of ~30,000 cases. J. Ovarian Res. 2020, 13, 50. [Google Scholar] [CrossRef]
- van Marcke, C.; Collard, A.; Vikkula, M.; Duhoux, F.P. Prevalence of pathogenic variants and variants of unknown significance in patients at high risk of breast cancer: A systematic review and meta-analysis of gene-panel data. Crit. Rev. Oncol. Hematol. 2018, 132, 138–144. [Google Scholar] [CrossRef]
- Radice, P.; De Summa, S.; Caleca, L.; Tommasi, S. Unclassified variants in BRCA genes: Guidelines for interpretation. Ann. Oncol. 2011, 22 (Suppl 1), i18–i23. [Google Scholar] [CrossRef]
- Eccles, D.M.; Mitchell, G.; Monteiro, A.N.A.; Schmutzler, R.; Couch, F.J.; Spurdle, A.B.; Gómez-García, E.B.; ENIGMA Clinical Working Group. BRCA1 and BRCA2 genetic testing—pitfalls and recommendations for managing variants of uncertain clinical significance. Ann. Oncol. 2015, 26, 2057–2065. [Google Scholar] [CrossRef]
- Meindl, A.; Hellebrand, H.; Wiek, C.; Erven, V.; Wappenschmidt, B.; Niederacher, D.; Freund, M.; Lichtner, P.; Hartmann, L.; Schaal, H.; et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010, 42, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Dicks, E.; Ramus, S.J.; Tyrer, J.P.; Intermaggio, M.P.; Hayward, J.; Edlund, C.K.; Conti, D.; Harrington, P.; Fraser, L.; et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J. Clin. Oncol. 2015, 33, 2901–2907. [Google Scholar] [CrossRef] [PubMed]
- Acedo, A.; Sanz, D.J.; Durán, M.; Infante, M.; Pérez-Cabornero, L.; Miner, C.; Velasco, E.A. Comprehensive splicing functional analysis of DNA variants of the BRCA2 gene by hybrid minigenes. Breast Cancer Res. 2012, 14, R87. [Google Scholar] [CrossRef] [PubMed]
- Brandão, R.D.; Mensaert, K.; López-Perolio, I.; Tserpelis, D.; Xenakis, M.; Lattimore, V.; Walker, L.C.; Kvist, A.; Vega, A.; Gutiérrez-Enríquez, S.; et al. Targeted RNA-seq successfully identifies normal and pathogenic splicing events in breast/ovarian cancer susceptibility and Lynch syndrome genes. Int. J. Cancer 2019, 145, 401–414. [Google Scholar] [CrossRef]
- Fackenthal, J.D.; Yoshimatsu, T.; Zhang, B.; de Garibay, G.R.; Colombo, M.; De Vecchi, G.; Ayoub, S.C.; Lal, K.; Olopade, O.I.; Vega, A.; et al. Naturally occurring BRCA2 alternative mRNA splicing events in clinically relevant samples. J. Med. Genet. 2016, 53, 548–558. [Google Scholar] [CrossRef]
- Lopez-Perolio, I.; Leman, R.; Behar, R.; Lattimore, V.; Pearson, J.F.; Castéra, L.; Martins, A.; Vaur, D.; Goardon, N.; Davy, G.; et al. Alternative splicing and ACMG-AMP-2015-based classification of PALB2 genetic variants: An ENIGMA report. J. Med. Genet. 2019, 56, 453–460. [Google Scholar] [CrossRef]
- Fraile-Bethencourt, E.; Valenzuela-Palomo, A.; Díez-Gómez, B.; Goina, E.; Acedo, A.; Buratti, E.; Velasco, E.A. Mis-splicing in breast cancer: Identification of pathogenic BRCA2 variants by systematic minigene assays. J. Pathol. 2019, 248, 409–420. [Google Scholar] [CrossRef]
