Dual Pharmacological Targeting of HDACs and PDE5 Inhibits Liver Disease Progression in a Mouse Model of Biliary Inflammation and Fibrosis

 , , ,
, , ,  , ,
, ,  ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
Simple Summary
Abstract
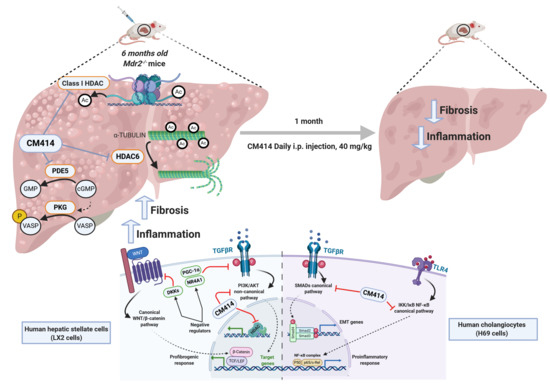
1. Introduction
2. Results
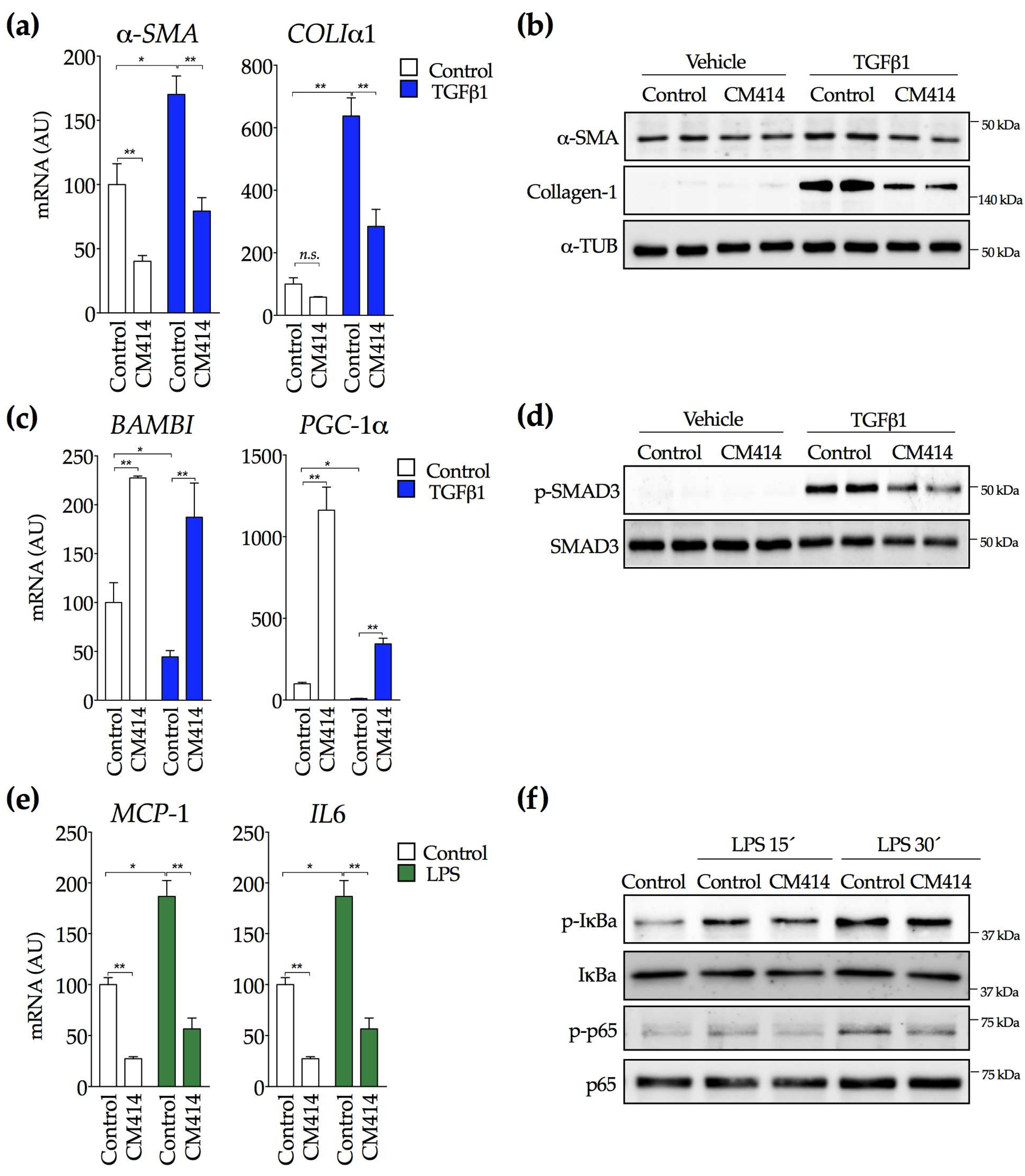
2.1. In Vivo Evaluation of the Anti-Fibrogenic Potential of CM414, a Dual HDAC-PDE5 Inhibitor
2.2. Mechanisms of CM414 Anti-Fibrogenic and Anti-Inflammatory Effects
3. Discussion
4. Materials and Methods
4.1. CM414
4.2. Animal Studies
4.3. Immunohistochemistry and Tissue Staining
4.4. Cell Culture and Treatments
4.5. Immunoblotting
4.6. RNA Isolation and Quantitative Real-Time RT-PCR
4.7. Histone Extraction
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Fw Sequence 5′-3′ | Rev Sequence 5′-3′ |
|---|---|---|
| Ck19 (mouse) | CTCGGATTGAGGAGCTGAAC | TCACGCTCTGGATCTGTGAC |
| ColIα1 (mouse) | CAGATTGAGAACATCCGCAG | GAATCCATCGGTCATGCTCTC |
| H3f3a (mouse) | AAAGCCGCTCGCAAGAGTGCG | ACTTGCCTCCTGCAAAGCAC |
| Hdac1 (mouse) | CTACTACGACGGGGATGTTGG | GAATCTCTGCATCTGCTTGCT |
| Hdac2 (mouse) | GAGGGATATTGGTGCTGGAA | AAGGGCAACTGCAGTCTCAT |
| Hdac3 (mouse) | GAGAGTCAGCCCCACCAATA | GACCCGGTCAGTGAGGTAGA |
| Hdac6 (mouse) | GCTGACTACATTGCTGCTTTCCT | ACTCCAGCATGGGGCAAG |
| Il-1β (mouse) | CATTGTGGCTGTGGAGAAGC | CCTTGTACAAAGCTCATGGAG |
| Il-10 (mouse) | CTGCACCCACTTCCCAG | AGAAATCGATGACAGCG |
| iNos (mouse) | GTGAGGATCAAAAACTGG | TTTGCCTCTTTGAAGGAGC |
| Lox (mouse) | TAGGGCGGATGTCAGAGACT | CCAGGACTCAATCCCTGTGT |
| Mcp1 (mouse) | CCACTCACCTGCTGCTACTC | TTCACATTCAAAGGTGCTGAAG |
| Mmp13 (mouse) | CCTGATGTGGGTGAATACAATG | GGTTTCATCATCATCAAAATGG |
| Pde5 (mouse) | GAAGGGGACCAGTGCTCAAG | CCACAATGCCTTTGTTCCA |
| Tenascin C (mouse) | GGATTGGTGTTTCTGCTGTCAA | TTTCAGACACCCGTAAGTCCTT |
| Tgfβ2 (mouse) | CAGATCCTGAGCAAGCTGAA | CTGAAGTAGGGTCTGTAGAAAGTGG |
| Timp1 (mouse) | AGACCACCTTATACCAGCG | AACAGGGAAACACTGTGCA |
| Tnfα (mouse) | GAGTGACAAGCCTGTAGCCC | CCCTTCTCCAGCTGGAAGAC |
| Wnt-5a (mouse) | GTCCTTTGAGATGGGTGGTATC | ACCTCTGGGTTAGGGAGTGTCT |
| α-SMA (human) | CCAGGGCTGTTTTCCCATCC | GTCATTTTCTCCCGGTTGGCC |
| BAMBI (human) | TGGATCGCCACTCCAGCTA | TGTCTTCATGACAGCATTCCA |
| c-MYC (human) | CTCAACGACAGCAGCTCGCC | CTTGAGGACCAGTGGGCTGTG |
| COLIα1 (human) | CAGATTGAGAACATCCGCAG | GAATCCATCGGTCATGCTCTC |
| DKK1 (human) | CAAAAATGTATCACACCAAAGG | TGGCTTGATGGTGATCTTTC |
| DKK2 (human) | TGCCACAGTCCCCACCAA | TACAGACTTCCCCCTGATGG |
| sGCβ1 (human) | GCAAGCATGCATCTGGAGAA | CCAGCAATTTCCATCATGTC |
| GFAP (human) | GAGATGATGGAGCTCAATGAC | TCCAGCCTCAGGTTGGTTTC |
| H3F3A (human) | AAAGCCGCTCGCAAGAGTGCG | ACTTGCCTCCTGCAAAGCAC |
| IL-6 (human) | CCTTCCAAAGATGGCTGAAA | AAAGCTGCGCAGAATGAGAT |
| MCP1 (human) | TCTGCCGCCCTTCTGTGCCTG | CTTCTTTGGGACACTTGCTGC |
| PDGFRβ (human) | GTGATGTGGGAACAGATGTCC | GAGGAAGCCCATGGTGGGATC |
| PGC-1α (human) | GCTGACAGATGGAGACGTGA | GTGTGAGGAGGGTCATCGTT |
| PI3K (human) | ATTGAACCAGTAGGCAACCG | GAAACTATTACCCAGATCACCAC |
| PKG (human) | CGCCAACCTGAAGCTGTCT | CCTTTGGAATGCAGATAGGC |
| PTEN (human) | GACTTAGACTTGACCTATATTTATCC | ACTCCCTTTTTGTCTCTGGTCC |
| TGFβ2 (human) | CAGATCCTGAGCAAGCTGAA | CTGAAGTAGGGTCTGTAGAAAGTGG |
| WNT-5A (human) | GGGTGGGAACCAAGAAAAAT | TGGAACCTACCCATCCCATA |
References
- Trautwein, C.; Friedman, S.L.; Schuppan, D.; Pinzani, M. Hepatic fibrosis: Concept to treatment. J. Hepatol. 2015, 62, S15–S24. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Byass, P. The global burden of liver disease: A challenge for methods and for public health. BMC Med. 2014, 12, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Kutala, B.K. Liver diseases: A major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int. 2018, 38 (Suppl. 1), 2–6. [Google Scholar] [CrossRef]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Pimpin, L.; Cortez-Pinto, H.; Negro, F.; Corbould, E.; Lazarus, J.V.; Webber, L.; Sheron, N. Burden of liver disease in Europe: Epidemiology and analysis of risk factors to identify prevention policies. J. Hepatol. 2018, 69, 718–735. [Google Scholar] [CrossRef]
