Combined CDK2 and CDK4/6 Inhibition Overcomes Palbociclib Resistance in Breast Cancer by Enhancing Senescence

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
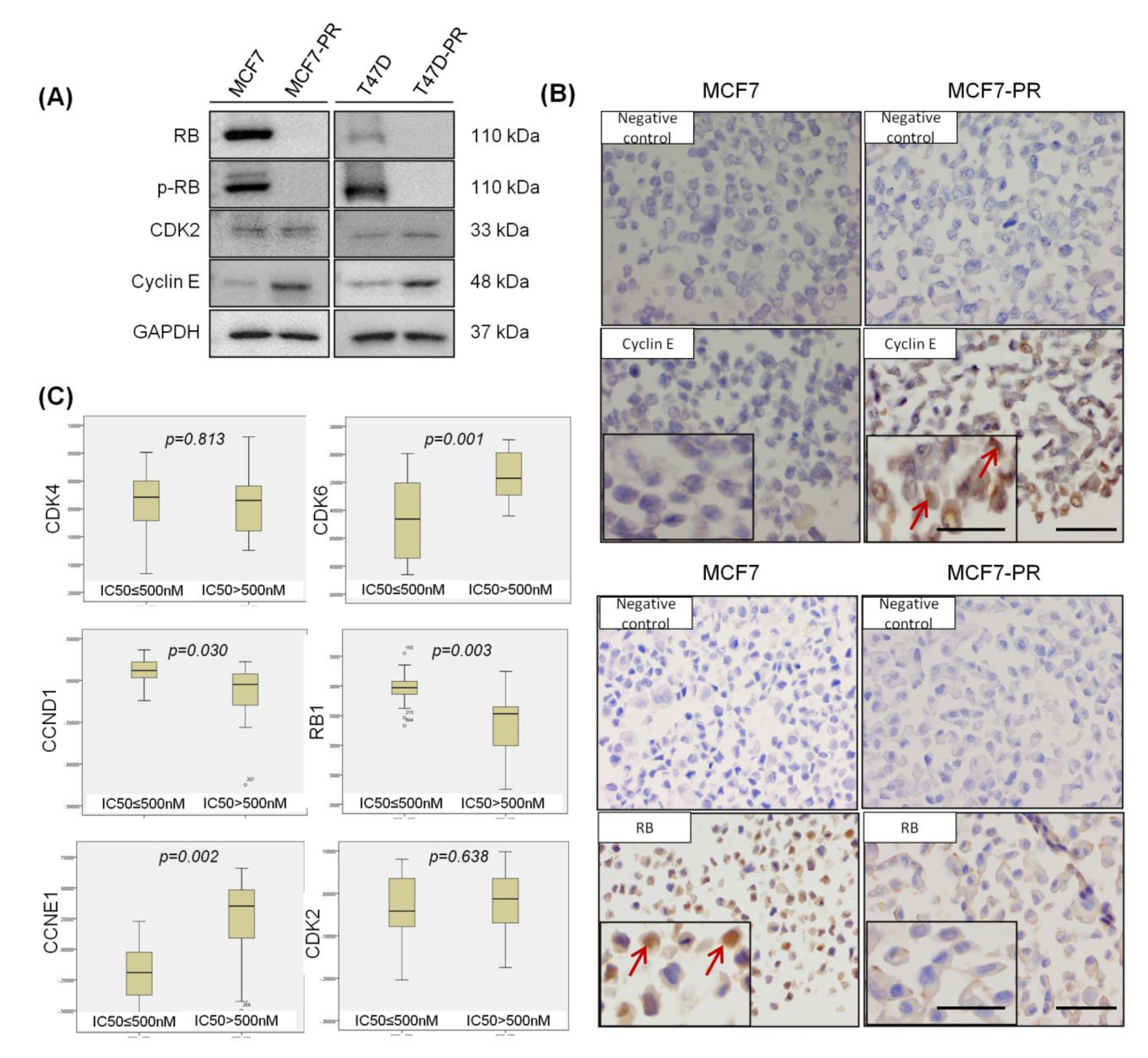
2.1. Generation and Confirmation of Palbociclib-Resistant Cell Lines
2.2. RB Loss and Cyclin E Overexpression Are Observed in CDK4/6 Inhibitor-Resistant Cells
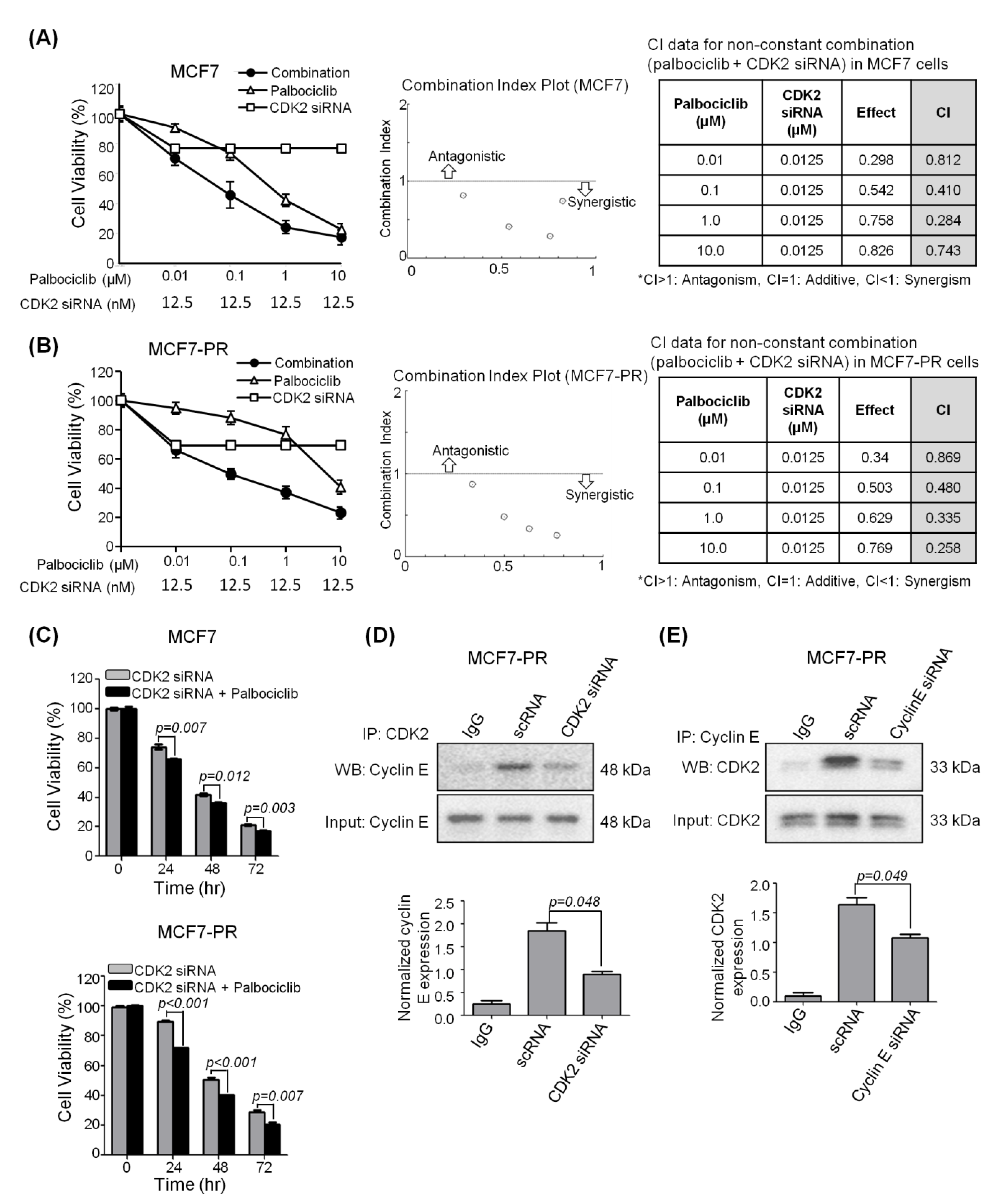
2.3. CDK2 Inhibitor Synergizes with Palbociclib to Inhibit Cell Proliferation
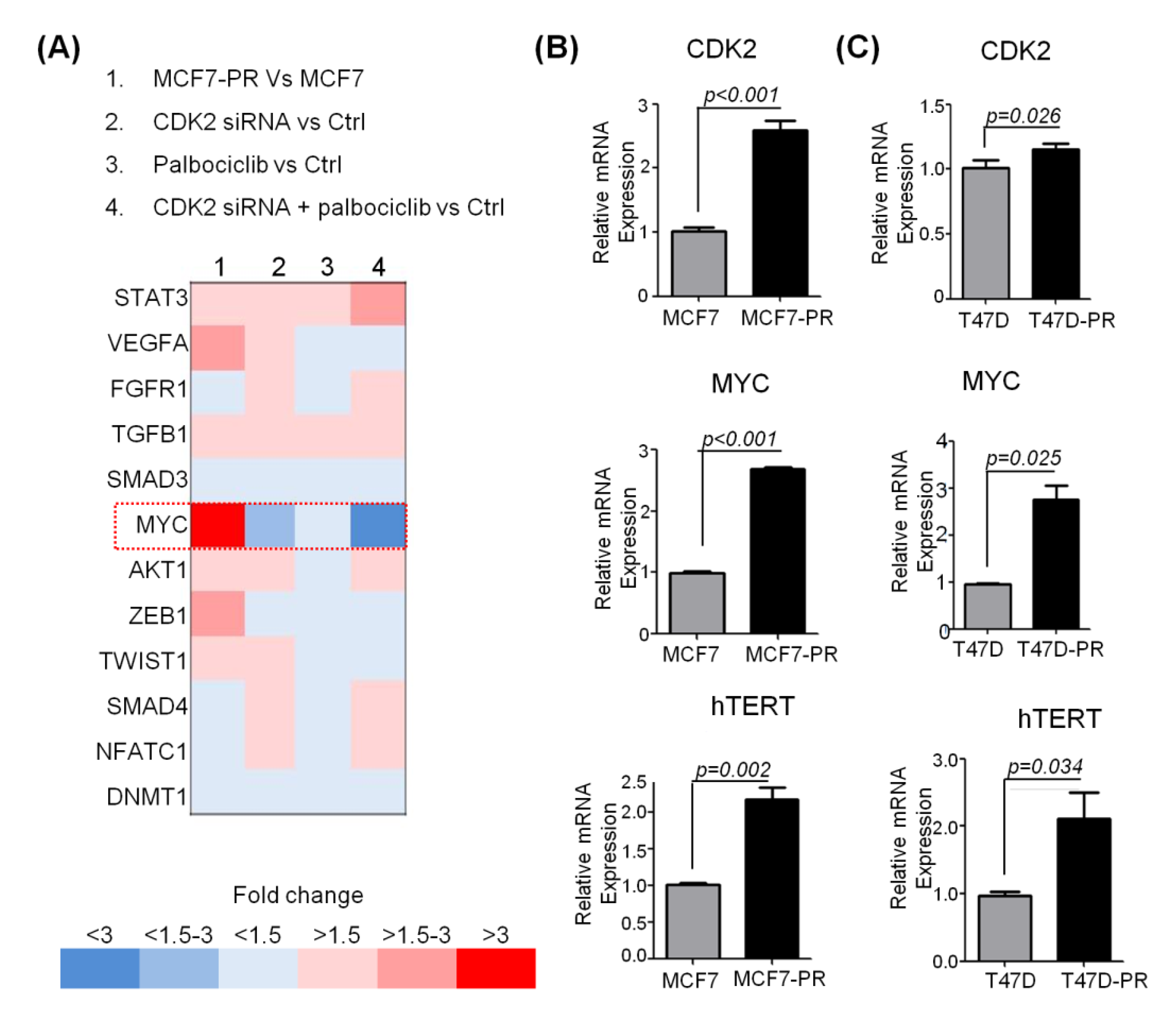
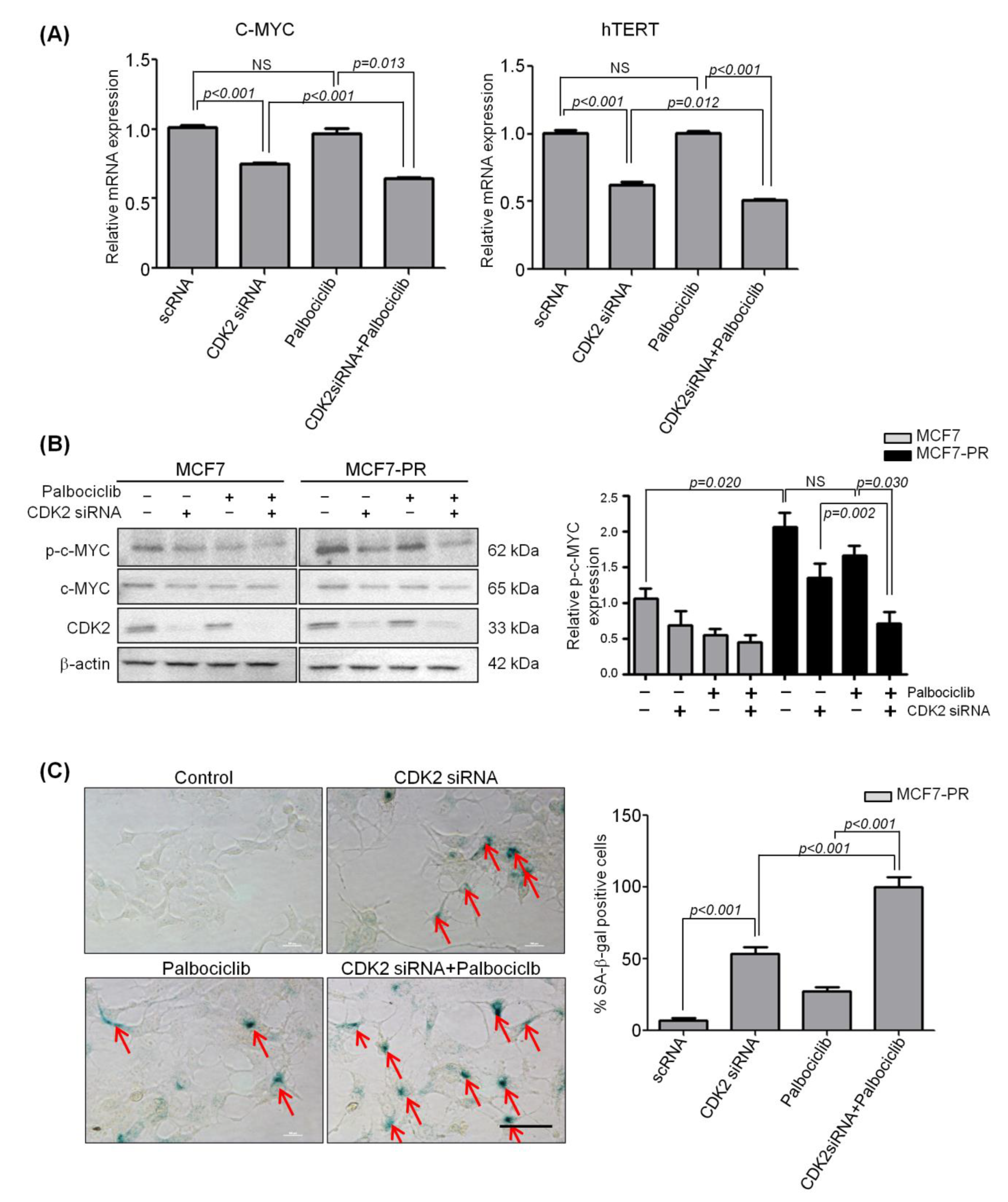
2.4. Inhibition of CDK2 Increases Senescence by Inhibiting Phospho-C-MYC, which Is Responsible for Acquired Resistance to Palbociclib
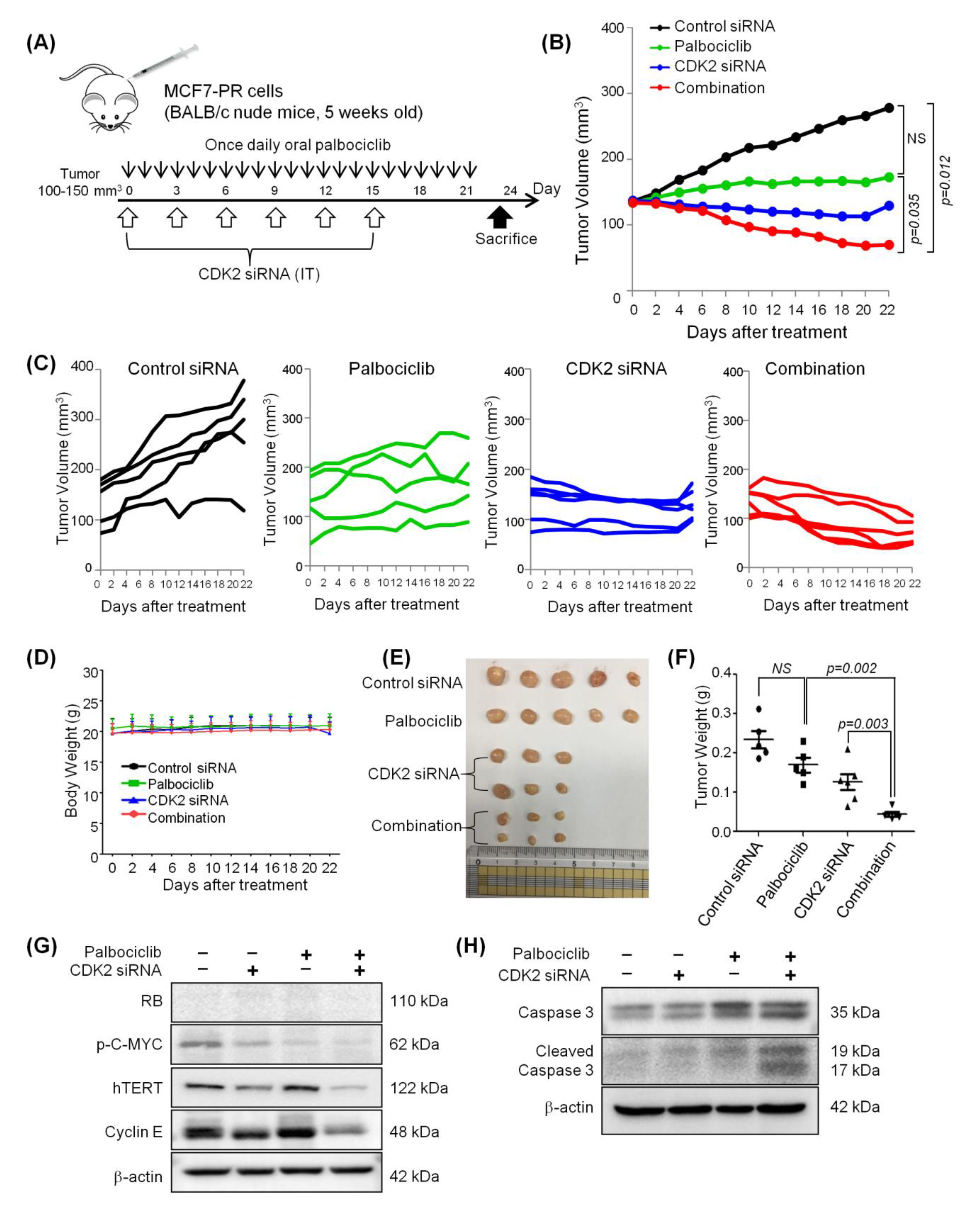
2.5. Combined Inhibition of CDK2 and CDK4/6 Overcomes Resistance to Palbociclib in a Palbociclib-Resistant Xenograft Model
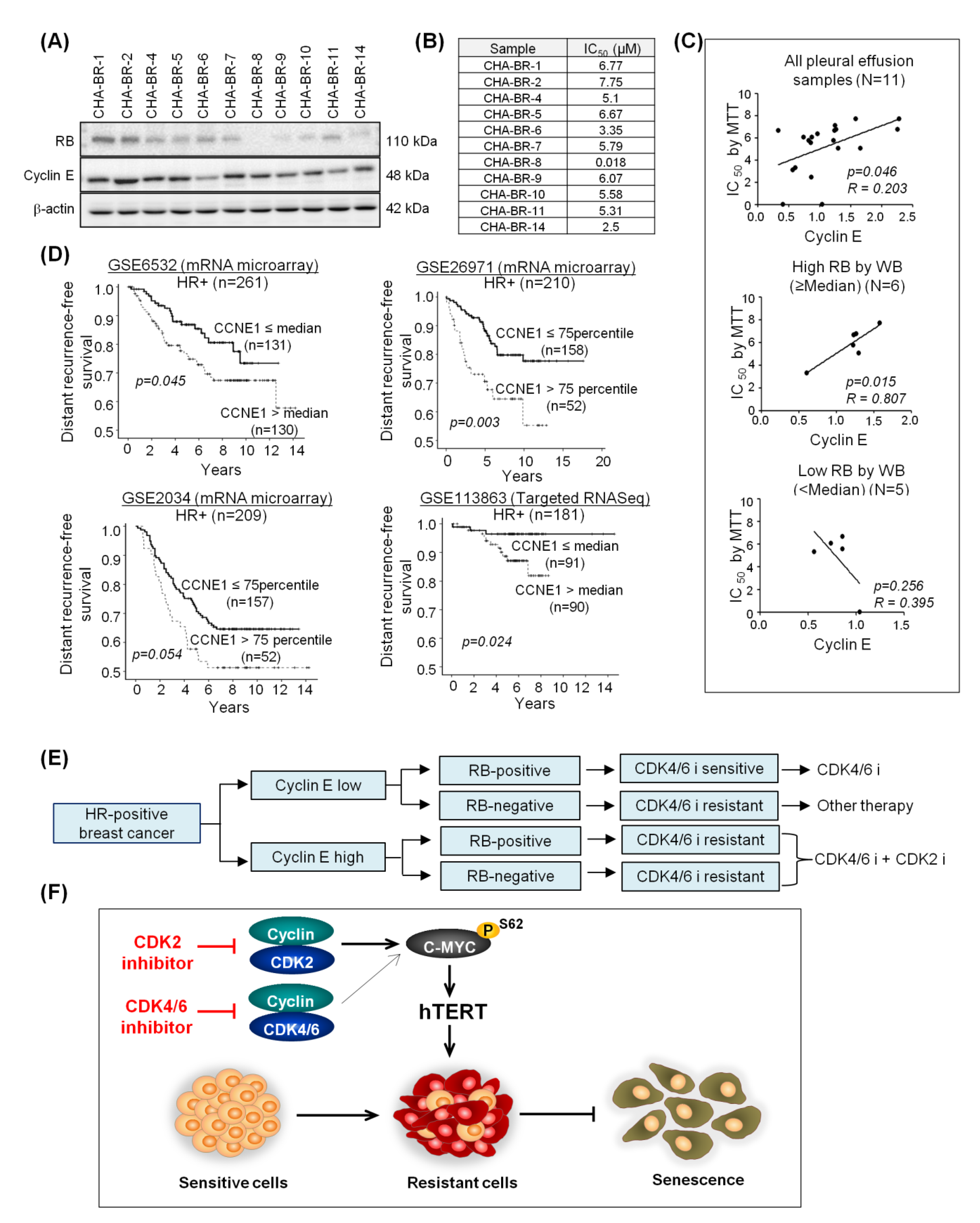
2.6. High Cyclin E Expression Predicts Palbociclib Resistance and Poor Prognosis in HR-Positive Breast Cancer Patients
3. Discussion
4. Materials and Methods
4.1. Resistant Cell Line Establishment
4.2. Cancer Cell Line Encyclopedia (CCLE) Analysis
4.3. Senescence Associated (SA)-β-Galactosidase Staining
4.4. Animal Studies
4.5. Clinical Samples
4.6. Public Gene Expression Profiling Data Sets in Breast Cancer Patients
4.7. Study Approval
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Momenimovahed, Z.; Salehiniya, H. Epidemiological characteristics of and risk factors for breast cancer in the world. Breast Cancer Targets Ther. 2019, 11, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, C.D.; Migliaccio, I.; Malorni, L.; Guarducci, C.; Biganzoli, L.; Di Leo, A. Challenges in the management of advanced, ER-positive, HER2-negative breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 541. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Tyson, J.J.; Dixon, J.M. Endocrine resistance in breast cancer–an overview and update. Mol. Cell. Endocrinol. 2015, 418, 220–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, J.; Bohlmann, R.; Heinrich, N.; Hofmeister, H.; Kroll, J.; Künzer, H.; Lichtner, R.B.; Nishino, Y.; Parczyk, K.; Sauer, G. Characterization of new estrogen receptor destabilizing compounds: Effects on estrogen-sensitive and tamoxifen-resistant breast cancer. J. Natl. Cancer Inst. 2004, 96, 210–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, N.; D’Angelo, A.; Pittacolo, M.; Roviello, G.; Miccoli, A.; Corona, S.P.; Bernocchi, O.; Generali, D.; Otto, T. Updates on the CDK4/6 inhibitory strategy and combinations in breast cancer. Cells 2019, 8, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S. Palbociclib and letrozole in advanced breast cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.-A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar]
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A. MONARCH 2: Abemaciclib in combination with fulvestrant in women with HR+/HER2− advanced breast cancer who had progressed while receiving endocrine therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.K.; Im, S.-A.; Fasching, P.A.; DeLaurentiis, M.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martin, M. Ribociclib (RIB)+ fulvestrant (FUL) in postmenopausal women with hormone receptor-positive (HR+), HER2-negative (HER2–) advanced breast cancer (ABC): Results from MONALEESA-3. J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.