Detection of Structural Variants in Circulating Cell-Free DNA from Sarcoma Patients Using Next Generation Sequencing

 , ,
, ,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Patient Information
2.2. Matched Tumour Tissue Sequencing Metrics
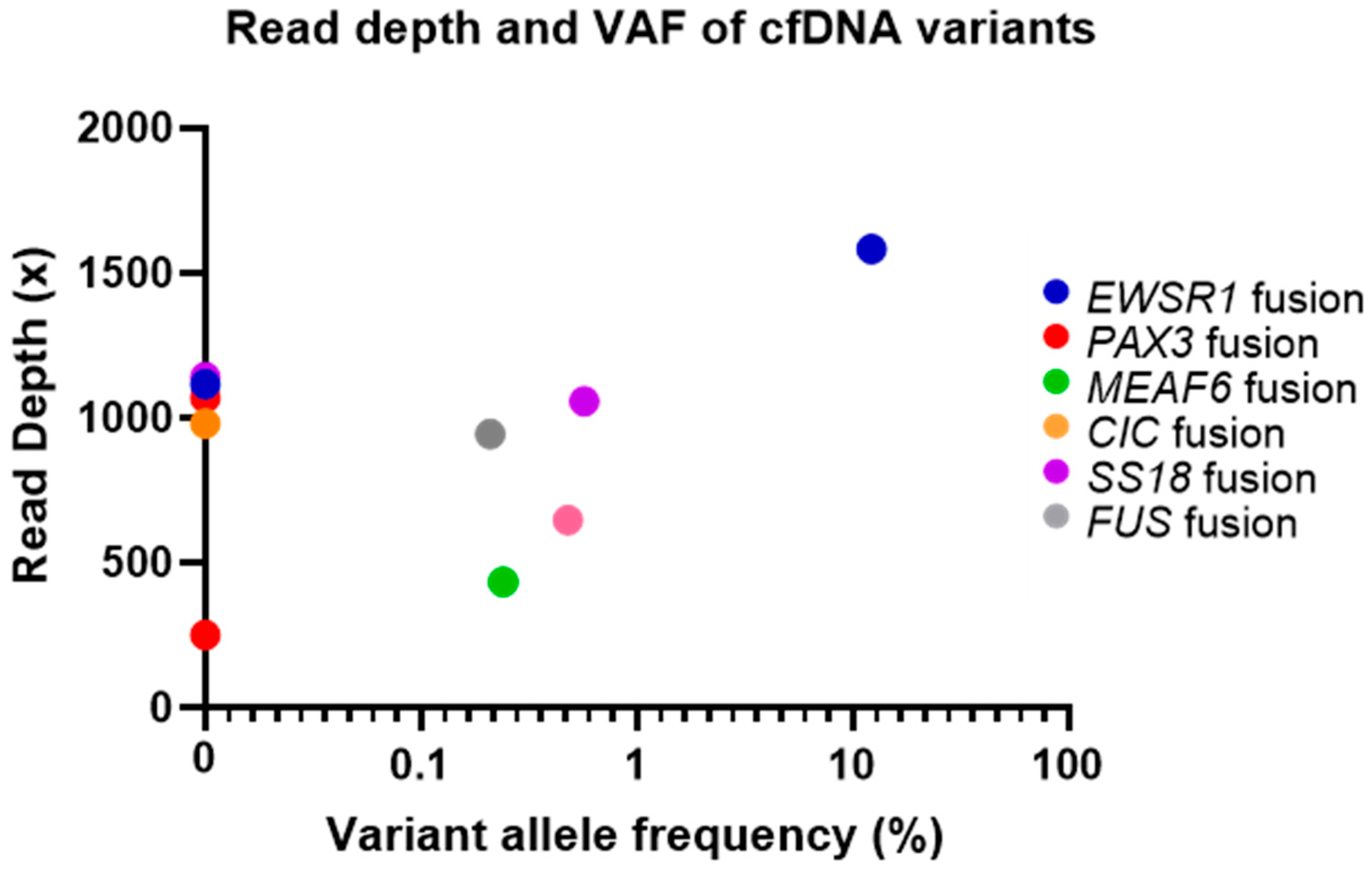
2.3. cfDNA and Matched Tumour Sample Variant Analysis
3. Discussion
4. Materials and Methods
4.1. Ethics Approval and Consent to Participate
4.2. Tumour Specimen Collection
Preparation of DNA Libraries from Matched Fresh Frozen Tumour Samples
4.3. Preparation of Libraries from Sarcoma cfDNA Samples
4.4. Sequencing and Structural Variant Data Analysis
4.5. Copy Number Variation Data Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fletcher, C.D.M.; Bridge, J.A.; Hogendoorn, P.C.W.; Mertens, F. (Eds.) WHO Classification of Tumours of Soft Tissue and Bone, 4th ed.; IARC: Lyon, France, 2013. [Google Scholar]
- Bleloch, J.S.; Ballim, R.D.; Kimani, S.; Parkes, J.; Panieri, E.; Willmer, T.; Prince, S. Managing sarcoma: Where have we come from and where are we going? Ther. Adv. Med. Oncol. 2017, 9, 637–659. [Google Scholar] [CrossRef] [PubMed]
- Ducimetière, F.; Lurkin, A.; Ranchère-Vince, D.; Decouvelaere, A.V.; Péoc’h, M.; Istier, L.; Chalabreysse, P.; Muller, C.; Alberti, L.; Bringuier, P.; et al. Incidence of Sarcoma Histotypes and Molecular Subtypes in a Prospective Epidemiological Study with Central Pathology Review and Molecular Testing. PLoS ONE 2011, 6, e20294. [Google Scholar] [CrossRef] [PubMed]
- Pingping, B.; Yuhong, Z.; Weiqi, L.; Chunxiao, W.; Chunfang, W.; Yuanjue, S.; Chenping, Z.; Jianru, X.; Jiade, L.; Lin, K.; et al. Incidence and Mortality of Sarcomas in Shanghai, China, During 2002–2014. Front. Oncol. 2019, 9, 662. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review (CSR), 1975–2016; National Cancer Institute: Bethesda, MD, USA, 2019.
- DeVita, V.T.J.; Lawrence, T.S.; Rosenberg, S.A. (Eds.) DeVita, Hellman, and Rosenberg’s Cancer: Principles & Practice of Oncology, 8th ed.; Wolters Kluwer: Philadelphia, PA, USA, 2008. [Google Scholar]
- Mercado, G.E.; Barr, F.G. Chromosomal Translocations in Sarcomas: New Perspectives. 2006. Available online: http://sarcomahelp.org/articles/chromosomal-translocations.html (accessed on 24 January 2020).
