NEK1 Phosphorylation of YAP Promotes Its Stabilization and Transcriptional Output

 ,
, 

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids and Antibodies
2.2. Cell Culture
2.3. Cell Treatment
2.4. Cell Transfection
2.5. Co-immunoprecipitation (co-IP)
2.6. Generation of NT1 NEK1 Knockout (KO) Cells Lines
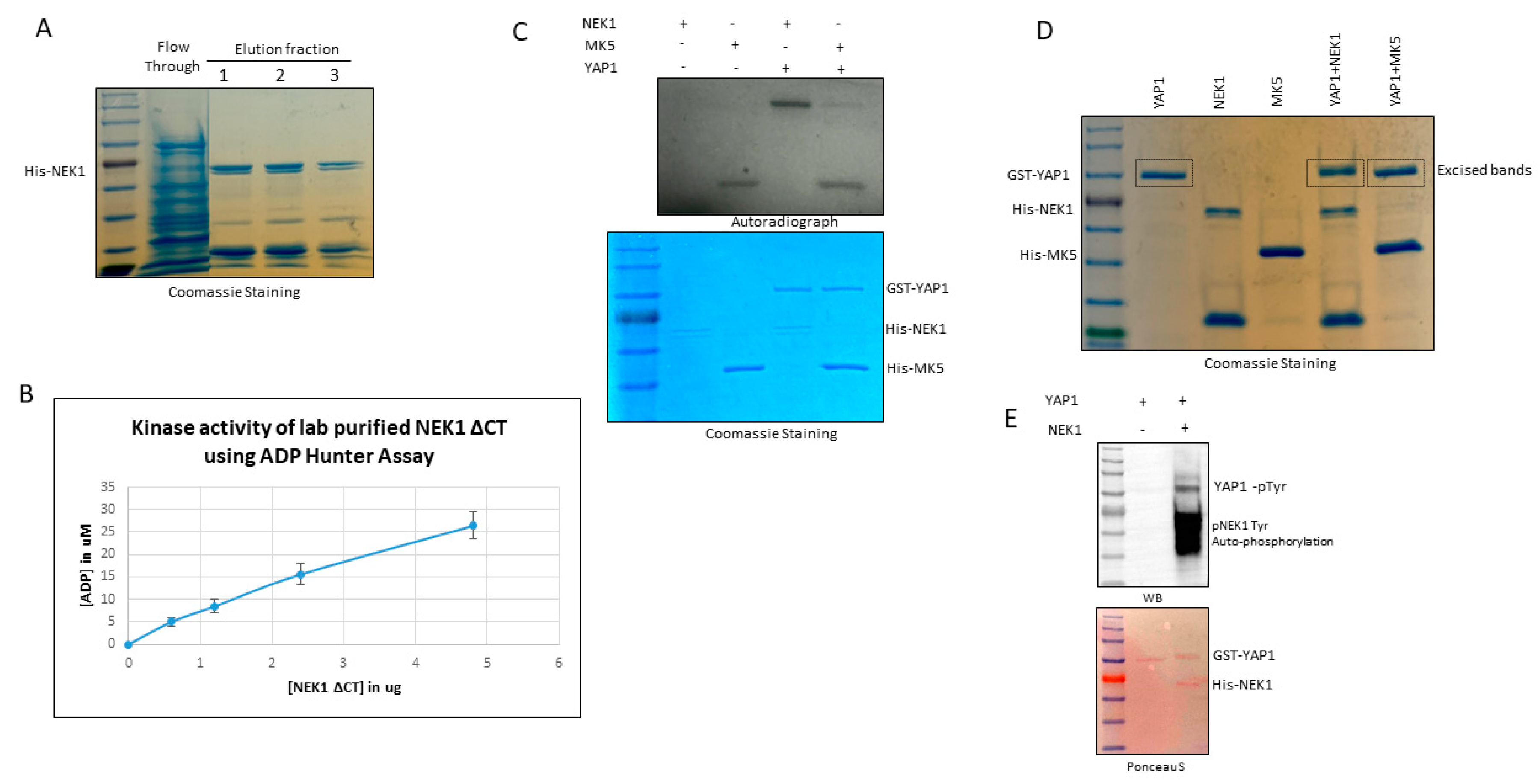
2.7. Protein Purification
2.8. ADP Hunter Assay
2.9. In Vitro Kinase Assay
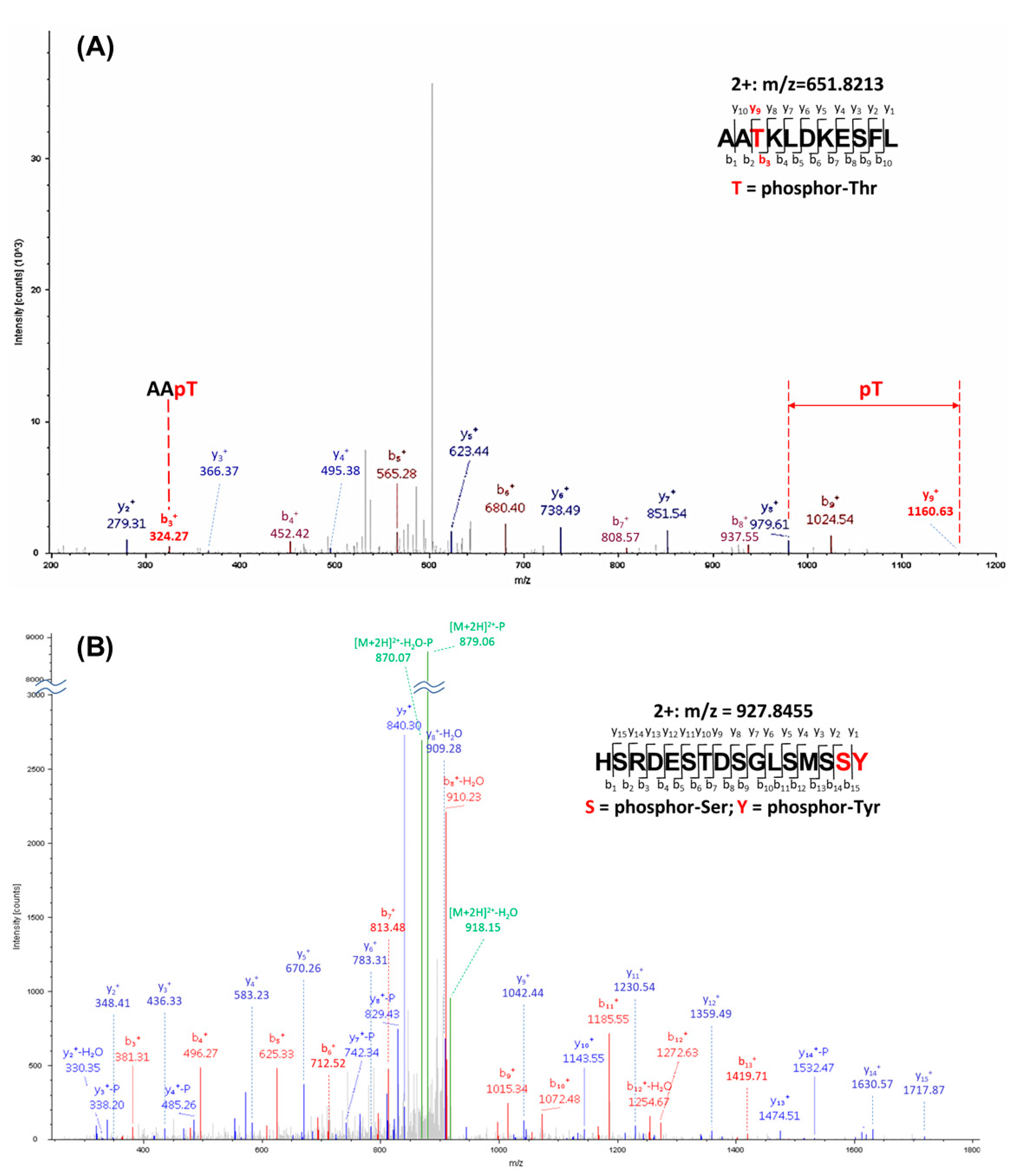
2.10. Identification of YAP1 Phosphorylation by Mass Spectrometry
2.11. Western Blotting
2.12. Real-time Quantitative PCR (RT-qPCR)
2.13. Bioinformatics Analysis
3. Results
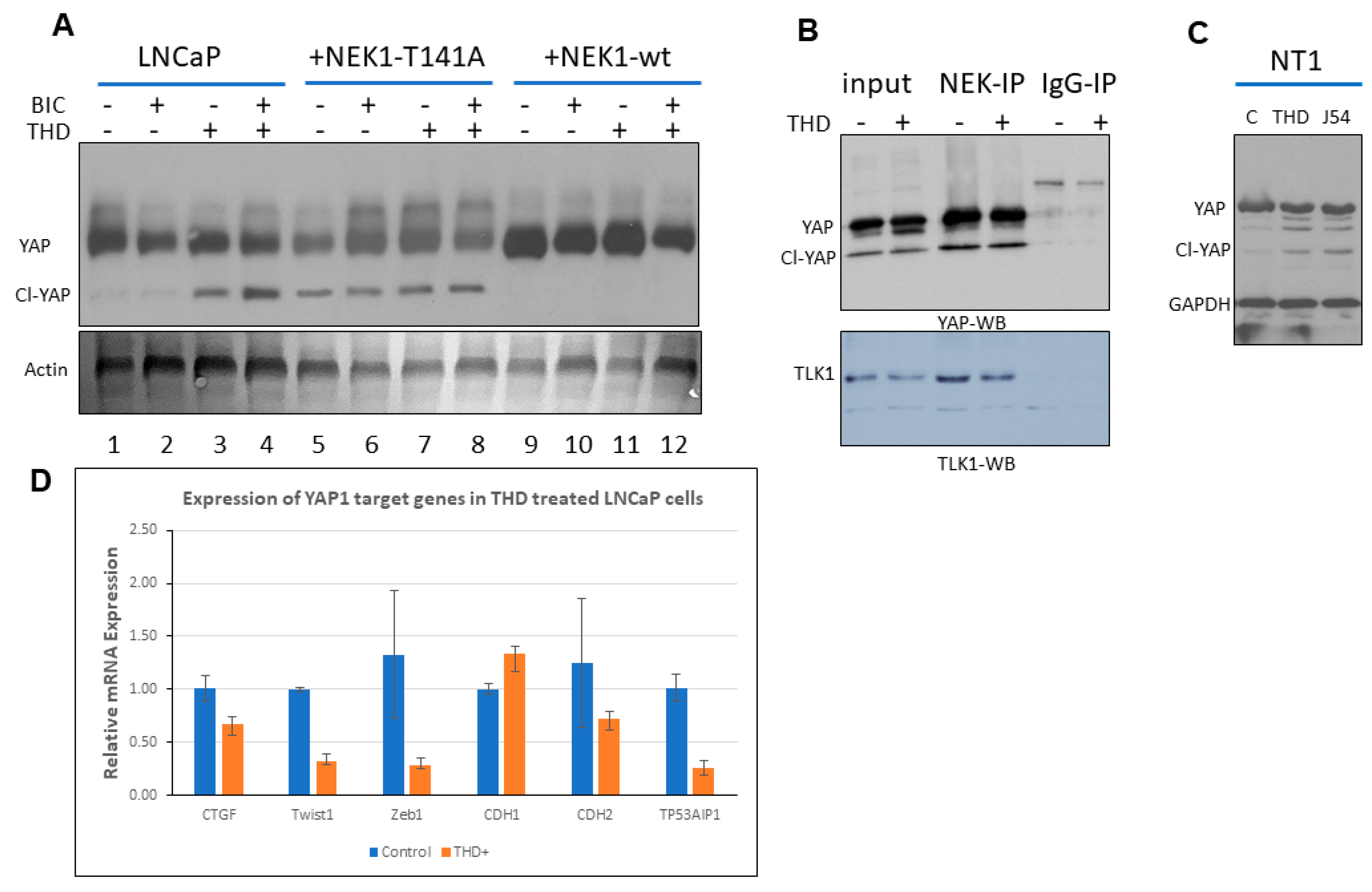
3.1. NEK1 Regulated the Stability of YAP
3.2. NEK1 KO in NeoTag1 Cells Resulted in Reduced YAP Levels and Expression of Several of Its Target Genes
3.3. NEK1 Phosphorylated YAP In Vitro on Several Residues
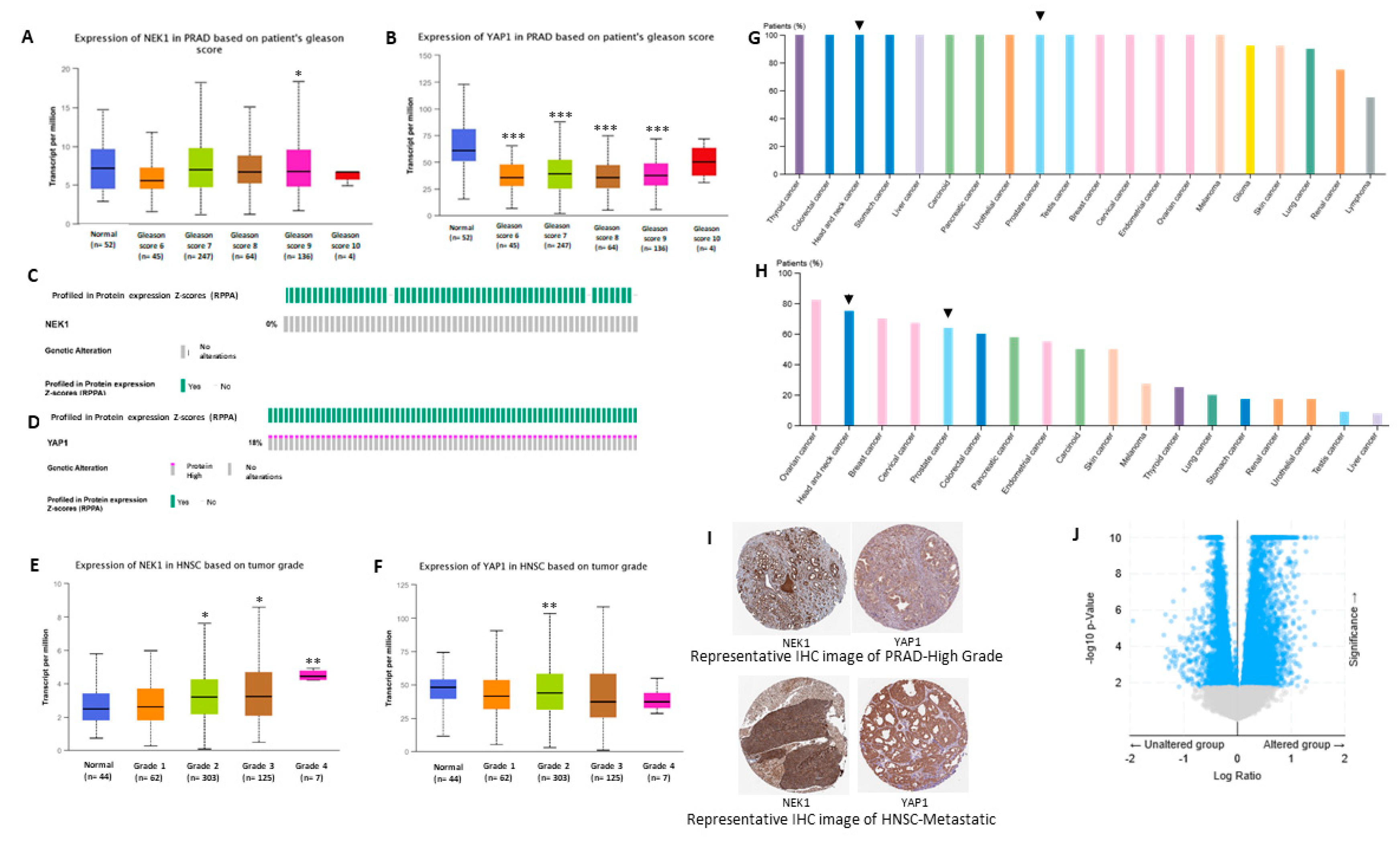
3.4. Bioinformatic Studies Suggest NEK1 Mediated Stabilization of YAP1 in Different Cancers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Osmani, S.A.; Pu, R.T.; Morris, N.R. Mitotic induction and maintenance by overexpression of a G2-specific gene that encodes a potential protein kinase. Cell 1988, 53, 237–244. [Google Scholar] [CrossRef]
- Lu, K.P.; Hunter, T. Evidence for a NIMA-like mitotic pathway in vertebrate cells. Cell 1995, 81, 413–424. [Google Scholar] [CrossRef][Green Version]
- Meirelles, G.V.; Perez, A.M.; de Souza, E.E.; Basei, F.L.; Papa, P.F.; Melo Hanchuk, T.D.; Cardoso, V.B.; Kobarg, J. “Stop Ne(c)king around”: How interactomics contributes to functionally characterize Nek family kinases. World J. Biol. Chem. 2014, 5, 141–160. [Google Scholar] [PubMed]