-S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.; Martin, M.; Di Leo, A.; Im, S.-A.; Awada, A.; Forrester, T.; Frenzel, M.; Hardebeck, M.C.; Cox, J.; Barriga, S. MONARCH 3 final PFS: A randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer 2019, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, K.; An, H.J.; Kim, S.K.; Lee, S.A.; Kim, S.; Lim, S.M.; Kim, G.M.; Sohn, J.; Moon, Y.W. Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: A review. Int. J. Cancer 2019, 145, 1179–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor–positive breast cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef] [Green Version]
- Dean, J.L.; McClendon, A.K.; Hickey, T.E.; Butler, L.M.; Tilley, W.D.; Witkiewicz, A.K.; Knudsen, E.S. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle 2012, 11, 2756–2761. [Google Scholar] [CrossRef]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017, 36, 2255–2264. [Google Scholar] [CrossRef] [Green Version]
- Mao, P.; Kusiel, J.; Cohen, O.; Wagle, N. Abstract PD4-01: The role of FGF/FGFR axis in resistance to SERDs and CDK4/6 inhibitors in ER+ breast cancer. AACR 2018, 78. [Google Scholar] [CrossRef]
- Sánchez-Martínez, C.; Lallena, M.J.; Sanfeliciano, S.G.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: Recent advances (2015–2019). Bioorganic Med. Chem. Lett. 2019, 29, 126637. [Google Scholar] [CrossRef]
- Guarducci, C.; Nardone, A.; Feiglin, A.; Migliaccio, I.; Malorni, L.; Bonechi, M.; Benelli, M.; Di Leo, A.; Hodgson, G.; Shapiro, G. Abstract PD7-12: Inhibition of CDK7 overcomes resistance to CDK4/6 inhibitors in hormone receptor positive breast cancer cells. AACR 2019, 79. [Google Scholar] [CrossRef]