- Shirts, B.H.; Casadei, S.; Jacobson, A.L.; Lee, M.K.; Gulsuner, S.; Bennett, R.L.; Miller, M.; Hall, S.A.; Hampel, H.; Hisama, F.M.; et al. Improving performance of multigene panels for genomic analysis of cancer predisposition. Genet. Med. 2016, 18, 974–981. [Google Scholar] [CrossRef]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.S.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef]
- Lhota, F.; Zemankova, P.; Kleiblova, P.; Soukupova, J.; Vocka, M.; Stranecky, V.; Janatova, M.; Hartmannova, H.; Hodanova, K.; Kmoch, S.; et al. Hereditary truncating mutations of DNA repair and other genes in BRCA1/BRCA2/PALB2 -negatively tested breast cancer patients. Clin. Genet. 2016, 90, 324–333. [Google Scholar] [CrossRef]
- Golmard, L.; Caux-Moncoutier, V.; Davy, G.; Al Ageeli, E.; Poirot, B.; Tirapo, C.; Michaux, D.; Barbaroux, C.; D’Enghien, C.D.; Nicolas, A.; et al. Germline mutation in the RAD51B gene confers predisposition to breast cancer. BMC Cancer 2013, 13, 484. [Google Scholar] [CrossRef] [PubMed]
- Coulet, F.; Fajac, A.; Colas, C.; Eyries, M.; Dion-Minière, A.; Rouzier, R.; Uzan, S.; Lefranc, J.-P.; Carbonnel, M.; Cornelis, F.; et al. Germline RAD51C mutations in ovarian cancer susceptibility. Clin. Genet. 2013, 83, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Pelttari, L.M.; Heikkinen, T.; Thompson, D.; Kallioniemi, A.; Schleutker, J.; Holli, K.; Blomqvist, C.; Aittomäki, K.; Bützow, R.; Nevanlinna, H. RAD51C is a susceptibility gene for ovarian cancer. Hum. Mol. Genet. 2011, 20, 3278–3288. [Google Scholar] [CrossRef] [PubMed]
- Neidhardt, G.; Becker, A.; Hauke, J.; Horváth, J.; Bogdanova Markov, N.; Heilmann-Heimbach, S.; Hellebrand, H.; Thiele, H.; Altmüller, J.; Nürnberg, P.; et al. The RAD51C exonic splice-site mutations c.404G>C and c.404G>T are associated with familial breast and ovarian cancer. Eur. J. Cancer Prev. 2017, 26, 165–169. [Google Scholar] [CrossRef]
- Hertel, K.J. Combinatorial control of exon recognition. J. Biol. Chem. 2008, 283, 1211–1215. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Brnich, S.E.; Abou Tayoun, A.N.; Couch, F.J.; Cutting, G.R.; Greenblatt, M.S.; Heinen, C.D.; Kanavy, D.M.; Luo, X.; McNulty, S.M.; Starita, L.M.; et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2020, 12, 3. [Google Scholar] [CrossRef]
- Tavtigian, S.V.; Greenblatt, M.S.; Harrison, S.M.; Nussbaum, R.L.; Prabhu, S.A.; Boucher, K.M.; Biesecker, L.G.; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet. Med. 2018, 20, 1054–1060. [Google Scholar] [CrossRef]
- Lee, K.; Krempely, K.; Roberts, M.E.; Anderson, M.J.; Carneiro, F.; Chao, E.; Dixon, K.; Figueiredo, J.; Ghosh, R.; Huntsman, D.; et al. Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Hum. Mutat. 2018, 39, 1553–1568. [Google Scholar] [CrossRef]