- Moniz, L.; Dutt, P.; Haider, N.; Stambolic, V. Nek family of kinases in cell cycle, checkpoint control and cancer. Cell Div. 2011, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, C.F.; Riley, D.J.; Chen, P.L. Nek1 kinase functions in DNA damage response and checkpoint control through a pathway independent of ATM and ATR. Cell Cycle 2011, 10, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ho, C.K.; Ouyang, J.; Zou, L. Nek1 kinase associates with ATR-ATRIP and primes ATR for efficient DNA damage signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 2175–2180. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, P.L.; Chen, C.F.; Jiang, X.; Riley, D.J. Never-in-mitosis related kinase 1 functions in DNA damage response and checkpoint control. Cell Cycle 2008, 7, 3194–3201. [Google Scholar] [CrossRef]
- Pelegrini, A.L.; Moura, D.J.; Brenner, B.L.; Ledur, P.F.; Maques, G.P.; Henriques, J.A.; Saffi, J.; Lenz, G. Nek1 silencing slows down DNA repair and blocks DNA damage-induced cell cycle arrest. Mutagenesis 2010, 25, 447–454. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, C.F.; Chiang, H.C.; Pena, M.; Polci, R.; Wei, R.L.; Edwards, R.A.; Hansel, D.E.; Chen, P.L.; Riley, D.J. Mutation of NIMA-related kinase 1 (NEK1) leads to chromosome instability. Mol. Cancer 2011, 10, 5. [Google Scholar] [CrossRef]
- Chen, Y.; Gaczynska, M.; Osmulski, P.; Polci, R.; Riley, D.J. Phosphorylation by Nek1 regulates opening and closing of voltage dependent anion channel 1. Biochem. Biophys. Res. Commun. 2010, 394, 798–803. [Google Scholar] [CrossRef]