- Barr, F.G.; Zhang, P.J. The impact of genetics on sarcoma diagnosis: An evolving science. Clin. Cancer Res. 2006, 12, 5256–5257. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef]
- Parker, B.C.; Zhang, W. Fusion genes in solid tumors: An emerging target for cancer diagnosis and treatment. Chin. J. Cancer 2013, 32, 594–603. [Google Scholar] [CrossRef]
- Thway, K.; Wang, J.; Swansbury, J.; Min, T.; Fisher, C. Fluorescence in Situ Hybridization for MDM2 Amplification as a Routine Ancillary Diagnostic Tool for Suspected Well-Differentiated and Dedifferentiated Liposarcomas: Experience at a Tertiary Center. Sarcoma 2015, 2015, 812089. [Google Scholar] [CrossRef]
- Demicco, E.G.; Maki, R.G.; Lev, D.C.; Lazar, A.J. New Therapeutic Targets in Soft Tissue Sarcoma. Adv. Anat. Pathol. 2012, 19, 170–180. [Google Scholar] [CrossRef]
- Smolle, M.A.; Andreou, D.; Tunn, P.; Szkandera, J.; Liegl-Atzwanger, B.; Leithner, A. Diagnosis and treatment of soft-tissue sarcomas of the extremities and trunk. EFORT Open Rev. 2017, 2, 421–431. [Google Scholar] [CrossRef]
- Iwasaki, H.; Nabeshima, K.; Nishio, J.; Jimi, S.; Aoki, M.; Koga, K.; Hamasaki, M.; Hayashi, H.; Mogi, A. Pathology of soft-tissue tumors: Daily diagnosis, molecular cytogenetics and experimental approach. Pathol. Int. 2009, 59, 501–521. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child. Educ. Pract. 2013, 98, 236–238. [Google Scholar] [CrossRef] [PubMed]
- Dilliott, A.A.; Farhan, S.M.; Ghani, M.; Sato, C.; Liang, E.; Zhang, M.; McIntyre, A.D.; Cao, H.; Racacho, L.; Robinson, J.F.; et al. Targeted Next-generation Sequencing and Bioinformatics Pipeline to Evaluate Genetic Determinants of Constitutional Disease. J. Vis. Exp. 2018, 2018, e57266. [Google Scholar] [CrossRef] [PubMed]
- García-García, G.; Baux, D.; Faugère, V.; Moclyn, M.; Koenig, M.; Claustres, M.; Roux, A.-F. Assessment of the latest NGS enrichment capture methods in clinical context. Sci. Rep. 2016, 6, 20948. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Mertens, F.; Antonescu, C.R.; Hohenberger, P.; Ladanyi, M.; Modena, P.; D’Incalci, M.; Casali, P.G.; Aglietta, M.; Alvegård, T. Translocation-Related Sarcomas. Semin. Oncol. 2009, 36, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Pandya, P.H.; Liu, E.; Chandra, P.; Wang, L.; Murray, M.E.; Carter, J.; Ferguson, M.; Saadatzadeh, M.R.; Bijangi-Visheshsaraei, K.; et al. Integration of genomic copy number variations and chemotherapy-response biomarkers in pediatric sarcoma. BMC Med. Genom. 2019, 12, 23. [Google Scholar] [CrossRef]