- Fraile-Bethencourt, E.; Valenzuela-Palomo, A.; Díez-Gómez, B.; Caloca, M.J.; Gómez-Barrero, S.; Velasco, E.A. Minigene Splicing Assays Identify 12 Spliceogenic Variants of BRCA2 Exons 14 and 15. Front. Genet. 2019, 10, 503. [Google Scholar] [CrossRef]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Bëroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Houdayer, C.; Caux-Moncoutier, V.; Krieger, S.; Barrois, M.; Bonnet, F.; Bourdon, V.; Bronner, M.; Buisson, M.; Coulet, F.; Gaildrat, P.; et al. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum. Mutat. 2012, 33, 1228–1238. [Google Scholar] [CrossRef]
- de Garibay, G.R.; Acedo, A.; García-Casado, Z.; Gutiérrez-Enríquez, S.; Tosar, A.; Romero, A.; Garre, P.; Llort, G.; Thomassen, M.; Díez, O.; et al. Capillary electrophoresis analysis of conventional splicing assays: IARC analytical and clinical classification of 31 BRCA2 genetic variants. Hum. Mutat. 2014, 35, 53–57. [Google Scholar] [CrossRef]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M.; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef]
- Lara, B.; Martínez, M.T.; Blanco, I.; Hernández-Moro, C.; Velasco, E.A.; Ferrarotti, I.; Rodriguez-Frias, F.; Perez, L.; Vazquez, I.; Alonso, J.; et al. Severe alpha-1 antitrypsin deficiency in composite heterozygotes inheriting a new splicing mutation QOMadrid. Respir. Res. 2014, 15, 125. [Google Scholar] [CrossRef]
- Gailite, L.; Valenzuela-Palomo, A.; Sanoguera-Miralles, L.; Rots, D.; Kreile, M.; Velasco, E.A. UGT1A1 Variants c.864+5G>T and c.996+2_996+5del of a Crigler-Najjar Patient Induce Aberrant Splicing in Minigene Assays. Front. Genet. 2020, 11, 169. [Google Scholar] [CrossRef]
- Villate, O.; Ibarluzea, N.; Fraile-Bethencourt, E.; Valenzuela, A.; Velasco, E.A.; Grozeva, D.; Raymond, F.L.; Botella, M.P.; Tejada, M.-I. Functional Analyses of a Novel Splice Variant in the CHD7 Gene, Found by Next Generation Sequencing, Confirm Its Pathogenicity in a Spanish Patient and Diagnose Him with CHARGE Syndrome. Front. Genet. 2018, 9, 26–31. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant (HGVS) 1 | Bioinformatics 2 | Transcripts | |||
|---|---|---|---|---|---|
| Canonical | PTC 3 | In-Frame | Uncharacterized | ||
| Wild type | 98.6% ± 0.2% | 1106-nt (1.4% ± 0.2%) | |||
| c.146-3C > T | [↓]3′SS (9.5→8.7) | 100% | |||
| c.404G > A | [−]5′SS (4.8→−3.5) | - | ▼(E2q27): 69.3% ± 2.9% Δ(E2q175): 19.9% ± 0.6% Δ(E2q22): 4.3% ± 0.5% Δ(E2): 2.4% ± 0.2% | 913-nt (4.1% ± 3.0%) | |
| c.405-6T > A | [−]3′SS (7.7→2.2) [+] 3′SS (8.6) 4-nt upstream | - | ▼(E3p4):95.2%± 1.6% Δ(E3): 4.8% ± 1.6% | ||
| c.571 + 4A > G | [↓]5′SS (10.5→8.1) [+] 5′SS (5.5) 4-nt downstream | 5.4% ± 0.1% | Δ(E3): 76.5% ± 0.3% ▼(E3q4): 11.6% ± 0.2% | Δ(E3q114): 4.0 ± 0.0% | 808-nt (1.4% ± 0.0%) 774-nt (1.1% ± 0.0%) |
| c.571 + 5G > A | [↓] 5′SS (10.5→5.8) | - | Δ(E3): 91.5% ± 0.3% | Δ(E3q114): 4.8 ± 0.2% | 808-nt (1.6% ± 0.0%) 917-nt (1.1% ± 0.1%) 774-nt (1.0% ± 0.0%) |
| c.572-1G > T | [−]3′SS (7.4→−1.2) | - | Δ(E4): 93.4% ± 0.2% | 1005-nt (3.3% ± 0.1%) 1058-nt (3.3% ± 0.1%) | |
| c.705G > T | [−]5′SS (9.1→2.6) | - | Δ(E4): 100% | ||
| c.705 + 5G > C | [↓]5′SS (9.1→7.2) | 51.6% ± 2.4% | Δ(E4): 48.4% ± 2.4% | ||