- Singh, V.; Khalil, M.I.; De Benedetti, A. The TLK1/Nek1 axis contributes to mitochondrial integrity and apoptosis prevention via phosphorylation of VDAC1. Cell Cycle 2020, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, C.F.; Polci, R.; Wei, R.; Riley, D.J.; Chen, P.L. Increased Nek1 expression in renal cell carcinoma cells is associated with decreased sensitivity to DNA-damaging treatment. Oncotarget 2014, 5, 4283–4294. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Higelin, J.; Catanese, A.; Semelink-Sedlacek, L.L.; Oeztuerk, S.; Lutz, A.K.; Bausinger, J.; Barbi, G.; Speit, G.; Andersen, P.M.; Ludolph, A.C.; et al. NEK1 loss-of-function mutation induces DNA damage accumulation in ALS patient-derived motoneurons. Stem Cell Res. 2018, 30, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Naruse, H.; Ishiura, H.; Mitsui, J.; Takahashi, Y.; Matsukawa, T.; Yoshimura, J.; Doi, K.; Morishita, S.; Goto, J.; Toda, T.; et al. Loss-of-function variants in NEK1 are associated with an increased risk of sporadic ALS in the Japanese population. J. Hum. Genet. 2020, 12, 020–00830. [Google Scholar] [CrossRef]
- Mahjoub, M.R.; Trapp, M.L.; Quarmby, L.M. NIMA-related kinases defective in murine models of polycystic kidney diseases localize to primary cilia and centrosomes. J. Am. Soc. Nephrol. 2005, 16, 3485–3489. [Google Scholar] [CrossRef]
- Shalom, O.; Shalva, N.; Altschuler, Y.; Motro, B. The mammalian Nek1 kinase is involved in primary cilium formation. FEBS Lett. 2008, 582, 1465–1470. [Google Scholar] [CrossRef]
- Surpili, M.J.; Delben, T.M.; Kobarg, J. Identification of proteins that interact with the central coiled-coil region of the human protein kinase NEK1. Biochemistry 2003, 42, 15369–15376. [Google Scholar] [CrossRef]