- Sepanlou, S.G.; Safiri, S.; Bisignano, C.; Ikuta, K.S.; Merat, S.; Saberifiroozi, M.; Poustchi, H.; Tsoi, D.; Colombara, D.V.; Abdoli, A.; et al. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 245–266. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Berasain, C.; Avila, M.A. Regulation of hepatocyte identity and quiescence. Cell. Mol. Life Sci. 2015, 72, 3831–3851. [Google Scholar] [CrossRef]
- Carloni, V.; Luong, T.V.; Rombouts, K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: More complicated than ever. Liver Int. 2014, 34, 834–843. [Google Scholar] [CrossRef]
- Filliol, A.; Schwabe, R.F. Contributions of Fibroblasts, Extracellular Matrix, Stiffness, and Mechanosensing to Hepatocarcinogenesis. Semin. Liver Dis. 2019, 39, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Baglieri, J.; Brenner, D.A.; Kisseleva, T. The role of fibrosis and liver-associated fibroblasts in the pathogenesis of hepatocellular carcinoma. Int. J. Mol. Sci. 2019, 20, 1723. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of Hepatic Fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Evolving challenges in hepatic fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Yoon, Y.J.; Friedman, S.L.; Lee, Y.A. Antifibrotic Therapies: Where Are We Now? Semin. Liver Dis. 2016, 36, 83–98. [Google Scholar] [CrossRef]
- Lemoinne, S.; Friedman, S.L. New and emerging anti-fibrotic therapeutics entering or already in clinical trials in chronic liver diseases. Curr. Opin. Pharmacol. 2019, 49, 60–70. [Google Scholar] [CrossRef]
- Barcena-Varela, M.; Paish, H.; Alvarez, L.; Uriarte, I.; Latasa, M.U.; Santamaria, E.; Recalde, M.; Garate, M.; Claveria, A.; Colyn, L.; et al. Epigenetic mechanisms and metabolic reprogramming in fibrogenesis: Dual targeting of G9a and DNMT1 for the inhibition of liver fibrosis. Gut 2020. [Google Scholar] [CrossRef]
- Barcena-Varela, M.; Colyn, L.; Fernandez-Barrena, M.G. Epigenetic mechanisms in hepatic stellate cell activation during liver fibrosis and carcinogenesis. Int. J. Mol. Sci. 2019, 20, 2507. [Google Scholar] [CrossRef] [PubMed]
- Hardy, T.; Mann, D.A. Epigenetics in liver disease: From biology to therapeutics. Gut 2016, 65, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.; Mann, D. Epigenetic regulation of wound healing and fibrosis. Curr. Opin. Rheumatol. 2013, 25, 101–107. [Google Scholar] [CrossRef]
- Eberharter, A.; Becker, P.B. Histone Acetylation: A Switch between Repressive and Permissive Chromatin. Second in Review Series on Chromatin Dynamics. EMBO Rep. 2002, 3. [Google Scholar] [CrossRef] [PubMed]
- Thiagalingam, S.; Cheng, K.H.; Lee, H.J.; Mineva, N.; Thiagalingam, A.; Ponte, J.F. Histone deacetylases: Unique players in shaping the epigenetic histone code. Ann. N. Y. Acad. Sci. 2003, 983, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-J.; Huang, C.; Meng, X.-M.; Li, J. Epigenetic modifications by histone deacetylases: Biological implications and therapeutic potential in liver fibrosis. Biochimie 2015, 116, 61–69. [Google Scholar] [CrossRef]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef]
- Mannaerts, I.; Eysackers, N.; Onyema, O.O.; Van Beneden, K.; Valente, S.; Mai, A.; Odenthal, M.; van Grunsven, L.A. Class II HDAC Inhibition Hampers Hepatic Stellate Cell Activation by Induction of MicroRNA-29. PLoS ONE 2013, 8, e55786. [Google Scholar] [CrossRef]
- Shaker, M.E.; Ghani, A.; Shiha, G.E.; Ibrahim, T.M.; Mehal, W.Z. Nilotinib induces apoptosis and autophagic cell death of activated hepatic stellate cells via inhibition of histone deacetylases. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 1992–2003. [Google Scholar] [CrossRef]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868–4873. [Google Scholar] [CrossRef] [PubMed]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Peng, L.; Seto, E.; Huang, S.; Qiu, Y. Modulation of histone deacetylase 6 (HDAC6) nuclear import and tubulin deacetylase activity through acetylation. J. Biol. Chem. 2012, 287, 29168–29174. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shin, D.; Kwon, S.H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013, 280, 775–793. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, L.; Wei, Z.; Zhang, X.; Wang, Y.; Li, F.; Fu, Y.; Liu, B. Inhibition of histone deacetylase reduces lipopolysaccharide-induced-inflammation in primary mammary epithelial cells by regulating ROS-NF-кB signaling pathways. Int. Immunopharmacol. 2018, 56, 230–234. [Google Scholar] [CrossRef]