- Guarducci, C.; Bonechi, M.; Benelli, M.; Biagioni, C.; Boccalini, G.; Romagnoli, D.; Verardo, R.; Schiff, R.; Osborne, C.K.; De Angelis, C. Cyclin E1 and Rb modulation as common events at time of resistance to palbociclib in hormone receptor-positive breast cancer. NPJ Breast Cancer 2018, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Taylor-Harding, B.; Aspuria, P.-J.; Agadjanian, H.; Cheon, D.-J.; Mizuno, T.; Greenberg, D.; Allen, J.R.; Spurka, L.; Funari, V.; Spiteri, E. Cyclin E1 and RTK/RAS signaling drive CDK inhibitor resistance via activation of E2F and ETS. Oncotarget 2015, 6, 696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.C.; Liu, Y.; Zhu, Z.; Loi, S.; Colleoni, M.; Loibl, S.; DeMichele, A.; Harbeck, N.; André, F.; Bayar, M.A. Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor–positive metastatic breast cancer. J. Clin. Oncol. 2019, 37, 1169. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Tsiperson, V.; Gottesman, S.R.; Somma, J.; Blain, S.W. Dual inhibition of CDK4 and CDK2 via targeting p27 tyrosine phosphorylation induces a potent and durable response in breast cancer cells. Mol. Cancer Res. 2018, 16, 361–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Lypova, N.; Lanceta, L.; Gipson, A.; Vega, S.; Garza-Morales, R.; McMasters, K.M.; Chesney, J.; Gomez-Gutierrez, J.G.; Imbert-Fernandez, Y. Targeting Palbociclib-Resistant Estrogen Receptor-Positive Breast Cancer Cells via Oncolytic Virotherapy. Cancers 2019, 11, 684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hydbring, P.; Castell, A.; Larsson, L.-G. MYC Modulation around the CDK2/p27/SKP2 axis. Genes 2017, 8, 174. [Google Scholar] [CrossRef]