- Yim, H.; Sung, C.K.; You, J.; Tian, Y.; Benjamin, T. Nek1 and TAZ interact to maintain normal levels of polycystin 2. J. Am. Soc. Nephrol. JASN 2011, 22, 832–837. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, S.; Chen, X.; Stauffer, S.; Yu, F.; Lele, S.M.; Fu, K.; Datta, K.; Palermo, N.; Chen, Y.; et al. The hippo pathway effector YAP regulates motility, invasion, and castration-resistant growth of prostate cancer cells. Mol. Cell. Biol. 2015, 35, 1350–1362. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.L. The Hippo-YAP pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef]
- Chang, L.; Azzolin, L.; Di Biagio, D.; Zanconato, F.; Battilana, G.; Lucon Xiccato, R.; Aragona, M.; Giulitti, S.; Panciera, T.; Gandin, A.; et al. The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature 2018, 563, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Prieto-Dominguez, N.; Yang, S.; Connelly, Z.M.; StPierre, S.; Rushing, B.; Watkins, A.; Shi, L.; Lakey, M.; Baiamonte, L.B.; et al. The expression of YAP1 is increased in high-grade prostatic adenocarcinoma but is reduced in neuroendocrine prostate cancer. Prostate Cancer Prostatic Dis. 2020, 23, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Connelly, Z.M.; Shen, X.; De Benedetti, A. Identification of the proteome complement of humanTLK1 reveals it binds and phosphorylates NEK1 regulating its activity. Cell Cycle 2017, 16, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Jaiswal, P.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. Targeting the TLK1/NEK1 DDR axis with Thioridazine suppresses outgrowth of Androgen Independent Prostate tumors. Int. J. Cancer 2019, 145, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. The TLK1-Nek1 axis promotes prostate cancer progression. Cancer Lett. 2019, 453, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- Kuser-Abali, G.; Alptekin, A.; Lewis, M.; Garraway, I.P.; Cinar, B. YAP1 and AR interactions contribute to the switch from androgen-dependent to castration-resistant growth in prostate cancer. Nat. Commun. 2015, 6, 8126. [Google Scholar] [CrossRef]
- Noh, M.-G.; Kim, S.S.; Hwang, E.C.; Kwon, D.D.; Choi, C. Yes-Associated Protein Expression Is Correlated to the Differentiation of Prostate Adenocarcinoma. J. Pathol. Transl. Med. 2017, 51, 365–373. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Tretiakova, M.S.; Silvis, M.R.; Lucas, J.; Klezovitch, O.; Coleman, I.; Bolouri, H.; Kutyavin, V.I.; Morrissey, C.; True, L.D.; et al. ERG Activates the YAP1 Transcriptional Program and Induces the Development of Age-Related Prostate Tumors. Cancer Cell 2015, 27, 797–808. [Google Scholar] [CrossRef]
- Wang, Y.; Kasper, S.; Yuan, J.; Jin, R.J.; Zhang, J.; Ishii, K.; Wills, M.L.; Hayward, S.W.; Matusik, R.J. Androgen-dependent prostate epithelial cell selection by targeting ARR(2)PBneo to the LPB-Tag model of prostate cancer. Lab. Investig. 2006, 86, 1074–1088. [Google Scholar] [CrossRef][Green Version]
- Singh, V.; Bhoir, S.; Chikhale, R.; Hussain, J.; Dwyer, D.; Bryce, R.; Kirubakaran, S.; De Benedetti, A. Generation of Phenothiazine with Potent Anti-TLK1 Activity for Prostate Cancer Therapy. iScience 2020, 23. [Google Scholar] [CrossRef] [PubMed]
- Kamelgarn, M.; Chen, J.; Kuang, L.; Arenas, A.; Zhai, J.; Zhu, H.; Gal, J. Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim. Biophys. Acta 2016, 1862, 2004–2014. [Google Scholar] [CrossRef] [PubMed]
- UALCAN. Available online: http://ualcan.path.uab.edu/ (accessed on 16 October 2020).