- Zhang, W.B.; Zhang, H.Y.; Jiao, F.Z.; Wang, L.W.; Zhang, H.; Gong, Z.J. Histone deacetylase 6 inhibitor ACY-1215 protects against experimental acute liver failure by regulating the TLR4-MAPK/NF-κB pathway. Biomed. Pharmacother. 2018, 97, 818–824. [Google Scholar] [CrossRef]
- Tao, H.; Yang, J.J.; Shi, K.H.; Li, J. Epigenetic factors MeCP2 and HDAC6 control α-tubulin acetylation in cardiac fibroblast proliferation and fibrosis. Inflamm. Res. 2016, 65, 415–426. [Google Scholar] [CrossRef]
- Saito, S.; Zhuang, Y.; Shan, B.; Danchuk, S.; Luo, F.; Korfei, M.; Guenther, A.; Lasky, J.A. Tubastatin ameliorates pulmonary fibrosis by targeting the TGFβ-PI3K-Akt pathway. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Shan, B.; Yao, T.P.; Nguyen, H.T.; Zhuo, Y.; Levy, D.R.; Klingsberg, R.C.; Tao, H.; Palmer, M.L.; Holder, K.N.; Lasky, J.A. Requirement of HDAC6 for transforming growth factor-β1-induced epithelial-mesenchymal transition. J. Biol. Chem. 2008, 283, 21065–21073. [Google Scholar] [CrossRef]
- Yoon, S.; Kang, G.; Eom, G.H. Hdac inhibitors: Therapeutic potential in fibrosis-associated human diseases. Int. J. Mol. Sci. 2019, 20, 1329. [Google Scholar] [CrossRef] [PubMed]
- Činčárová, L.; Zdráhal, Z.; Fajkus, J. New perspectives of valproic acid in clinical practice. Expert Opin. Investig. Drugs 2013, 22, 1535–1547. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [PubMed]
- Ikura, Y.; Iwasa, Y.; Ueda, M. Valproic acid administration for hepatic fibrosis: A balance between antifibrotic efficacy and hepatotoxicity. Hepatology 2010, 51, 2227–2228. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.E.M.; Azouz, A.A.; Bakr, A.G.; Abo-youssef, A.M.; Hemeida, R.A.M. Hepatoprotective effects of diosmin and/or sildenafil against cholestatic liver cirrhosis: The role of Keap-1/Nrf-2 and P38-MAPK/NF-κB/iNOS signaling pathway. Food Chem. Toxicol. 2018, 120, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Mansour, H.M.; Salama, A.A.A.; Abdel-Salam, R.M.; Ahmed, N.A.; Yassen, N.N.; Zaki, H.F. The anti-inflammatory and anti-fibrotic effects of tadalafil in thioacetamide-induced liver fibrosis in rats. Can. J. Physiol. Pharmacol. 2018, 96, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Andersson, K.E. Mechanisms of penile erection and basis for pharmacological treatment of erectile dysfunction. Pharmacol. Rev. 2011, 63, 811–859. [Google Scholar] [CrossRef]
- Andersson, K.E. PDE5 inhibitors—Pharmacology and clinical applications 20 years after sildenafil discovery. Br. J. Pharmacol. 2018, 175, 2554–2565. [Google Scholar] [CrossRef]
- Friebe, A.; Sandner, P.; Schmidtko, A. cGMP: A unique 2nd messenger molecule—Recent developments in cGMP research and development. Naunyn. Schmiedebergs. Arch. Pharmacol. 2020, 393, 287–302. [Google Scholar] [CrossRef]
- Shah, V.; Haddad, F.G.; Garcia-Cardena, G.; Frangos, J.A.; Mennone, A.; Groszmann, R.J.; Sessa, W.C. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J. Clin. Investig. 1997, 100, 2923–2930. [Google Scholar] [CrossRef]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Perri, R.E.; Langer, D.A.; Chatterjee, S.; Gibbons, S.J.; Gadgil, J.; Cao, S.; Farrugia, G.; Shah, V.H. Defects in cGMP-PKG pathway contribute to impaired NO-dependent responses in hepatic stellate cells upon activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.C.; Bernier, S.G.; Jacobson, S.; Liu, G.; Zhang, P.Y.; Sarno, R.; Catanzano, V.; Currie, M.G.; Masferrer, J.L. SGC stimulator praliciguat suppresses stellate cell fibrotic transformation and inhibits fibrosis and inflammation in models of NASH. Proc. Natl. Acad. Sci. USA 2019, 166, 11057–11062. [Google Scholar] [CrossRef] [PubMed]