- La, S.-H.; Kim, S.-J.; Kang, H.-G.; Lee, H.-W.; Chun, K.-H. Ablation of human telomerase reverse transcriptase (hTERT) induces cellular senescence in gastric cancer through a galectin-3 dependent mechanism. Oncotarget 2016, 7, 57117. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cunningham, M.; Zhang, X.; Tokarz, S.; Laraway, B.; Troxell, M.; Sears, R.C. Phosphorylation regulates c-Myc’s oncogenic activity in the mammary gland. Cancer Res. 2011, 71, 925–936. [Google Scholar] [CrossRef] [Green Version]
- Tarrado-Castellarnau, M.; de Atauri, P.; Tarragó-Celada, J.; Perarnau, J.; Yuneva, M.; Thomson, T.M.; Cascante, M. De novo MYC addiction as an adaptive response of cancer cells to CDK4/6 inhibition. Mol. Syst. Biol. 2017, 13, 940. [Google Scholar] [CrossRef]
- Hydbring, P.; Bahram, F.; Su, Y.; Tronnersjö, S.; Högstrand, K.; von der Lehr, N.; Sharifi, H.R.; Lilischkis, R.; Hein, N.; Wu, S. Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc. Natl. Acad. Sci. USA 2010, 107, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Ling, X.; Yang, W.; Zou, P.; Zhang, G.; Wang, Z.; Zhang, X.; Chen, H.; Peng, K.; Han, F.; Liu, J. TERT regulates telomere-related senescence and apoptosis through DNA damage response in male germ cells exposed to BPDE in vitro and to B [a] P in vivo. Environ. Pollut. 2018, 235, 836–849. [Google Scholar] [CrossRef] [PubMed]
- Malorni, L.; Piazza, S.; Ciani, Y.; Guarducci, C.; Bonechi, M.; Biagioni, C.; Hart, C.D.; Verardo, R.; Di Leo, A.; Migliaccio, I. A gene expression signature of retinoblastoma loss-of-function is a predictive biomarker of resistance to palbociclib in breast cancer cell lines and is prognostic in patients with ER positive early breast cancer. Oncotarget 2016, 7, 68012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formisano, L.; Lu, Y.; Servetto, A.; Hanker, A.B.; Jansen, V.M.; Bauer, J.A.; Sudhan, D.R.; Guerrero-Zotano, A.L.; Croessmann, S.; Guo, Y. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat. Commun. 2019, 10, 1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilgelm, A.E.; Saleh, N.; Shattuck-Brandt, R.; Riemenschneider, K.; Slesur, L.; Chen, S.-C.; Johnson, C.A.; Yang, J.; Blevins, A.; Yan, C. MDM2 antagonists overcome intrinsic resistance to CDK4/6 inhibition by inducing p21. Sci. Transl. Med. 2019, 11, eaav7171. [Google Scholar] [CrossRef]
- Teh, J.L.; Aplin, A.E. Arrested developments: CDK4/6 inhibitor resistance and alterations in the tumor immune microenvironment. Clin. Cancer Res. 2019, 25, 921–927. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-H.; Van Riggelen, J.; Yetil, A.; Fan, A.C.; Bachireddy, P.; Felsher, D.W. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc. Natl. Acad. Sci. USA 2007, 104, 13028–13033. [Google Scholar] [CrossRef] [Green Version]
- Elbadawy, M.; Usui, T.; Yamawaki, H.; Sasaki, K. Emerging roles of C-Myc in Cancer stem cell-related signaling and resistance to cancer chemotherapy: A potential therapeutic target against colorectal cancer. Int. J. Mol. Sci. 2019, 20, 2340. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Wang, L.; Mao, C.; Duraki, D.; Kim, J.E.; Huang, R.; Helferich, W.G.; Nelson, E.R.; Park, B.H.; Shapiro, D.J. Estrogen-independent Myc overexpression confers endocrine therapy resistance on breast cancer cells expressing ERαY537S and ERαD538G mutations. Cancer Lett. 2019, 442, 373–382. [Google Scholar] [CrossRef]