- cBIOPORTAL. Available online: https://www.cbioportal.org/ (accessed on 16 October 2020).
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 16 October 2020).
- Lu, S.; Tan, Z.; Wortman, M.; Dong, Z. Preferential induction of G1 arrest in androgen-responsive human prostate cancer cells by androgen receptor signaling antagonists DL3 and antiandrogen bicalutamide. Cancer Lett. 2010, 298, 250–257. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Vander Griend, D.J.; Antony, L.; Dalrymple, S.; De Marzo, A.M.; Drake, C.G.; Isaacs, J.T. Androgen receptor as a licensing factor for DNA replication in androgen-sensitive prostate cancer cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15085–15090. [Google Scholar] [CrossRef] [PubMed]
- Ronald, S.; Awate, S.; Rath, A.; Carroll, J.; Galiano, F.; Dwyer, D.; Kleiner-Hancock, H.; Mathis, J.M.; Vigod, S.; De Benedetti, A. Phenothiazine Inhibitors of TLKs Affect Double-Strand Break Repair and DNA Damage Response Recovery and Potentiate Tumor Killing with Radiomimetic Therapy. Genes Cancer 2013, 4, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Li, J.; Luo, Z.; Zhang, S.; Xue, S.; Wang, K.; Shi, Y.; Zhang, C.; Chen, H.; Li, Z. Roles of dopamine receptors and their antagonist thioridazine in hepatoma metastasis. Onco Targets Ther. 2015, 8, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Melo-Hanchuk, T.D.; Slepicka, P.F.; Meirelles, G.V.; Basei, F.L.; Lovato, D.V.; Granato, D.C.; Pauletti, B.A.; Domingues, R.R.; Leme, A.F.P.; Pelegrini, A.L.; et al. NEK1 kinase domain structure and its dynamic protein interactome after exposure to Cisplatin. Sci. Rep. 2017, 7, 5445. [Google Scholar] [CrossRef]
- Seo, J.; Kim, M.H.; Hong, H.; Cho, H.; Park, S.; Kim, S.K.; Kim, J. MK5 Regulates YAP Stability and Is a Molecular Target in YAP-Driven Cancers. Cancer Res. 2019, 79, 6139–6152. [Google Scholar] [CrossRef]
- Letwin, K.; Mizzen, L.; Motro, B.; Ben-David, Y.; Bernstein, A.; Pawson, T. A mammalian dual specificity protein kinase, Nek1, is related to the NIMA cell cycle regulator and highly expressed in meiotic germ cells. EMBO J. 1992, 11, 3521–3531. [Google Scholar] [CrossRef]
- Salem, O.; Hansen, C.G. The Hippo Pathway in Prostate Cancer. Cells 2019, 8, 370. [Google Scholar] [CrossRef]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol. Cell 2008, 29, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Totty, N.F.; Irwin, M.S.; Sudol, M.; Downward, J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell 2003, 11, 11–23. [Google Scholar] [CrossRef]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Yuan, L.; Yu, W.; Sun, Y.; Jiang, D.; Wang, X.; Feng, X.; Wang, Z.; Xu, J.; Yang, R.; et al. A Regulation Loop between YAP and NR4A1 Balances Cell Proliferation and Apoptosis. Cell Rep. 2020, 33, 108284. [Google Scholar] [CrossRef]
- Zhang, L.; Tang, F.; Terracciano, L.; Hynx, D.; Kohler, R.; Bichet, S.; Hess, D.; Cron, P.; Hemmings, B.A.; Hergovich, A.; et al. NDR functions as a physiological YAP1 kinase in the intestinal epithelium. Curr. Biol. 2015, 25, 296–305. [Google Scholar] [CrossRef]
- Li, B.; He, J.; Lv, H.; Liu, Y.; Lv, X.; Zhang, C.; Zhu, Y.; Ai, D. C-Abl regulates YAPY357 phosphorylation to activate endothelial atherogenic responses to disturbed flow. J. Clin. Investig. 2019, 129, 1167–1179. [Google Scholar] [CrossRef]