- Beyer, C.; Zenzmaier, C.; Palumbo-Zerr, K.; Mancuso, R.; Distler, A.; Dees, C.; Zerr, P.; Huang, J.; Maier, C.; Pachowsky, M.L.; et al. Stimulation of the soluble guanylate cyclase (sGC) inhibits fibrosis by blocking non-canonical TGFβ signalling. Ann. Rheum. Dis. 2015, 74, 1408–1416. [Google Scholar] [CrossRef]
- Schinner, E.; Wetzl, V.; Schlossmann, J. Cyclic nucleotide signalling in kidney fibrosis. Int. J. Mol. Sci. 2015, 16, 2320–2351. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado-Tejedor, M.; Garcia-Barroso, C.; Sánchez-Arias, J.A.; Rabal, O.; Pérez-González, M.; Mederos, S.; Ugarte, A.; Franco, R.; Segura, V.; Perea, G.; et al. A First-in-Class Small-Molecule that Acts as a Dual Inhibitor of HDAC and PDE5 and that Rescues Hippocampal Synaptic Impairment in Alzheimer’s Disease Mice. Neuropsychopharmacology 2017, 42, 524–539. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado-Tejedor, M.; Garcia-Barroso, C.; Sanzhez-Arias, J.; Mederos, S.; Rabal, O.; Ugarte, A.; Franco, R.; Pascual-Lucas, M.; Segura, V.; Perea, G.; et al. Concomitant histone deacetylase and phosphodiesterase 5 inhibition synergistically prevents the disruption in synaptic plasticity and it reverses cognitive impairment in a mouse model of Alzheimer’s disease. Clin. Epigenetics 2015. [Google Scholar] [CrossRef]
- Rabal, O.; Sánchez-Arias, J.A.; Cuadrado-Tejedor, M.; De Miguel, I.; Pérez-González, M.; García-Barroso, C.; Ugarte, A.; Estella-Hermoso De Mendoza, A.; Sáez, E.; Espelosin, M.; et al. Design, Synthesis, and Biological Evaluation of First-in-Class Dual Acting Histone Deacetylases (HDACs) and Phosphodiesterase 5 (PDE5) Inhibitors for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2016, 59, 8967–9004. [Google Scholar] [CrossRef]
- Fickert, P.; Fuchsbichler, A.; Wagner, M.; Zollner, G.; Kaser, A.; Tilg, H.; Krause, R.; Lammert, F.; Langner, C.; Zatloukal, K.; et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 2004, 127, 261–274. [Google Scholar] [CrossRef]
- Nakken, K.E.; Nygård, S.; Haaland, T.; Berge, K.E.; Arnkværn, K.; Ødegaard, A.; Labori, K.J.; Ræder, M.G. Multiple inflammatory-, tissue remodelling- and fibrosis genes are differentially transcribed in the livers of Abcb4 (-/-) mice harbouring chronic cholangitis. Scand. J. Gastroenterol. 2007, 42, 1245–1255. [Google Scholar] [CrossRef]
- Mauad, T.H.; Van Nieuwkerk, C.M.J.; Dingemans, K.P.; Smit, J.J.M.; Schinkel, A.H.; Notenboom, R.G.E.; Van den Bergh Weerman, M.A.; Verkruisen, R.P.; Groen, A.K.; Elferink, R.P.J.O.; et al. Mice with homozygous disruption of the mdr2 P-glycoprotein gene: A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am. J. Pathol. 1994, 145, 1237–1245. [Google Scholar] [PubMed]
- Nishio, T.; Hu, R.; Koyama, Y.; Liang, S.; Rosenthal, S.B.; Yamamoto, G.; Karin, D.; Baglieri, J.; Ma, H.Y.; Xu, J.; et al. Activated hepatic stellate cells and portal fibroblasts contribute to cholestatic liver fibrosis in MDR2 knockout mice. J. Hepatol. 2019, 71, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Uriell-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Popov, Y.; Patsenker, E.; Fickert, P.; Trauner, M.; Schuppan, D. Mdr2 (Abcb4)-/- mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J. Hepatol. 2005, 43, 1045–1054. [Google Scholar] [CrossRef]
- Latasa, M.U.; Gil-Puig, C.; Fernández-Barrena, M.G.; Rodríguez-Ortigosa, C.M.; Banales, J.M.; Urtasun, R.; Goñi, S.; Méndez, M.; Arcelus, S.; Juanarena, N.; et al. Oral methylthioadenosine administration attenuates fibrosis and chronic liver disease progression in Mdr2-/- mice. PLoS ONE 2010, 5, e15690. [Google Scholar] [CrossRef]
- Yavuz, B.G.; Pestana, R.C.; Abugabal, Y.I.; Krishnan, S.; Chen, J.; Hassan, M.M.; Wolff, R.A.; Rashid, A.; Amin, H.M.; Kaseb, A.O. Origin and role of hepatic myofibroblasts in hepatocellular carcinoma. Oncotarget 2020, 11, 1186–1201. [Google Scholar] [CrossRef]
