High-Plex and High-Throughput Digital Spatial Profiling of Non-Small-Cell Lung Cancer (NSCLC)

 ,
, 

Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Tissue Microarray
2.2. Nanostring GeoMX Digital Spatial Profiler: Tissue Microarray
2.3. Nanostring GeoMX Digital Spatial Profiler: Data Analysis
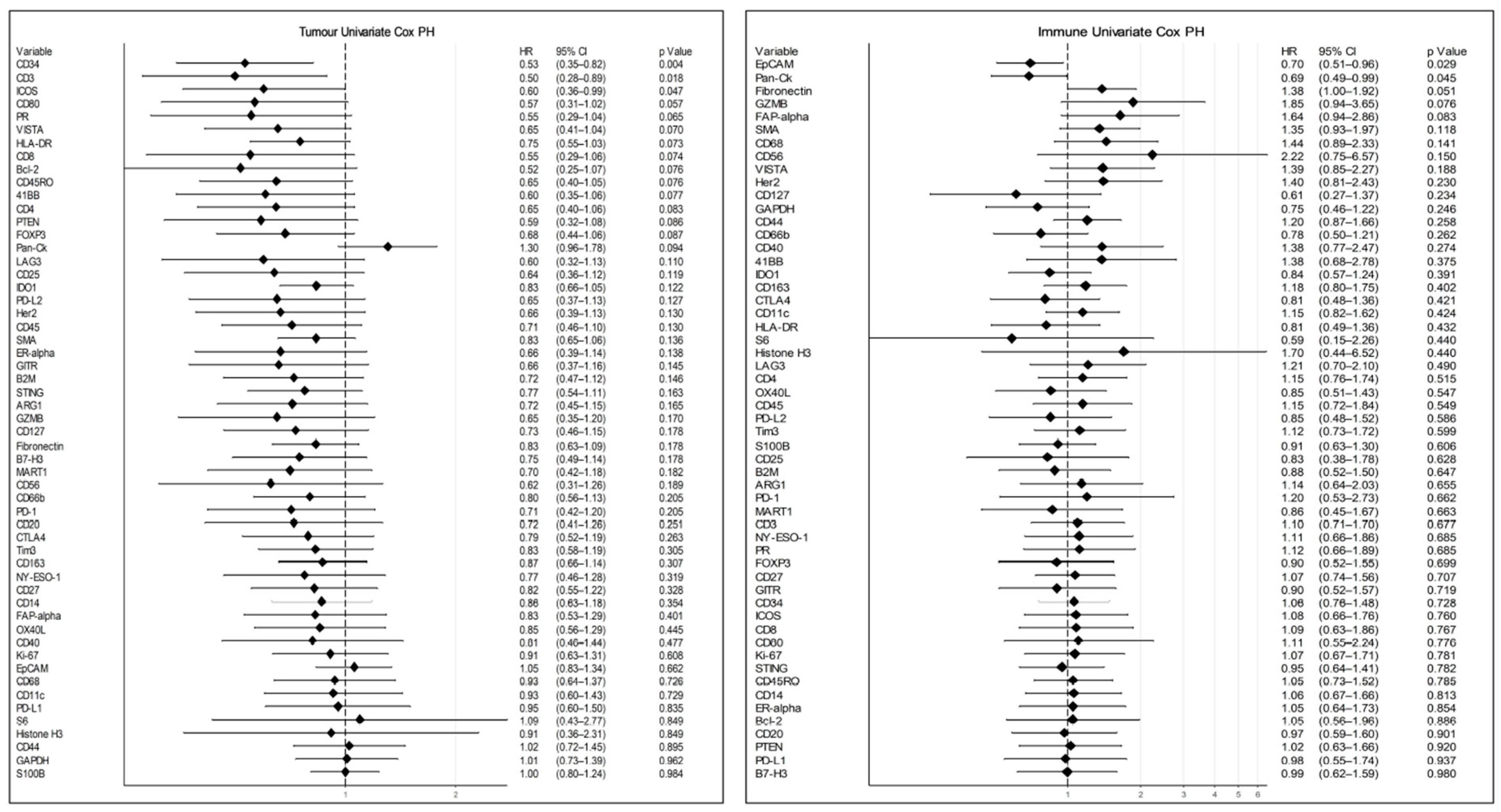
3. Results
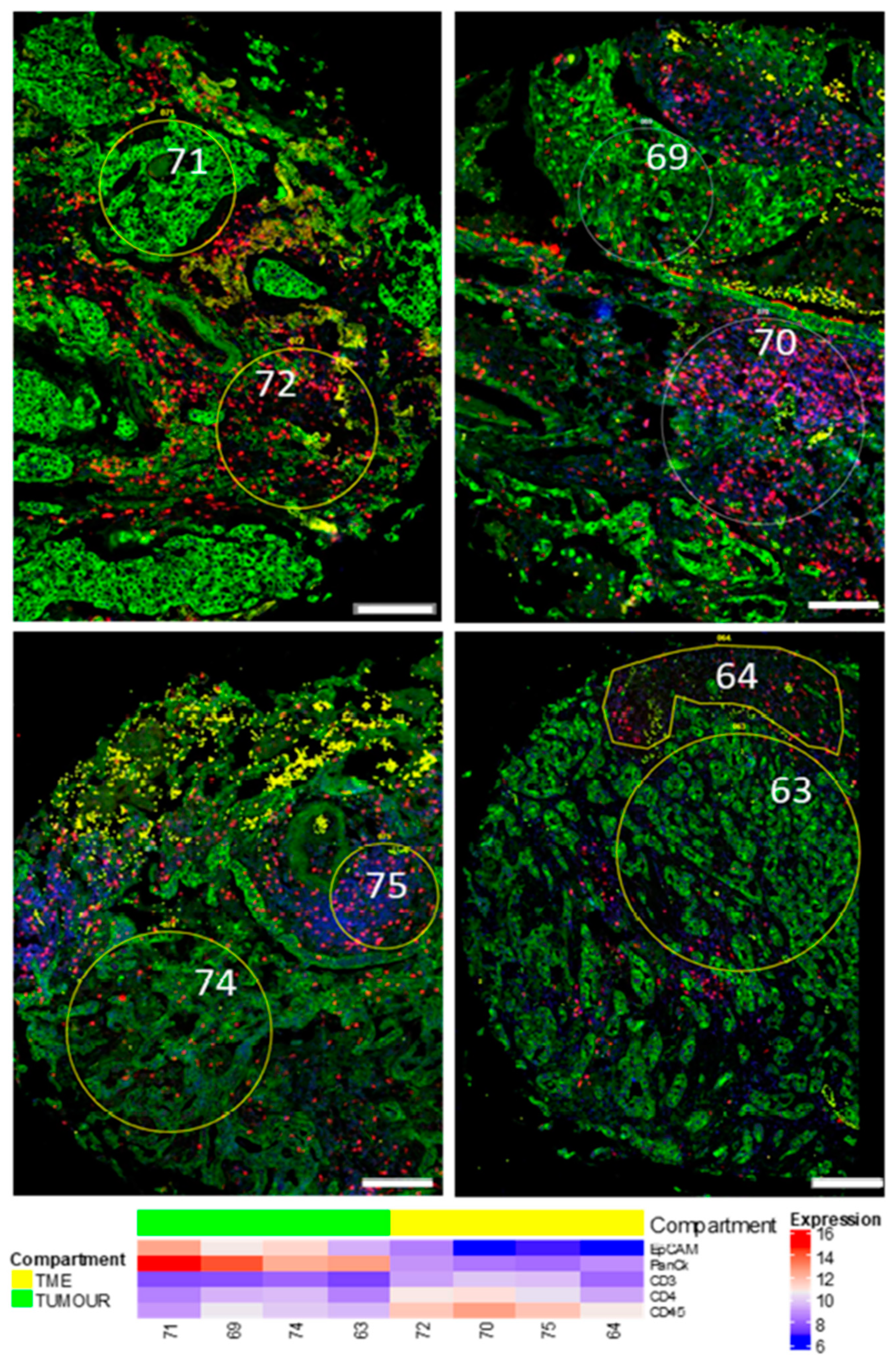
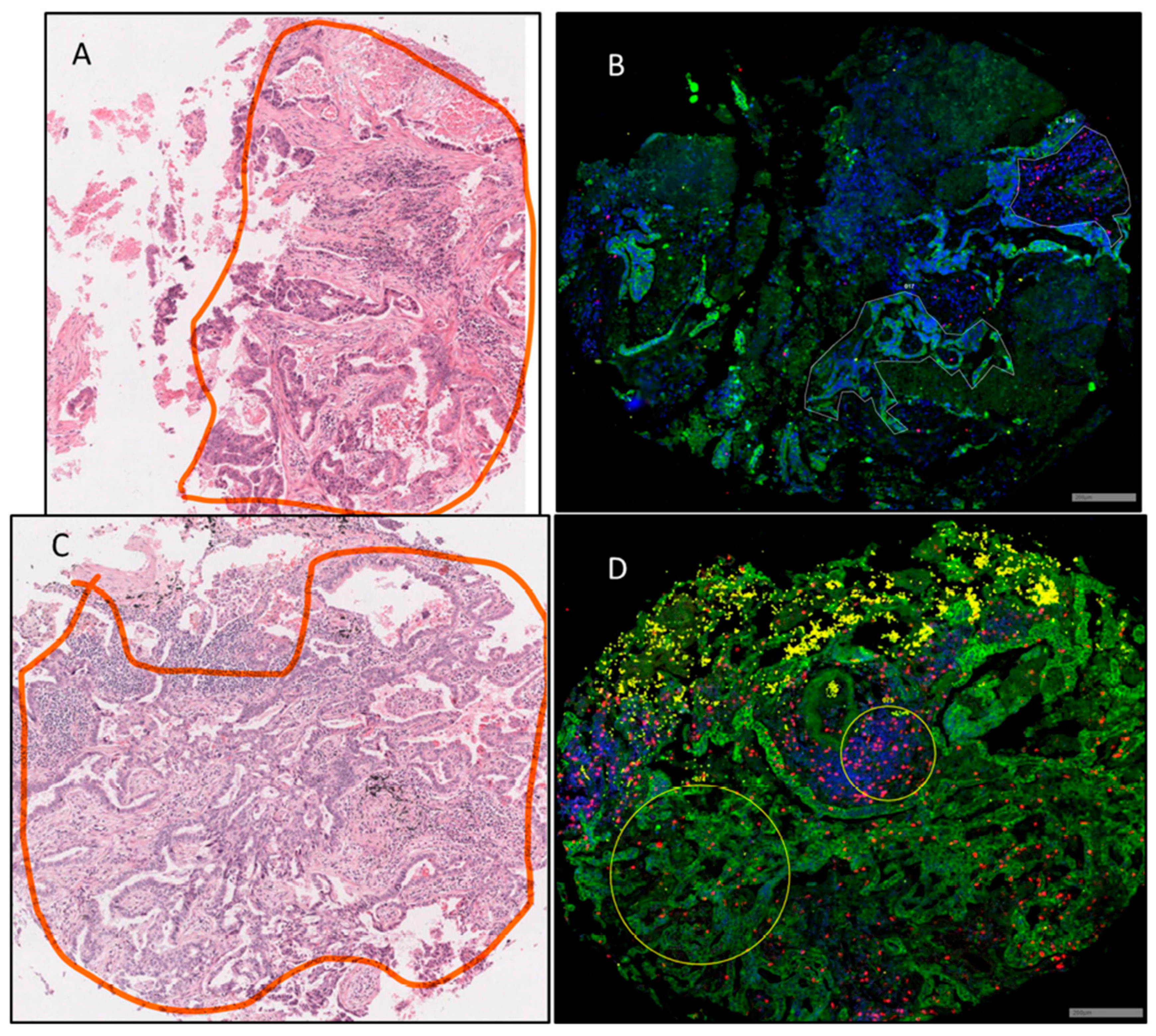
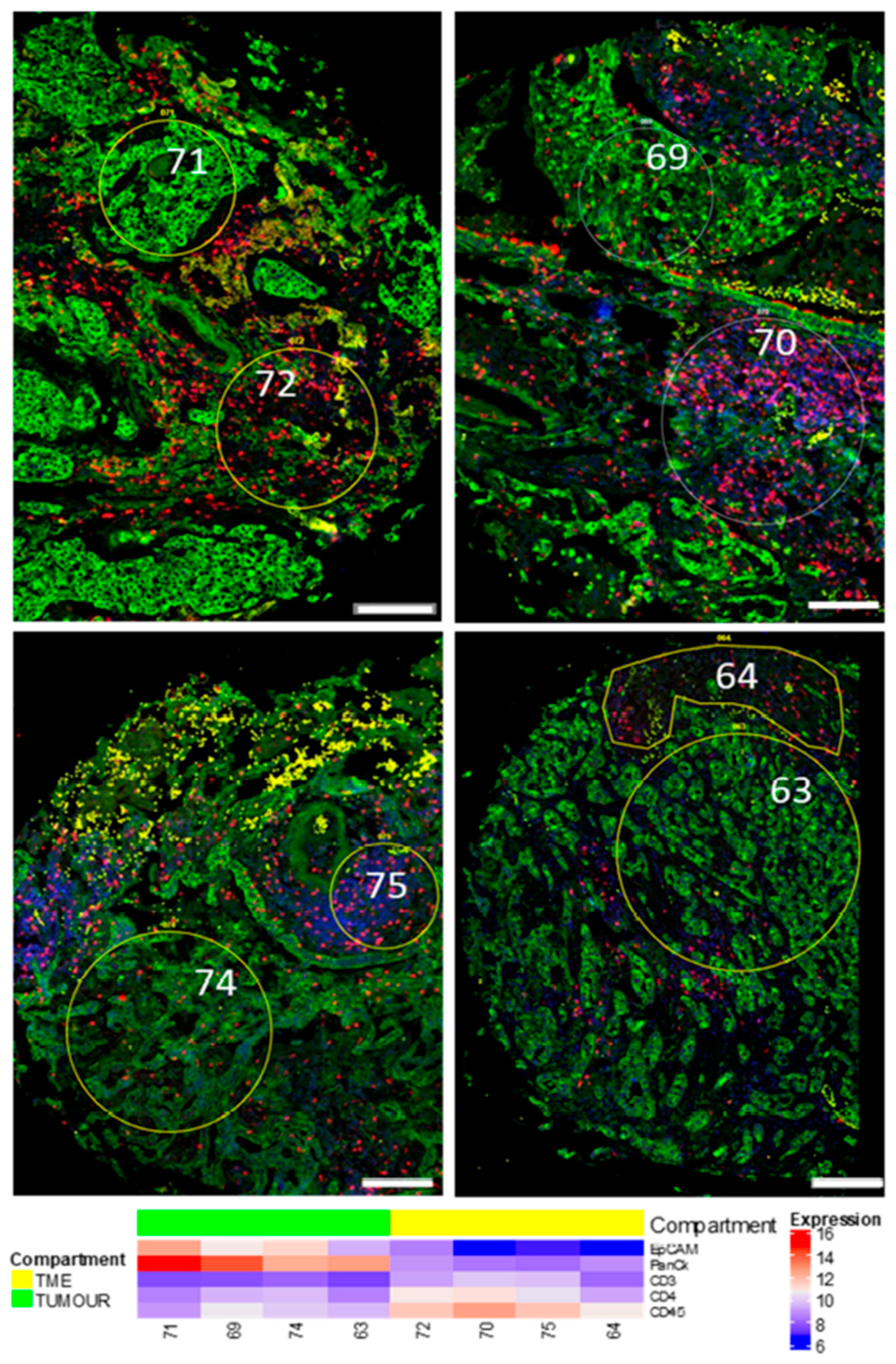
3.1. Region of Interest (ROI) Selection
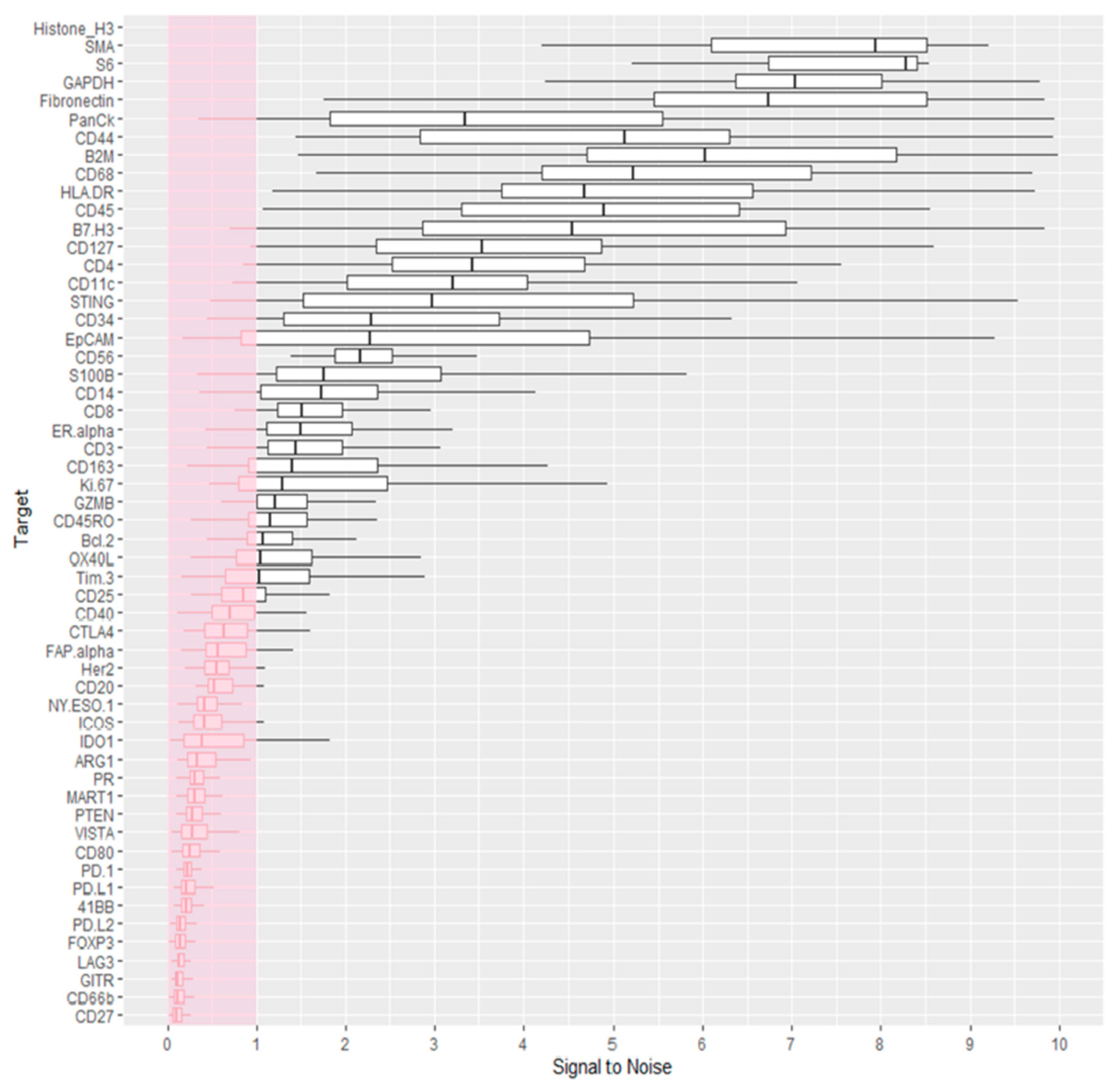