- Sugihara, T.; Werneburg, N.W.; Hernandez, M.C.; Yang, L.; Kabashima, A.; Hirsova, P.; Yohanathan, L.; Sosa, C.; Truty, M.J.; Vasmatzis, G.; et al. YAP Tyrosine Phosphorylation and Nuclear Localization in Cholangiocarcinoma Cells Are Regulated by LCK and Independent of LATS Activity. Mol. Cancer Res. 2018, 16, 1556–1567. [Google Scholar] [CrossRef]
- Vlahov, N.; Scrace, S.; Soto Manuel, S.; Grawenda Anna, M.; Bradley, L.; Pankova, D.; Papaspyropoulos, A.; Yee Karen, S.; Buffa, F.; Goding Colin, R.; et al. Alternate RASSF1 Transcripts Control SRC Activity, E-Cadherin Contacts, and YAP-Mediated Invasion. Curr. Biol. 2015, 25, 3019–3034. [Google Scholar] [CrossRef]
- Tomlinson, V.; Gudmundsdottir, K.; Luong, P.; Leung, K.Y.; Knebel, A.; Basu, S. JNK phosphorylates Yes-associated protein (YAP) to regulate apoptosis. Cell Death Dis. 2010, 1, e29. [Google Scholar] [CrossRef]
- Moon, S.; Kim, W.; Kim, S.; Kim, Y.; Song, Y.; Bilousov, O.; Kim, J.; Lee, T.; Cha, B.; Kim, M.; et al. Phosphorylation by NLK inhibits YAP-14-3-3-interactions and induces its nuclear localization. EMBO Rep. 2017, 18, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Jang, E.J.; Jeong, H.; Han, K.H.; Kwon, H.M.; Hong, J.H.; Hwang, E.S. TAZ suppresses NFAT5 activity through tyrosine phosphorylation. Mol. Cell. Biol. 2012, 32, 4925–4932. [Google Scholar] [CrossRef] [PubMed]
- Danovi, S.A.; Rossi, M.; Gudmundsdottir, K.; Yuan, M.; Melino, G.; Basu, S. Yes-associated protein (YAP) is a critical mediator of c-Jun-dependent apoptosis. Cell Death Differ. 2008, 15, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Iliuk, A.B.; Martin, V.A.; Alicie, B.M.; Geahlen, R.L.; Tao, W.A. In-depth analyses of kinase-dependent tyrosine phosphoproteomes based on metal ion-functionalized soluble nanopolymers. Mol. Cell Proteom. 2010, 9, 2162–2172. [Google Scholar] [CrossRef]
- Freund, I.; Hehlgans, S.; Martin, D.; Ensminger, M.; Fokas, E.; Rödel, C.; Löbrich, M.; Rödel, F. Fractionation-Dependent Radiosensitization by Molecular Targeting of Nek1. Cells 2020, 9, 1235. [Google Scholar] [CrossRef]
- Jiang, N.; Hjorth-Jensen, K.; Hekmat, O.; Iglesias-Gato, D.; Kruse, T.; Wang, C.; Wei, W.; Ke, B.; Yan, B.; Niu, Y.; et al. In vivo quantitative phosphoproteomic profiling identifies novel regulators of castration-resistant prostate cancer growth. Oncogene 2015, 34, 2764–2776. [Google Scholar] [CrossRef]
- Kang, W.; Tong, J.H.; Chan, A.W.; Lee, T.L.; Lung, R.W.; Leung, P.P.; So, K.K.; Wu, K.; Fan, D.; Yu, J.; et al. Yes-associated protein 1 exhibits oncogenic property in gastric cancer and its nuclear accumulation associates with poor prognosis. Clin. Cancer Res. 2011, 17, 2130–2139. [Google Scholar] [CrossRef]
- Lee, S.E.; Lee, J.U.; Lee, M.H.; Ryu, M.J.; Kim, S.J.; Kim, Y.K.; Choi, M.J.; Kim, K.S.; Kim, J.M.; Kim, J.W.; et al. RAF kinase inhibitor-independent constitutive activation of Yes-associated protein 1 promotes tumor progression in thyroid cancer. Oncogenesis 2013, 2, e55. [Google Scholar] [CrossRef]
- Xu, C.M.; Liu, W.W.; Liu, C.J.; Wen, C.; Lu, H.F.; Wan, F.S. Mst1 overexpression inhibited the growth of human non-small cell lung cancer in vitro and in vivo. Cancer Gene Ther. 2013, 20, 453–460. [Google Scholar] [CrossRef]
- Steinhardt, A.A.; Gayyed, M.F.; Klein, A.P.; Dong, J.; Maitra, A.; Pan, D.; Montgomery, E.A.; Anders, R.A. Expression of Yes-associated protein in common solid tumors. Hum. Pathol. 2008, 39, 1582–1589. [Google Scholar] [CrossRef]