| c.706-2A > C | [−]3′SS (11.1→3.1) [+]3′SS (3.3) 10-nt downstream | - | Δ(E5p10): 91.4% ± 1.5% Δ(E5p52): 1.8% ± 0.9% | Δ(E5): 4.0% ± 0.1% | 886-nt (2.8% ± 1.6%) |
| c.706-2A > G | [−]3′SS (11.1→3.1) [+]3′SS (3.2) 10-nt downstream | - | Δ(E5p10): 33.5% ± 0.2% | Δ(E5): 65.4% ± 0.3% | 972-nt (1.1% ± 0.1%) |
| c.837 + 2T > C | [−]5′SS (8.6→0.8) | - | Δ(E4_5): 2.2% ± 0.1% | Δ(E5): 89.3% ± 0.2% | 972-nt (8.5% ± 0.1%) |
| c.905-3C > G | [−]3′SS (8.2→−4.9) | - | Δ(E7): 98.1% ± 1.0% Δ(E7_8): 1.9% ± 1.0% | ||
| c.905-2A > C | [−]3′SS (8.2→0.1) | - | Δ(E7): 97.4% ± 0.4% | 660-nt (2.6% ± 0.4%) | |
| c.905-2_905-1del | [−]3′SS(8.2→0.6) | - | Δ(E7): 100% | ||
| c.965 + 5G > A | [↓]5′SS(8.7→3.8) | - | Δ(E7): 100% | ||
| c.966-3C > A | [−]3′SS (7.3→4.4) | 2% ± 1.7% | Δ(E8): 86.8% ± 3.2% | ▼(E8p3)-a: 9.7% ± 0.4% | 881-nt (1.5% ± 1.4%) |
| c.966-2A > G | [−]3′SS (7.3→−0.7) [+]3′SS(7) 3-nt upstream | - | Δ(E8): 86.7% ± 0.5% | ▼(E8p3)-b:11.0% ± 0.4% | 881-nt (1.2% ± 0.0%) 940-nt (1.1% ± 0.2%) |
| c.966-2A > T | [−]3′SS (7.3→−1.1) [+]3′SS(7.6) 3-nt upstream | - | Δ(E8): 89.1% ± 0.3% | ▼(E8p3)-c:5.9% ± 0.1% | 881-nt (2.8% ± 0.3%) 940-nt (2.2% ± 0.0%) |
| c.1026 + 5_1026 + 7del | [−]5′SS(2→?) (NNSplice: 0.8→ <0.1) | - | Δ(E8): 79.5% ± 1.4% ▼(E8q41): 3.3% ± 0.2% | Δ(E8q18):13.8% ± 0.7% | 881-nt (2% ± 0.6%) 778-nt (1.4% ± 1.6%) |
| c.1026 + 5G > T | [−]5′SS(2→?) (NNSplice: 0.8→ <0.1) | - | Δ(E8): 78.0% ± 0.5% ▼(E8q44): 1.4% ± 0.2% | Δ(E8q18):18.7% ± 0.5% | 881-nt (1.9% ± 0.2%) |
| c.HGVS 1 | Clinvar 2 | PVS1 3 | PP3/BP4 4 | PS3/BS3 5 | PS4 6 | PM2 7 | PM 8 | Proposed pSAD-Based ACMG/AMP-Like Variant Classification 9 |
|---|---|---|---|---|---|---|---|---|
| c.146-3C > T | Conflicting (*) LB (2), VUS, (2) | N/A | (−4%) N/A | BS3 | N/A | (4/303,851) N/A | N/A | (BS3 only) Uncertain Significance |
| c.404G > A | LP (**) | N/A | (−99.5%) PP3 | PS3_VS | N/A | (1/300,225) PM2 | N/A | (PS3_VS + PM2) Likely Pathogenic |
| c.405-6T > A | VUS (*) | N/A | (−79%) PP3 | PS3_VS | N/A | (0/304,932) PM2 | N/A | (PS3_VS + PM2) Likely Pathogenic |
| c.571 + 4A > G | Conflicting (*) LB (1), LP(1), VUS (6) | N/A | (−30.5%) PP3 | (88%VS + 4%S + 5%N/A) PS3 | N/A | (1/84,873) PM2 | N/A | (PS3 + PM2) Likely Pathogenic 10 |
| c.571 + 5G > A | VUS (**) | N/A | (−33.9%) PP3 | (95% vs. + 5%S) PS3_VS | PS4 | (8/336,321) N/A | PM3 11 | (PS3_VS + PS4 + PM3) Pathogenic |
| c.572-1G > T | not reported | PVS1 | N/A | PS3_VS | N/A | (1/304,681) PM2 | N/A | (PS3_VS + PM2) Likely Pathogenic |
| c.705G > T | VUS (**) | N/A | (−75.8%) PP3 | PS3_VS | N/A | (2/304,499) PM2 | N/A | (PS3_VS + PM2) Likely Pathogenic 10 |
| c.705 + 5G > C | not reported | N/A | (−16.8%) PP3 | (48%VS + 52% N/A) N/A | N/A | (1/304,406) PM2 | N/A | (PM2 only) Uncertain Significance |
| c.706-2A > C | LP (**) | PVS1 | N/A | (95%VS + 5% S) PS3_VS | N/A | (0/336,207) PM2 | N/A | (PS3_VS + PM2) Likely Pathogenic |