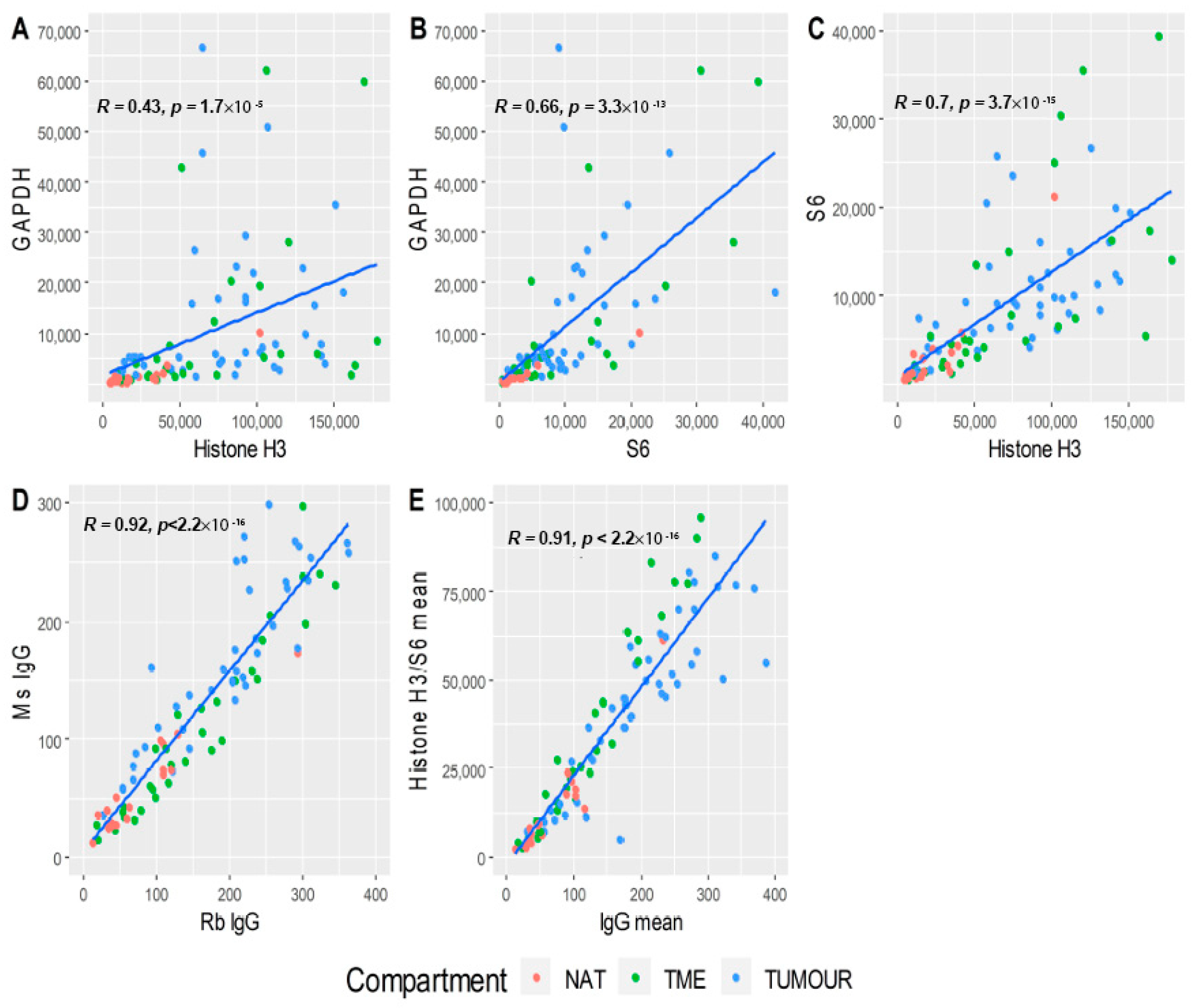
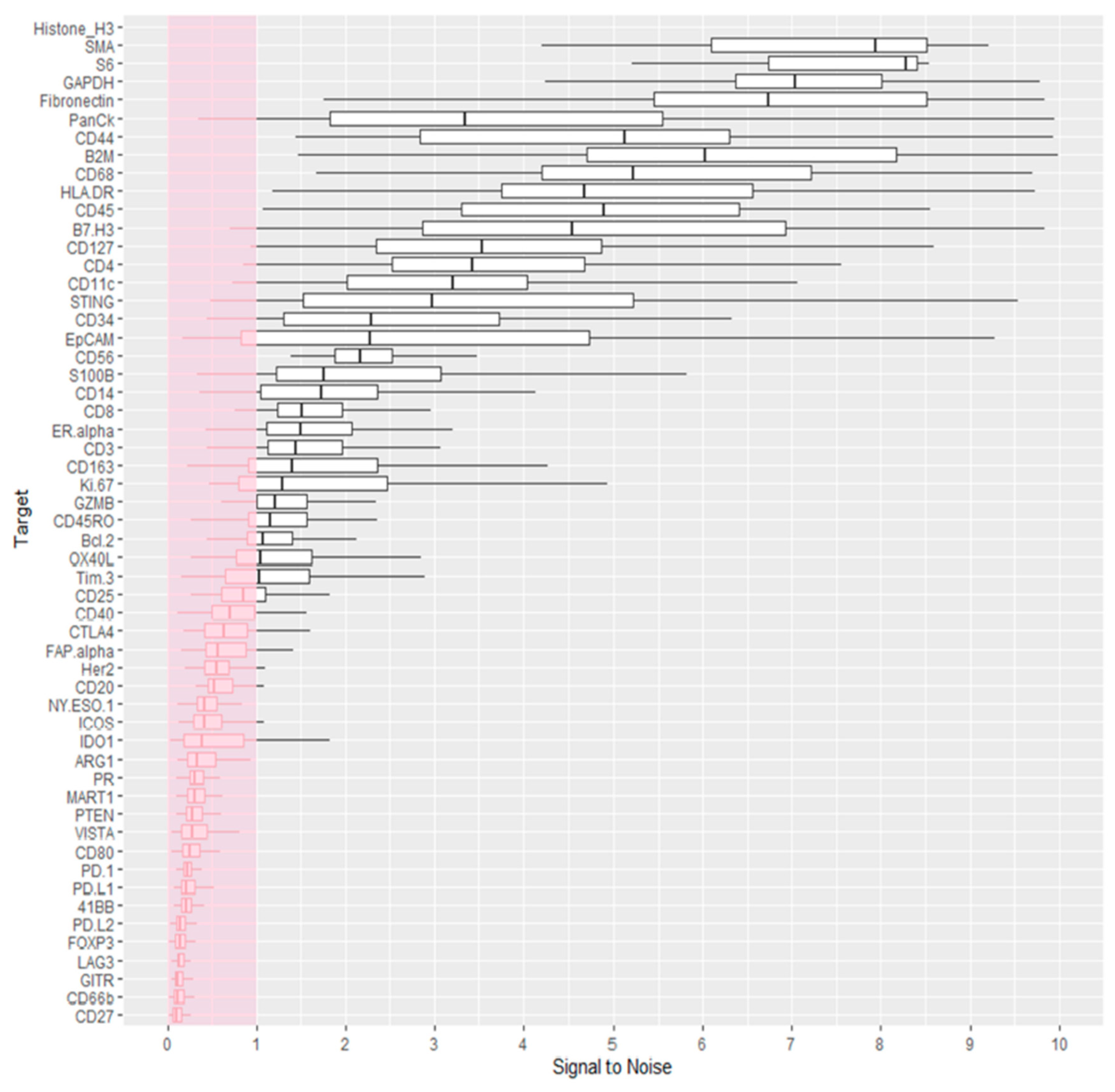
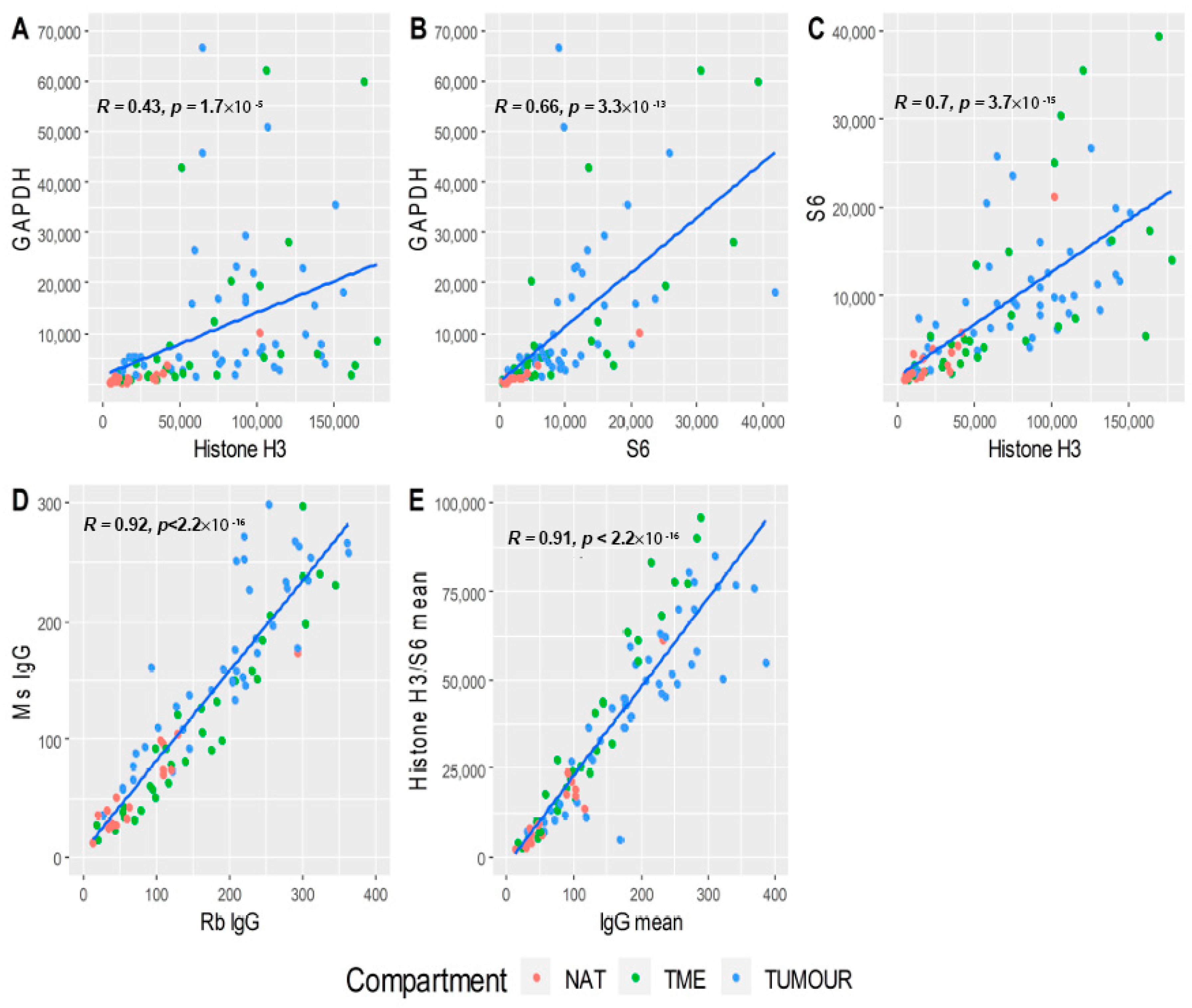
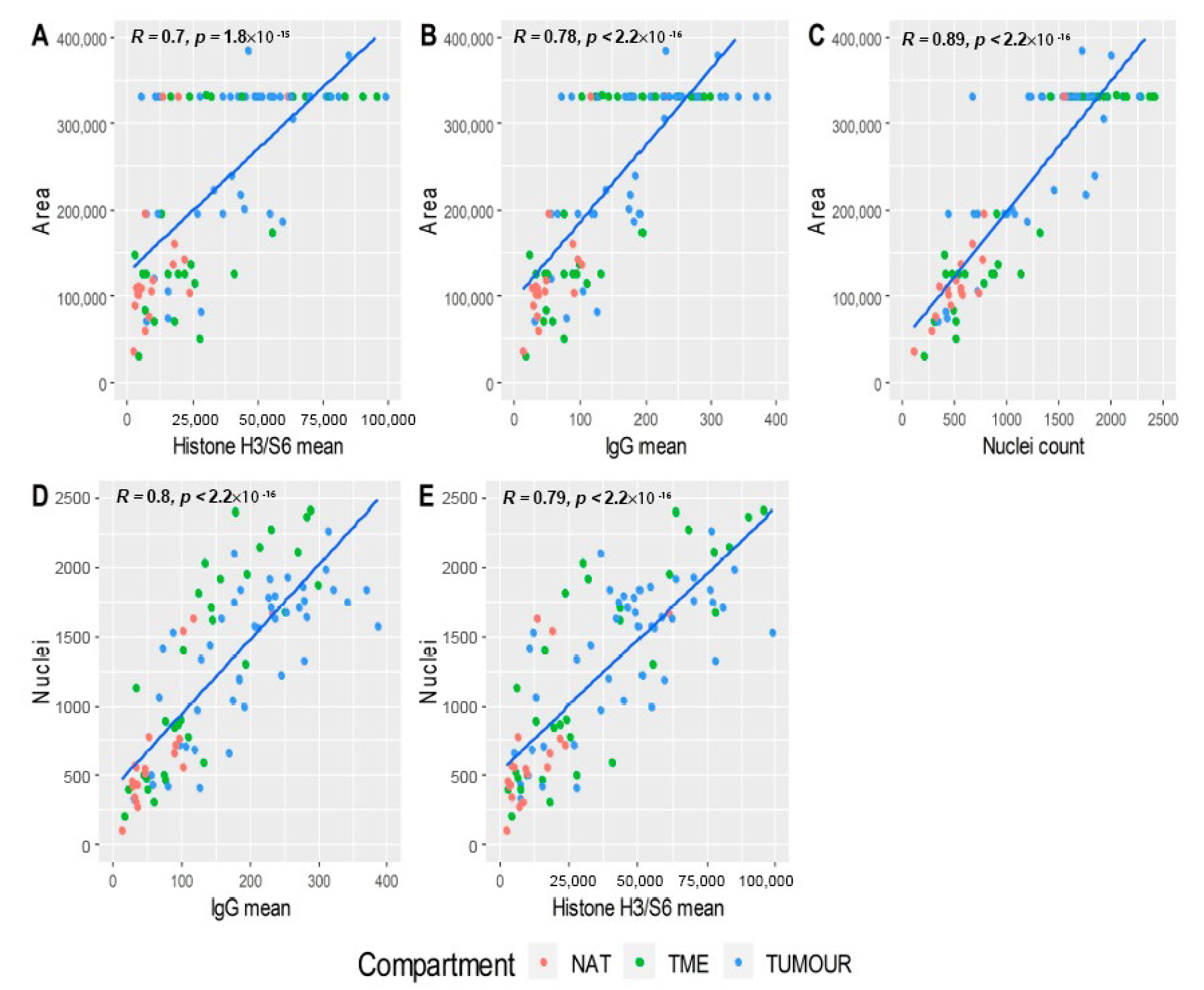
3.2. Data Quality Control
3.3. Data Normalisation
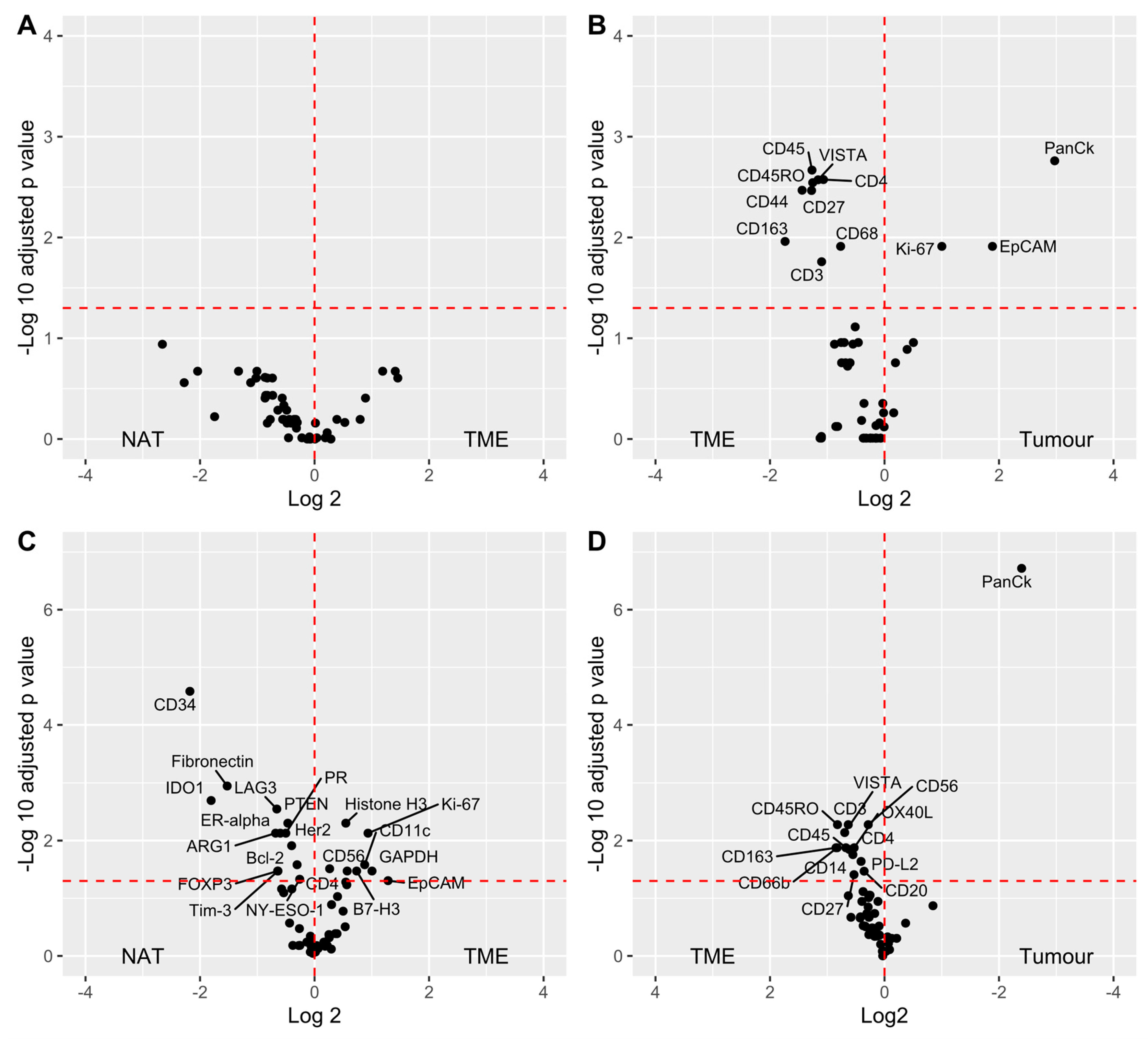
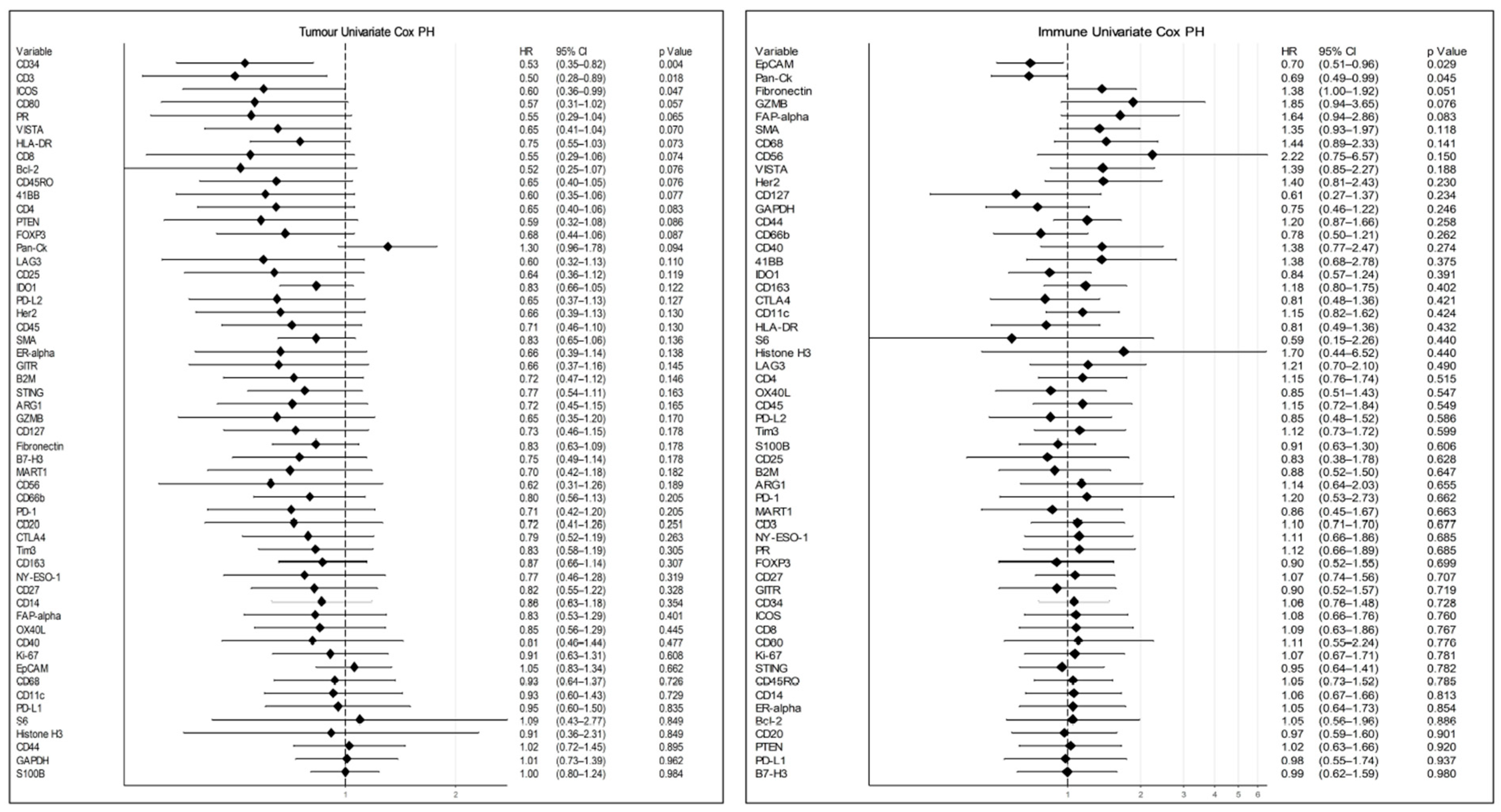
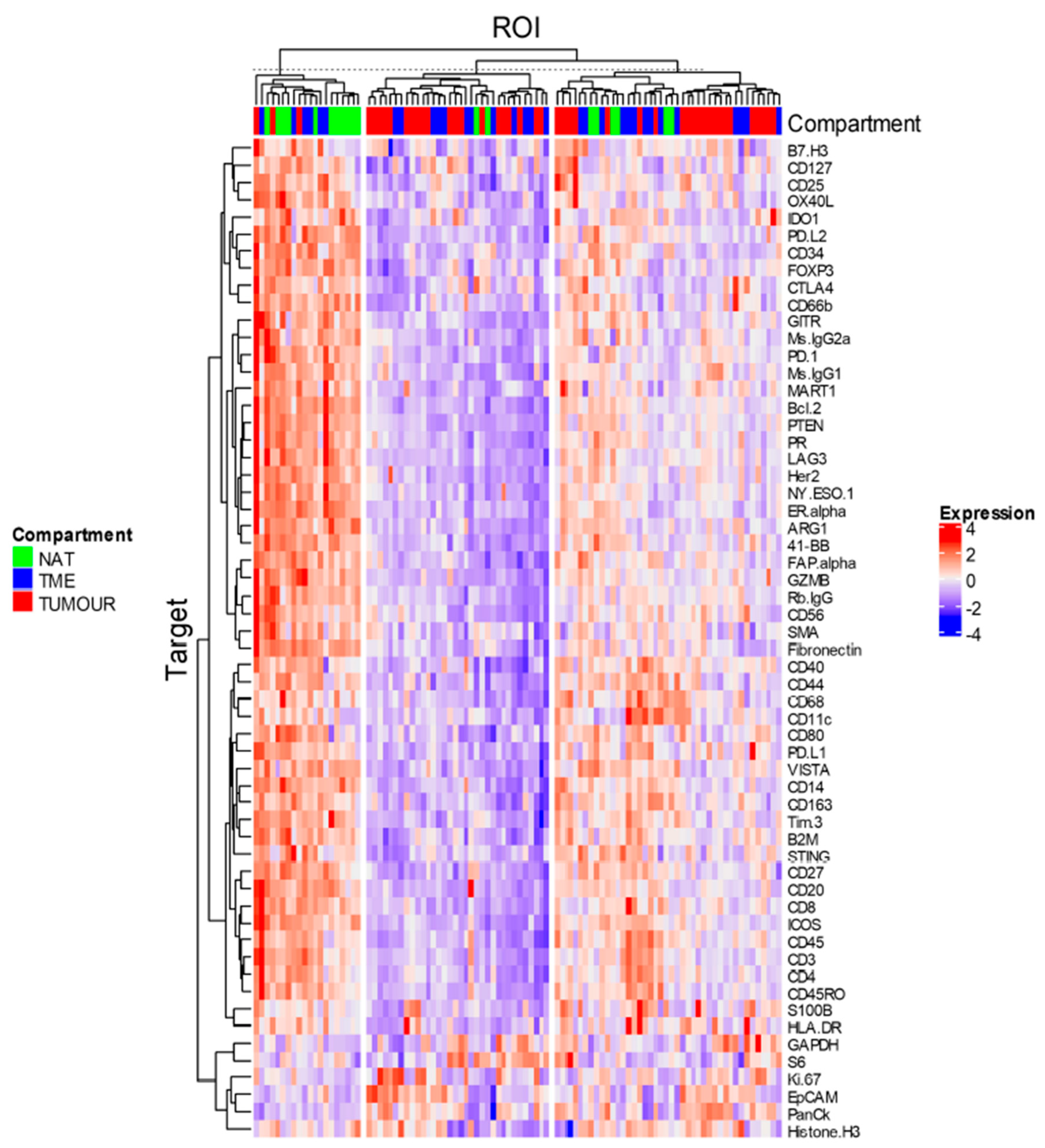
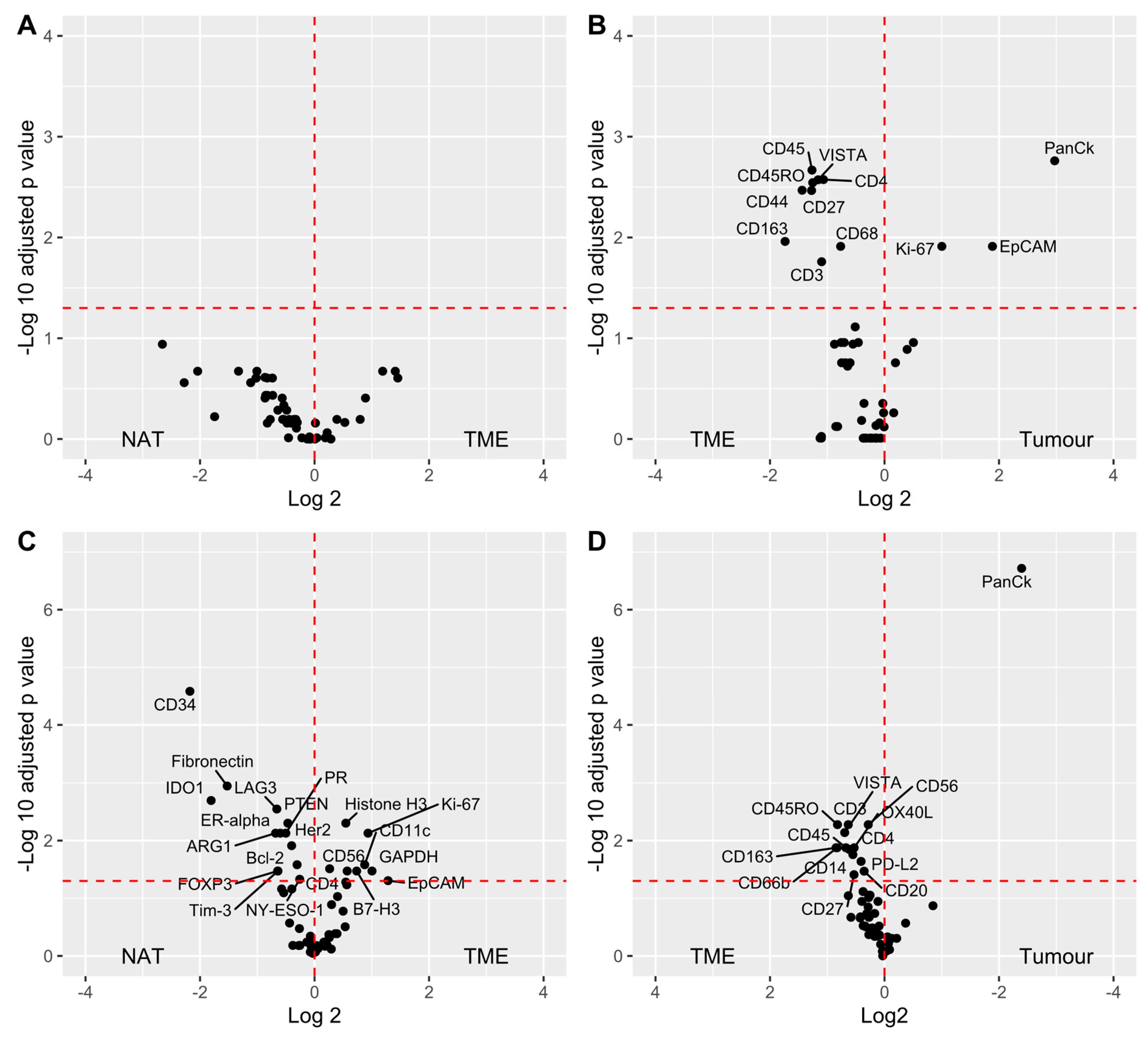
3.4. Data Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, W.; Yu, X.; Mao, Q.; Xia, W.; Wang, A.; Dong, G.; Chen, B.; Ma, W.; Xu, L.; Jiang, F. Improvement of survival for non-small cell lung