- McDaniel, K.; Wu, N.; Zhou, T.; Huang, L.; Sato, K.; Venter, J.; Ceci, L.; Chen, D.; Ramos-Lorenzo, S.; Invernizzi, P.; et al. Amelioration of Ductular Reaction by Stem Cell Derived Extracellular Vesicles in MDR2 Knockout Mice via Lethal-7 microRNA. Hepatology 2019, 69, 2562–2578. [Google Scholar] [CrossRef]
- Dropmann, A.; Dooley, S.; Dewidar, B.; Hammad, S.; Dediulia, T.; Werle, J.; Hartwig, V.; Ghafoory, S.; Woelfl, S.; Korhonen, H.; et al. TGF-β2 silencing to target biliary-derived liver diseases. Gut 2020. [Google Scholar] [CrossRef]
- Midwood, K.S.; Hussenet, T.; Langlois, B.; Orend, G. Advances in tenascin-C biology. Cell. Mol. Life Sci. 2011, 68, 3175–3199. [Google Scholar] [CrossRef]
- Mohs, A.; Kuttkat, N.; Reißing, J.; Zimmermann, H.W.; Sonntag, R.; Proudfoot, A.; Youssef, S.A.; de Bruin, A.; Cubero, F.J.; Trautwein, C. Functional role of CCL5/RANTES for HCC progression during chronic liver disease. J. Hepatol. 2017, 66, 743–753. [Google Scholar] [CrossRef]
- Xu, L.; Hui, A.Y.; Albanis, E.; Arthur, M.J.; O’Byrne, S.M.; Blaner, W.S.; Mukherjee, P.; Friedman, S.L.; Eng, F.J. Human hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, J.H.; Trussoni, C.E.; O’Hara, S.P.; Splinter, P.L.; Heimbach, J.K.; LaRusso, N.F. Characterization of cultured cholangiocytes isolated from livers of patients with primary sclerosing cholangitis. Lab. Investig. 2014, 94, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Ørstavik, S.; Natarajan, V.; Taskén, K.; Jahnsen, T.; Sandberg, M. Characterization of the human gene encoding the type Iα and type Iβ cGMP-dependent protein kinase (PRKG1). Genomics 1997, 42, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, T.M.; Cornwell, T.L. Intracellular cyclic GMP receptor proteins. FASEB J. 1993, 7, 328–338. [Google Scholar] [CrossRef]
- Chettimada, S.; Rawat, D.K.; Dey, N.; Kobelja, R.; Simms, Z.; Wolin, M.S.; Lincoln, T.M.; Gupte, S.A. Glc-6-PD and PKG contribute to hypoxia-induced decrease in smooth muscle cell contractile phenotype proteins in pulmonary artery. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303. [Google Scholar] [CrossRef]
- Franko, A.; Kovarova, M.; Feil, S.; Feil, R.; Wagner, R.; Heni, M.; Königsrainer, A.; Ruoß, M.; Nüssler, A.K.; Weigert, C.; et al. cGMP-dependent protein kinase I (cGKI) modulates human hepatic stellate cell activation. Metabolism 2018, 88, 22–30. [Google Scholar] [CrossRef]
- Jones, D.L.; Haak, A.J.; Caporarello, N.; Choi, K.M.; Ye, Z.; Yan, H.; Varelas, X.; Ordog, T.; Ligresti, G.; Tschumperlin, D.J. TGFβ-induced fibroblast activation requires persistent and targeted HDAC-mediated gene repression. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Palumbo-Zerr, K.; Zerr, P.; Distler, A.; Fliehr, J.; Mancuso, R.; Huang, J.; Mielenz, D.; Tomcik, M.; Fürnrohr, B.G.; Scholtysek, C.; et al. Orphan nuclear receptor NR4A1 regulates transforming growth factor-β 2 signaling and fibrosis. Nat. Med. 2015, 21, 150–158. [Google Scholar] [CrossRef]
- Ligresti, G.; Caporarello, N.; Meridew, J.A.; Jones, D.L.; Tan, Q.; Choi, K.M.; Haak, A.J.; Aravamudhan, A.; Roden, A.C.; Prakash, Y.S.; et al. CBX5/G9a/H3K9me-mediated gene repression is essential to fibroblast activation during lung fibrosis. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiang, S.; Liu, Q.; Gu, T.; Yao, Y.; Lu, X. PPAR γ Antagonizes Hypoxia-Induced Activation of Hepatic Stellate Cell Through Cross Mediating PI3K/AKT and cGMP/PKG Signaling. PPAR Res. 2018, 2018. [Google Scholar] [CrossRef]
- Kumar, P.; Raeman, R.; Chopyk, D.M.; Smith, T.; Verma, K.; Liu, Y.; Anania, F.A. Adiponectin inhibits hepatic stellate cell activation by targeting the PTEN/AKT pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3537–3545. [Google Scholar] [CrossRef] [PubMed]