- Shukla, N.; Patel, J.A.; Magnan, H.; Zehir, A.; You, D.; Tang, J.; Meng, F.; Samoila, A.; Slotkin, E.K.; Ambati, S.R.; et al. Plasma DNA-Based Molecular Diagnosis, Prognostication, and Monitoring of Patients With EWSR1 Fusion-Positive Sarcomas. JCO Precis. Oncol. 2017, 1, 1–11. [Google Scholar] [CrossRef]
- Shulman, D.S.; Klega, K.; Imamovic-Tuco, A.; Clapp, A.; Nag, A.; Thorner, A.R.; van Allen, E.; Ha, G.; Lessnick, S.L.; Gorlick, R.; et al. Detection of circulating tumour DNA is associated with inferior outcomes in Ewing sarcoma and osteosarcoma: A report from the Children’s Oncology Group. Br. J. Cancer 2018, 119, 615–621. [Google Scholar] [CrossRef]
- McConnell, L.; Houghton, O.; Stewart, P.; Gazdova, J.; Srivastava, S.; Kim, C.; Catherwood, M.; Strobl, A.; Flanagan, A.M.; Oniscu, A.; et al. A novel next generation sequencing approach to improve sarcoma diagnosis. Mod. Pathol. 2020, 33, 1350–1359. [Google Scholar] [CrossRef]
- Heining, C.; Horak, P.; Uhrig, S.; Codo, P.L.; Klink, B.; Hutter, B.; Fröhlich, M.; Bonekamp, D.; Richter, D.; Steiger, K.; et al. NRG1 Fusions in KRAS Wild-Type Pancreatic Cancer. Cancer Discov. 2018, 8, 1087–1095. [Google Scholar] [CrossRef]
- Gröschel, S.; Hübschmann, D.; Raimondi, F.; Horak, P.; Warsow, G.; Fröhlich, M.; Klink, B.; Gieldon, L.; Hutter, B.; Kleinhenz, K.; et al. Defective homologous recombination DNA repair as therapeutic target in advanced chordoma. Nat. Commun. 2019, 10, 1635. [Google Scholar] [CrossRef] [PubMed]
- Horak, P.; Klink, B.; Heining, C.; Gröschel, S.; Hutter, B.; Fröhlich, M.; Uhrig, S.; Hübschmann, D.; Schlesner, M.; Eils, R.; et al. Precision oncology based on omics data: The NCT Heidelberg experience. Int. J. Cancer 2017, 141, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. 2011. Available online: https://www.nature.com/articles/nbt.1754 (accessed on 23 January 2018).
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Oliviera, C.; Wolf, T. CNVPanelizer: Reliable CNV Detection in Targeted Sequencing Applications. R Package Version 1.4.0. 2016. Available online: https://bioconductor.riken.jp/packages/3.4/bioc/vignettes/CNVPanelizer/inst/doc/CNVPanelizer.pdf (accessed on 20 January 2019).