- Klauber-DeMore, N.; Schulte, B.A.; Wang, G.Y. Targeting MYC for triple-negative breast cancer treatment. Oncoscience 2018, 5, 120. [Google Scholar] [CrossRef]
- Hydbring, P.; Larsson, L.-G. Cdk2: A key regulator of the senescence control function of Myc. Aging (Albany NY) 2010, 2, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hydbring, P.; Larsson, L.-G. Tipping the balance: Cdk2 enables Myc to suppress senescence. Cancer Res. 2010, 70, 6687–6691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNair, C.; Xu, K.; Mandigo, A.C.; Benelli, M.; Leiby, B.; Rodrigues, D.; Lindberg, J.; Gronberg, H.; Crespo, M.; De Laere, B. Differential impact of RB status on E2F1 reprogramming in human cancer. J. Clin. Investig. 2018, 128, 341–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, E.S.; McClendon, A.K.; Franco, J.; Ertel, A.; Fortina, P.; Witkiewicz, A.K. RB loss contributes to aggressive tumor phenotypes in MYC-driven triple negative breast cancer. Cell Cycle 2015, 14, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Loi, S.; Haibe-Kains, B.; Desmedt, C.; Wirapati, P.; Lallemand, F.; Tutt, A.M.; Gillet, C.; Ellis, P.; Ryder, K.; Reid, J.F. Predicting prognosis using molecular profiling in estrogen receptor-positive breast cancer treated with tamoxifen. BMC Genom. 2008, 9, 239. [Google Scholar] [CrossRef] [Green Version]
- Filipits, M.; Rudas, M.; Jakesz, R.; Dubsky, P.; Fitzal, F.; Singer, C.F.; Dietze, O.; Greil, R.; Jelen, A.; Sevelda, P. A new molecular predictor of distant recurrence in ER-positive, HER2-negative breast cancer adds independent information to conventional clinical risk factors. Clin. Cancer Res. 2011, 17, 6012–6020. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Klijn, J.G.; Zhang, Y.; Sieuwerts, A.M.; Look, M.P.; Yang, F.; Talantov, D.; Timmermans, M.; Meijer-van Gelder, M.E.; Yu, J. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 2005, 365, 671–679. [Google Scholar] [CrossRef]
- Yang, B.; Chou, J.; Tao, Y.; Wu, D.; Wu, X.; Li, X.; Li, Y.; Chu, Y.; Tang, F.; Shi, Y. An assessment of prognostic immunity markers in breast cancer. NPJ Breast Cancer 2018, 4, 35. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, K.; Park, N.; Park, K.-S.; Hur, J.; Cho, Y.B.; Kang, M.; An, H.-J.; Kim, S.; Hwang, S.; Moon, Y.W. Combined CDK2 and CDK4/6 Inhibition Overcomes Palbociclib Resistance in Breast Cancer by Enhancing Senescence. Cancers 2020, 12, 3566. https://doi.org/10.3390/cancers12123566
Pandey K, Park N, Park K-S, Hur J, Cho YB, Kang M, An H-J, Kim S, Hwang S, Moon YW. Combined CDK2 and CDK4/6 Inhibition Overcomes Palbociclib Resistance in Breast Cancer by Enhancing Senescence. Cancers. 2020; 12(12):3566. https://doi.org/10.3390/cancers12123566
Chicago/Turabian StylePandey, Kamal, Nahee Park, Kyung-Soon Park, Jin Hur, Yong Bin Cho, Minsil Kang, Hee-Jung An, Sewha Kim, Sohyun Hwang, and Yong Wha Moon. 2020. "Combined CDK2 and CDK4/6 Inhibition Overcomes Palbociclib Resistance in Breast Cancer by Enhancing Senescence" Cancers 12, no. 12: 3566. https://doi.org/10.3390/cancers12123566
APA StylePandey, K., Park, N., Park, K.-S., Hur, J., Cho, Y. B., Kang, M., An, H.-J., Kim, S., Hwang, S., & Moon, Y. W. (2020). Combined CDK2 and CDK4/6 Inhibition Overcomes Palbociclib Resistance in Breast Cancer by Enhancing Senescence. Cancers, 12(12), 3566. https://doi.org/10.3390/cancers12123566