- Zhou, D.; Conrad, C.; Xia, F.; Park, J.S.; Payer, B.; Yin, Y.; Lauwers, G.Y.; Thasler, W.; Lee, J.T.; Avruch, J.; et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 2009, 16, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Su, X.; Qin, Q.; Hou, Y.; Zhang, X.; Zhang, H.; Jia, M.; Chen, Y. Yes-associated protein and transcriptional coactivator with PDZ-binding motif as new targets in cardiovascular diseases. Pharmacol. Res. 2020, 159, 105009. [Google Scholar] [CrossRef] [PubMed]
- Tumaneng, K.; Schlegelmilch, K.; Russell, R.C.; Yimlamai, D.; Basnet, H.; Mahadevan, N.; Fitamant, J.; Bardeesy, N.; Camargo, F.D.; Guan, K.L. YAP mediates crosstalk between the Hippo and PI(3)K–TOR pathways by suppressing PTEN via miR-29. Nat. Cell Biol. 2012, 14, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Qi, X.; McAnally, J.; Schwartz, R.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci. Signal. 2011, 4, ra70. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Z.; Chan, S.W.; Liu, A.M.; Wong, K.F.; Fan, S.T.; Chen, J.; Poon, R.T.; Zender, L.; Lowe, S.W.; Hong, W.; et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene 2011, 30, 1229–1240. [Google Scholar] [CrossRef]
- Yan, B.; Jiang, Z.; Cheng, L.; Chen, K.; Zhou, C.; Sun, L.; Qian, W.; Li, J.; Cao, J.; Xu, Q.; et al. Paracrine HGF/c-MET enhances the stem cell-like potential and glycolysis of pancreatic cancer cells via activation of YAP/HIF-1α. Exp. Cell Res. 2018, 371, 63–71. [Google Scholar] [CrossRef]
- Sheng, X.; Li, W.B.; Wang, D.L.; Chen, K.H.; Cao, J.J.; Luo, Z.; He, J.; Li, M.C.; Liu, W.J.; Yu, C. YAP is closely correlated with castration-resistant prostate cancer, and downregulation of YAP reduces proliferation and induces apoptosis of PC-3 cells. Mol. Med. Rep. 2015, 12, 4867–4876. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Mean Log2 mRNA Expression ± SD in NEK1-Overexpressed Group | p-Value | q-Value |
|---|---|---|---|
| Zeb1 | 8.57 ± 1.00 | 3.68 × 10−7 | 5.153 × 10−6 |
| Zeb2 | 8.80 ± 1.05 | 7.237 × 10−6 | 6.617 × 10−5 |
| Ankrd36B | 4.33 ± 0.93 | 1.410 × 10−4 | 8.367 × 10−4 |
| Ankrd11 | 11.55 ± 0.49 | 1.159 × 10−3 | 4.984 × 10−3 |
| BirC2 | 10.46 ± 0.96 | 0.0187 | 0.0498 |
| BirC6 | 11.13 ± 0.49 | 3.91 × 10−12 | 2.80 × 10−10 |
| HoxB3 | 6.44 ± 2.11 | 0.0169 | 0.0458 |
| ARID1B | 10.84 ± 0.46 | 4.14 × 10−10 | 1.57 × 10−8 |
| WSB2 | 11.05 ± 0.45 | 2.016 × 10−4 | 1.136 × 10−3 |
| CAT | 10.01 ± 0.68 | 3.621 × 10−4 | 1.872 × 10−3 |
| ABCB1 | 5.11 ± 1.45 | 0.0111 | 0.0327 |
| PTX3 | 5.33 ± 2.12 | 1.994 × 10−4 | 1.126 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalil, M.I.; Ghosh, I.; Singh, V.; Chen, J.; Zhu, H.; De Benedetti, A. NEK1 Phosphorylation of YAP Promotes Its Stabilization and Transcriptional Output. Cancers 2020, 12, 3666. https://doi.org/10.3390/cancers12123666
Khalil MI, Ghosh I, Singh V, Chen J, Zhu H, De Benedetti A. NEK1 Phosphorylation of YAP Promotes Its Stabilization and Transcriptional Output. Cancers. 2020; 12(12):3666. https://doi.org/10.3390/cancers12123666
Chicago/Turabian StyleKhalil, Md Imtiaz, Ishita Ghosh, Vibha Singh, Jing Chen, Haining Zhu, and Arrigo De Benedetti. 2020. "NEK1 Phosphorylation of YAP Promotes Its Stabilization and Transcriptional Output" Cancers 12, no. 12: 3666. https://doi.org/10.3390/cancers12123666
APA StyleKhalil, M. I., Ghosh, I., Singh, V., Chen, J., Zhu, H., & De Benedetti, A. (2020). NEK1 Phosphorylation of YAP Promotes Its Stabilization and Transcriptional Output. Cancers, 12(12), 3666. https://doi.org/10.3390/cancers12123666