cancer over time. OncoTargets Ther. 2017, 10, 4295–4303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulasinghe, A.; Kapeleris, J.; Cooper, C.; Warkiani, M.E.; O’Byrne, K.J.; Punyadeera, C. Phenotypic Characterization of Circulating Lung Cancer Cells for Clinically Actionable Targets. Cancers 2019, 11, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punyadeera, C.; Lim, Y.; Kapeleris, J.; Warkiani, M.E.; O’Byrne, K.J.; Punyadeera, C. The Use of Three-Dimensional DNA Fluorescent in Situ Hybridization (3D DNA FISH) for the Detection of Anaplastic Lymphoma Kinase (ALK) in Non-Small Cell Lung Cancer (NSCLC) Circulating Tumor Cells. Cells 2020, 9, 1465. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef]
- O’Callaghan, D.S.; Rexhepaj, E.; Gately, K.; Coate, L.; Delaney, D.; O’Donnell, D.M.; Kay, E.; O’Connell, F.; Gallagher, W.M.; O’Byrne, K.J. Tumour islet Foxp3+ T-cell infiltration predicts poor outcome in nonsmall cell lung cancer. Eur. Respir. J. 2015, 46, 1762–1772. [Google Scholar] [CrossRef] [Green Version]
- Swinson, D.E.; Jones, J.L.; Richardson, D.; Wykoff, C.; Turley, H.; Pastorek, J.; Taub, N.; Harris, A.L.; O’Byrne, K.J. Carbonic