{kind=link}
| Sample ID | Gender | Age at Diagnosis | Diagnosis | Structural Variant Detected | Date of Diagnosis (Month/Year) | Primary Tumour Location | Location of Metastases | Last Treatment Prior to Sampling | Time Elapsed between Last Treatment and cfDNA Sampling |
|---|---|---|---|---|---|---|---|---|---|
| M1 | M | 21 | Alveolar Rhabdomyosarcoma | PAX3-FOXO1 | 11/2017 | Hand | Intraabdominal, pleural sarcomatosis | Irinotecan | Ongoing |
| M2 | M | 65 | Extraskeletal myxoid chondrosarcoma | MEAF6-PHF1 | 11/2018 | Chest wall | Lung | Radiotherapy | 3 months |
| M3 | M | 26 | Ewing′s Sarcoma | EWSR1-FLI1 | 10/2016 | Thigh | Lung, lymph nodes | Irinotecan, Temozolomide | Ongoing |
| M4 | F | 19 | Undifferentiated round cell sarcoma | CIC-DUX4 | 11/2018 | Abdomen | Lung, CNS | Vincristine, Actinomycin D, Cyclophosphamide | Ongoing |
| M5 | F | 40 | Alveolar soft part cell Sarcoma | ASPSCR1-TFE3 | 04/2004 | Thigh | Lung, heart, liver, abdomen, CNS | Pazopanib | Ongoing |
| M6 | M | 59 | Dedifferentiated liposarcoma | MDM2/CDK4 amp | 05/2018 | Abdomen | Liver, colon, lymph nodes, lung | Eribulin | Ongoing |
| M7 | F | 56 | Synovial Sarcoma | SS18-SSX2 | 09/2017 | Lung | Lung, pleural sarcomatosis, abdominal | Radiotherapy | 1 year |
| M8 | F | 38 | Alveolar Rhabdomyosarcoma | PAX3-FOXO1 | 07/2017 | Abdomen | Abdominal | Gemcitabine, Docetaxel | One month |
| M9 | M | 65 | Synovial Sarcoma | SS18-SSX2 | 03/2016 | Lung | Pleural sarcomatosis, lymph nodes, bone | Trabectedin | 3 weeks |
| M10 | M | 54 | Myxoid Liposarcoma | FUS-DDIT3 | 01/07/2013 | Retroperitoneum | N/A | Doxorubicin, Olaratumab, surgery | 4 months |
| M11 | M | 54 | Ewing′s Sarcoma | EWSR1-FLI1 | 10/05/2017 | Thigh | Lung | Trabectedin | Ongoing |
| M12 | M | 56 | Clear cell sarcoma | EWSR1-ATF1 | 16/03/2017 | Thigh | Lung, Kidney, lymph nodes | Cisplatin, Etoposide, Ifosfamide | One month |
| Metrics | Tumour Tissue | cfDNA | ||
|---|---|---|---|---|
| Average | Range | Average | Range | |
| Total PF Reads | 27,924,669 | 13,533,026–44,137,640 | 156,632,804 | 138,511,518–187,883,946 |
| Unique PF Reads | 12,220,362 | 3,814,200–15,097,928 | 12,677,104 | 10,214,908–15,493,964 |
| Mean Target Coverage Depth | 2390× | 675×–4374× | 1026× | 842×–1270× |
| Percentage of Duplicates | 57.63% | 42.88–76.30% | 93.62% | 92.31–95.06% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mc Connell, L.; Gazdova, J.; Beck, K.; Srivastava, S.; Harewood, L.; Stewart, J.; Hübschmann, D.; Stenzinger, A.; Glimm, H.; Heilig, C.E.; et al. Detection of Structural Variants in Circulating Cell-Free DNA from Sarcoma Patients Using Next Generation Sequencing. Cancers 2020, 12, 3627. https://doi.org/10.3390/cancers12123627
Mc Connell L, Gazdova J, Beck K, Srivastava S, Harewood L, Stewart J, Hübschmann D, Stenzinger A, Glimm H, Heilig CE, et al. Detection of Structural Variants in Circulating Cell-Free DNA from Sarcoma Patients Using Next Generation Sequencing. Cancers. 2020; 12(12):3627. https://doi.org/10.3390/cancers12123627
Chicago/Turabian StyleMc Connell, Lauren, Jana Gazdova, Katja Beck, Shambhavi Srivastava, Louise Harewood, JP Stewart, Daniel Hübschmann, Albrecht Stenzinger, Hanno Glimm, Christoph E. Heilig, and et al. 2020. "Detection of Structural Variants in Circulating Cell-Free DNA from Sarcoma Patients Using Next Generation Sequencing" Cancers 12, no. 12: 3627. https://doi.org/10.3390/cancers12123627
APA StyleMc Connell, L., Gazdova, J., Beck, K., Srivastava, S., Harewood, L., Stewart, J., Hübschmann, D., Stenzinger, A., Glimm, H., Heilig, C. E., Fröhling, S., & Gonzalez, D. (2020). Detection of Structural Variants in Circulating Cell-Free DNA from Sarcoma Patients Using Next Generation Sequencing. Cancers, 12(12), 3627. https://doi.org/10.3390/cancers12123627