| c.706-2A > G | P/LP (**) | PVS1 | N/A | (34%VS + 65% S) PS3 | PS4 12 | (10/336,207) N/A | N/A | (PS3 + PS4) Pathogenic |
| c.837 + 2T > C | LP (**) | PVS1 | N/A | (90% S + 2% VS) PS3 | N/A | (0/304,832) PM2 | N/A | (PS3 + PM2) Likely Pathogenic |
| c.905-3C > G | not reported | N/A | (−92.8%) PP3 | PS3 | N/A | (1/336,187) PM2 | N/A | (PS3 + PM2) Likely Pathogenic |
| c.905-2A > C | P/LP (**) | PVS1 | N/A | PS3 | PS4 | (5/336,191) N/A | N/A | (PS3 + PS4) Pathogenic |
| c.905-2_905-1del | P/LP (**) | PVS1 | N/A | PS3 | PS4 | (4/304,579) N/A | N/A | (PS3 + PS4) Pathogenic |
| c.965 + 5G > A | LP(1); VUS(2) | N/A | (−59.9%) PP3 | PS3 | N/A | (2/304,579) PM2 | N/A | (PS3 + PM2) Likely Pathogenic |
| c.966-3C > A | not reported | N/A | (−35.3%) PP3 | (90%S + 10%N/A) N/A | N/A | (1/304,818) PM2 | N/A | (PM2 only) Uncertain Significance |
| c.966-2A > G | LP (*) | PVS1 | N/A | (90%S + 10%N/A) N/A | N/A | (0/304,818) PM2 | N/A | (PM2 only) Uncertain Significance |
| c.966-2A > T | not reported | PVS1 | N/A | (90%S + 10%N/A) N/A | N/A | (0/304,818) PM2 | N/A | (PM2 only) Uncertain Significance |
| c.1026 + 5_1026 + 7del | P/LP (**) | N/A | (−98.8%) PP3 | PS3 | PS4 | (6/304,853) N/A | N/A | (PS3 + PS4) Pathogenic |
| c.1026 + 5G > T | not reported | N/A | (−98.8%) PP3 | PS3 | N/A | (0/304,840) PM2 | N/A | (PS3 + PM2) Likely Pathogenic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanoguera-Miralles, L.; Valenzuela-Palomo, A.; Bueno-Martínez, E.; Llovet, P.; Díez-Gómez, B.; Caloca, M.J.; Pérez-Segura, P.; Fraile-Bethencourt, E.; Colmena, M.; Carvalho, S.; et al. Comprehensive Functional Characterization and Clinical Interpretation of 20 Splice-Site Variants of the RAD51C Gene. Cancers 2020, 12, 3771. https://doi.org/10.3390/cancers12123771
Sanoguera-Miralles L, Valenzuela-Palomo A, Bueno-Martínez E, Llovet P, Díez-Gómez B, Caloca MJ, Pérez-Segura P, Fraile-Bethencourt E, Colmena M, Carvalho S, et al. Comprehensive Functional Characterization and Clinical Interpretation of 20 Splice-Site Variants of the RAD51C Gene. Cancers. 2020; 12(12):3771. https://doi.org/10.3390/cancers12123771
Chicago/Turabian StyleSanoguera-Miralles, Lara, Alberto Valenzuela-Palomo, Elena Bueno-Martínez, Patricia Llovet, Beatriz Díez-Gómez, María José Caloca, Pedro Pérez-Segura, Eugenia Fraile-Bethencourt, Marta Colmena, Sara Carvalho, and et al. 2020. "Comprehensive Functional Characterization and Clinical Interpretation of 20 Splice-Site Variants of the RAD51C Gene" Cancers 12, no. 12: 3771. https://doi.org/10.3390/cancers12123771
APA StyleSanoguera-Miralles, L., Valenzuela-Palomo, A., Bueno-Martínez, E., Llovet, P., Díez-Gómez, B., Caloca, M. J., Pérez-Segura, P., Fraile-Bethencourt, E., Colmena, M., Carvalho, S., Allen, J., Easton, D. F., Devilee, P., Vreeswijk, M. P. G., de la Hoya, M., & Velasco, E. A. (2020). Comprehensive Functional Characterization and Clinical Interpretation of 20 Splice-Site Variants of the RAD51C Gene. Cancers, 12(12), 3771. https://doi.org/10.3390/cancers12123771