- Conte, E.; Fruciano, M.; Fagone, E.; Gili, E.; Caraci, F.; Iemmolo, M.; Crimi, N.; Vancheri, C. Inhibition of PI3K prevents the proliferation and differentiation of human lung fibroblasts into myofibroblasts: The role of class I P110 isoforms. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Conte, E.; Gili, E.; Fruciano, M.; Korfei, M.; Fagone, E.; Iemmolo, M.; Lo Furno, D.; Giuffrida, R.; Crimi, N.; Guenther, A.; et al. PI3K p110γ overexpression in idiopathic pulmonary fibrosis lung tissue and fibroblast cells: In vitro effects of its inhibition. Lab. Investig. 2013, 93, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Kypta, R. Secreted antagonists of the Wnt signalling pathway. J. Cell Sci. 2003, 116, 2627–2634. [Google Scholar] [CrossRef] [PubMed]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef]
- Xiong, W.-J.; Hu, L.-J.; Jian, Y.-C.; Wang, L.-J.; Jiang, M.; Li, W.; He, Y. Wnt5a participates in hepatic stellate cell activation observed by gene expression profile and functional assays. World J. Gastroenterol. 2012, 18, 1745. [Google Scholar] [CrossRef]
- Beljaars, L.; Daliri, S.; Dijkhuizen, C.; Poelstra, K.; Gosens, R. WNT-5A regulates TGF-β-related activities in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G219–G227. [Google Scholar] [CrossRef]
- Jones, H.; Hargrove, L.; Kennedy, L.; Meng, F.; Graf-Eaton, A.; Owens, J.; Alpini, G.; Johnson, C.; Bernuzzi, F.; Demieville, J.; et al. Inhibition of mast cell-secreted histamine decreases biliary proliferation and fibrosis in primary sclerosing cholangitis Mdr2−/− mice. Hepatology 2016, 64, 1202–1216. [Google Scholar] [CrossRef]
- Fabris, L.; Brivio, S.; Cadamuro, M.; Strazzabosco, M. Revisiting Epithelial-to-Mesenchymal Transition in Liver Fibrosis: Clues for a Better Understanding of the “Reactive” Biliary Epithelial Phenotype. Stem Cells Int. 2016, 2016. [Google Scholar] [CrossRef]
- Greenbaum, L.E.; Wells, R.G. The role of stem cells in liver repair and fibrosis. Int. J. Biochem. Cell Biol. 2011, 43, 222–229. [Google Scholar] [CrossRef]
- Doyle, S.L.; O’Neill, L.A.J. Toll-like receptors: From the discovery of NFκB to new insights into transcriptional regulations in innate immunity. Biochem. Pharmacol. 2006, 72, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, Y.; Guidotti, L.G.; Kuhlen, C.V.; Fowler, P.; Chisari, F.V. Immune pathogenesis of hepatocellular carcinoma. J. Exp. Med. 1998, 188, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Erstad, D.J.; Razavi, A.A.; Li, S.; Tanabe, K.K.; Fuchs, B.C. Prevention Strategies for Hepatocellular Carcinoma. In Hepatocellular Carcinoma; Humana Press: Totowa, NJ, USA, 2019; pp. 255–289. [Google Scholar]
- Yamauchi, M.; Mizuhara, Y.; Maezawa, Y.; Toda, G. Serum tenascin levels in chronic liver disease. Liver 1994, 14, 148–153. [Google Scholar] [CrossRef]
- Benbow, J.H.; Thompson, K.J.; Cope, H.L.; Brandon-Warner, E.; Culberson, C.R.; Bossi, K.L.; Li, T.; Russo, M.W.; Gersin, K.S.; McKillop, I.H.; et al. Diet-Induced Obesity Enhances Progression of Hepatocellular Carcinoma through Tenascin-C/Toll-Like Receptor 4 Signaling. Am. J. Pathol. 2016, 186, 145–158. [Google Scholar] [CrossRef]
- Boege, Y.; Malehmir, M.; Healy, M.E.; Bettermann, K.; Lorentzen, A.; Vucur, M.; Ahuja, A.K.; Böhm, F.; Mertens, J.C.; Shimizu, Y.; et al. A Dual Role of Caspase-8 in Triggering and Sensing Proliferation-Associated DNA Damage, a Key Determinant of Liver Cancer Development. Cancer Cell 2017, 32, 342–359.e10. [Google Scholar] [CrossRef]
- Brusilovskaya, K.; Königshofer, P.; Lampach, D.; Szodl, A.; Supper, P.; Bauer, D.; Beer, A.; Stift, J.; Timelthaler, G.; Oberhuber, G.; et al. Soluble guanylyl cyclase stimulation and phosphodiesterase-5 inhibition improve portal hypertension and reduce liver fibrosis in bile duct–ligated rats. United Eur. Gastroenterol. J. 2020. [Google Scholar] [CrossRef]
- Guixé-Muntet, S.; Zhu, C.P.; Xie, W.F.; Gracia-Sancho, J. Novel therapeutics for portal hypertension and fibrosis in chronic liver disease. Pharmacol. Ther. 2020, 215, 107626. [Google Scholar] [CrossRef]
- Lechuga, C.G.; Hernández-Nazara, Z.H.; Domínguez Rosales, J.A.; Morris, E.R.; Rincón, A.R.; Rivas-Estilla, A.M.; Esteban-Gamboa, A.; Rojkind, M. TGF-β1 modulates matrix metalloproteinase-13 expression in hepatic stellate cells by complex mechanisms involving p38MAPK, PI3-kinase, AKT, and p70S6k. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287. [Google Scholar] [CrossRef]
- Reif, S.; Lang, A.; Lindquist, J.N.; Yata, Y.; Gäbele, E.; Scanga, A.; Brenner, D.A.; Rippe, R.A. The role of focal adhesion kinase-phosphatidylinositol 3-kinase-Akt signaling in hepatic stellate cell proliferation and type I collagen expression. J. Biol. Chem. 2003, 278, 8083–8090. [Google Scholar] [CrossRef]
- Parsons, C.J.; Takashima, M.; Rippe, R.A. Molecular mechanisms of hepatic fibrogenesis. J. Gastroenterol. Hepatol. 2007, 22, S79–S84. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Z.; Wang, J.; Lam, W.; Kwong, S.; Li, F.; Friedman, S.L.; Zhou, S.; Ren, Q.; Xu, Z.; et al. A histone deacetylase inhibitor, largazole, decreases liver fibrosis and angiogenesis by inhibiting transforming growth factor-β and vascular endothelial growth factor signalling. Liver Int. 2013, 33, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Sotolongo, A.; Mónica, F.Z.; Kots, A.; Xiao, H.; Liu, J.; Seto, E.; Bian, K.; Murad, F. Epigenetic regulation of soluble guanylate cyclase (sGC) β1 in breast cancer cells. FASEB J. 2016, 30, 3171–3180. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Krämer, S.; Loof, T.; Martini, S.; Kron, S.; Kawachi, H.; Shimizu, F.; Neumayer, H.H.; Peters, H. Enhancing cGMP in experimental progressive renal fibrosis: Soluble guanylate cyclase stimulation vs. phosphodiesterase inhibition. Am. J. Physiol. Ren. Physiol. 2006, 290. [Google Scholar] [CrossRef]
- Matei, A.E.; Beyer, C.; Györfi, A.H.; Soare, A.; Chen, C.W.; Dees, C.; Bergmann, C.; Ramming, A.; Friebe, A.; Hofmann, F.; et al. Protein kinases G are essential downstream mediators of the antifibrotic effects of sGC stimulators. Ann. Rheum. Dis. 2018, 77, 459. [Google Scholar] [CrossRef]
- Liu, C.; Chen, X.; Yang, L.; Kisseleva, T.; Brenner, D.A.; Seki, E. Transcriptional repression of the transforming growth factor β (TGF-β) pseudoreceptor BMP and activin membrane-bound inhibitor (BAMBI) by nuclear factor κB (NF-κB) p50 enhances TGF-β signaling in hepatic stellate cells. J. Biol. Chem. 2014, 289, 7082–7091. [Google Scholar] [CrossRef]
- Miao, C.; Yang, Y.; He, X.; Huang, C.; Huang, Y.; Zhang, L.; Lv, X.-W.; Jin, Y.; Li, J. Wnt signaling in liver fibrosis: Progress, challenges and potential directions. Biochimie 2013, 95, 2326–2335. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, X.X.; Li, W.X.; Wu, X.Q.; Huang, C.; Xie, J.; Zhao, Y.X.; Meng, X.M.; Li, J. EZH2-mediated repression of Dkk1 promotes hepatic stellate cell activation and hepatic fibrosis. J. Cell. Mol. Med. 2017, 21, 2317–2328. [Google Scholar] [CrossRef]
- Zhao, Y.L.; Zhu, R.T.; Sun, Y.L. Epithelial-mesenchymal transition in liver fibrosis (Review). Biomed. Rep. 2016, 4, 269–274. [Google Scholar] [CrossRef]
- Lei, W.; Zhang, K.; Pan, X.; Hu, Y.; Wang, D.; Yuan, X.; Shu, G.; Song, J. Histone deacetylase 1 is required for transforming growth factor-β1-induced epithelial-mesenchymal transition. Int. J. Biochem. Cell Biol. 2010, 42, 1489–1497. [Google Scholar] [CrossRef]
- Gu, S.; Liu, Y.; Zhu, B.; Ding, K.; Yao, T.P.; Chen, F.; Zhan, L.; Xu, P.; Ehrlich, M.; Liang, T.; et al. Loss of α-tubulin acetylation is associated with TGF-β-induced epithelial-mesenchymal transition. J. Biol. Chem. 2016, 291, 5396–5405. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, X.; Zhu, W.; Wang, Y.; Zhao, D.; Wang, X.; Gurley, E.C.; Liang, G.; Chen, W.; Lai, G.; et al. Cholangiocyte-Derived Exosomal Long Noncoding RNA H19 Promotes Hepatic Stellate Cell Activation and Cholestatic Liver Fibrosis. Hepatology 2019, 70, 1317–1335. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Porrello, A.; Jung, Y.; Yang, L.; Popov, Y.; Choi, S.S.; Witek, R.P.; Alpini, G.; Venter, J.; Vandongen, H.M.; et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J. Clin. Investig. 2008, 118, 3331–3342. [Google Scholar] [CrossRef] [PubMed]
- EE, H.; MR, M.; KJ, L. HDAC Inhibitors as Epigenetic Regulators of the Immune System: Impacts on Cancer Therapy and Inflammatory Diseases. Biomed Res. Int. 2016, 2016. [Google Scholar] [CrossRef]
- Elizalde, M.; Urtasun, R.; Azkona, M.; Latasa, M.U.; Goñi, S.; García-Irigoyen, O.; Uriarte, I.; Segura, V.; Collantes, M.; Di Scala, M.; et al. Splicing regulator SLU7 is essential for maintaining liver homeostasis. J. Clin. Investig. 2014. [Google Scholar] [CrossRef]
- Bárcena-Varela, M.; Caruso, S.; Llerena, S.; Álvarez-Sola, G.; Uriarte, I.; Latasa, M.U.; Urtasun, R.; Rebouissou, S.; Alvarez, L.; Jimenez, M.; et al. Dual Targeting of Histone Methyltransferase G9a and DNA-Methyltransferase 1 for the Treatment of Experimental Hepatocellular Carcinoma. Hepatology 2019, 69, 587–603. [Google Scholar] [CrossRef]
- Garcia-Irigoyen, O.; Carotti, S.; Latasa, M.U.; Uriarte, I.; Fernández-Barrena, M.G.; Elizalde, M.; Urtasun, R.; Vespasiani-Gentilucci, U.; Morini, S.; Banales, J.M.; et al. Matrix metalloproteinase-10 expression is induced during hepatic injury and plays a fundamental role in liver tissue repair. Liver Int. 2013. [Google Scholar] [CrossRef]
- Santamaría, E.; Rodríguez-Ortigosa, C.M.; Uriarte, I.; Latasa, M.U.; Urtasun, R.; Alvarez-Sola, G.; Bárcena-Varela, M.; Colyn, L.; Arcelus, S.; Jiménez, M.; et al. The Epidermal Growth Factor Receptor Ligand Amphiregulin Protects From Cholestatic Liver Injury and Regulates Bile Acids Synthesis. Hepatology 2019, 69, 1632–1647. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Uriarte, I.; Banales, J.M.; Śaez, E.; Arenas, F.; Elferink, R.P.J.O.; Prieto, J.; Medina, J.F. Bicarbonate secretion of mouse cholangiocytes involves na-hco3 cotransport in addition to na-independent cl/hco3 exchange. Hepatology 2010, 51, 891–902. [Google Scholar] [CrossRef]
- Salter, K.D.; Roman, R.M.; Larusso, N.R.; Fitz, J.G.; Doctor, R.B. Modified culture conditions enhance expression of differentiated phenotypic properties of normal rat cholangiocytes. Lab. Investig. 2000, 80, 1775–1778. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rodriguez-Collazo, P.; Leuba, S.H.; Zlatanova, J. Robust methods for purification of histones from cultured mammalian cells with the preservation of their native modifications. Nucleic Acids Res. 2009, 37, e81. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claveria-Cabello, A.; Colyn, L.; Uriarte, I.; Latasa, M.U.; Arechederra, M.; Herranz, J.M.; Alvarez, L.; Urman, J.M.; Martinez-Chantar, M.L.; Banales, J.M.; et al. Dual Pharmacological Targeting of HDACs and PDE5 Inhibits Liver Disease Progression in a Mouse Model of Biliary Inflammation and Fibrosis. Cancers 2020, 12, 3748. https://doi.org/10.3390/cancers12123748
Claveria-Cabello A, Colyn L, Uriarte I, Latasa MU, Arechederra M, Herranz JM, Alvarez L, Urman JM, Martinez-Chantar ML, Banales JM, et al. Dual Pharmacological Targeting of HDACs and PDE5 Inhibits Liver Disease Progression in a Mouse Model of Biliary Inflammation and Fibrosis. Cancers. 2020; 12(12):3748. https://doi.org/10.3390/cancers12123748
Chicago/Turabian StyleClaveria-Cabello, Alex, Leticia Colyn, Iker Uriarte, Maria Ujue Latasa, Maria Arechederra, Jose M. Herranz, Laura Alvarez, Jesus M. Urman, Maria L. Martinez-Chantar, Jesus M. Banales, and et al. 2020. "Dual Pharmacological Targeting of HDACs and PDE5 Inhibits Liver Disease Progression in a Mouse Model of Biliary Inflammation and Fibrosis" Cancers 12, no. 12: 3748. https://doi.org/10.3390/cancers12123748
APA StyleClaveria-Cabello, A., Colyn, L., Uriarte, I., Latasa, M. U., Arechederra, M., Herranz, J. M., Alvarez, L., Urman, J. M., Martinez-Chantar, M. L., Banales, J. M., Sangro, B., Rombouts, K., Oyarzabal, J., Marin, J. J. G., Berasain, C., Avila, M. A., & Fernandez-Barrena, M. G. (2020). Dual Pharmacological Targeting of HDACs and PDE5 Inhibits Liver Disease Progression in a Mouse Model of Biliary Inflammation and Fibrosis. Cancers, 12(12), 3748. https://doi.org/10.3390/cancers12123748