anhydrase IX expression, a novel surrogate marker of tumor hypoxia, is associated with a poor prognosis in non-small-cell lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2003, 21, 473–482. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, Y.; Zheng, L.; Zheng, C.; Song, J.; Zhang, Q.; Kang, B.; Liu, Z.; Jin, L.; Xing, R.; et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018, 24, 978–985. [Google Scholar] [CrossRef]
- Welsh, T.J.; Green, R.H.; Richardson, D.; Waller, D.A.; O’Byrne, K.J.; Bradding, P. Macrophage and Mast-Cell Invasion of Tumor Cell Islets Confers a Marked Survival Advantage in Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2005, 23, 8959–8967. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, N.A.; Nguyen, P.; Engle, E.L.; Kaunitz, G.J.; Cottrell, T.R.; Berry, S.; Green, B.; Soni, A.; Cuda, J.D.; Stein, J.E.; et al. Multidimensional, quantitative assessment of PD-1/PD-L1 expression in patients with Merkel cell carcinoma and association with response to pembrolizumab. J. Immunother. Cancer 2018, 6, 99. [Google Scholar] [CrossRef] [PubMed]
- Beechem, J. High-Plex Spatially Resolved RNA and Protein Detection Using Digital Spatial Profiling: A Technology Designed for Immuno-oncology Biomarker Discovery and Translational Research. In Methods in Molecular Biology; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2019; Volume 2055, pp. 563–583. [Google Scholar]
- RStudio Team. RStudio: Integrated Development for R; RStudio: Boston, MA, USA, 2020. [Google Scholar]
- Daniel, D.; Sjoberg, M.H.; Whiting, K.; Zabor, E.C. Gtsummary: Presentation-Ready Data Summary and Analytic Result Tables. 2020. Available online: http://www.danieldsjoberg.com/gtsummary/ (accessed on 27 November 2017).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Kassambara, A. ggpubr: ‘ggplot2′ Based Publication Ready Plots. 2020. Available online: https://rpkgs.datanovia.com/ggpubr/ (accessed on 27 November 2017).
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesweg, M. SurvivalAnalysis: High-Level Interface for Survival Analysis and Associated Plots. 2020. Available online: https://rdrr.io/cran/survivalAnalysis/ (accessed on 27 November 2017).
- Murtagh, F.; Legendre, P. Ward’s hierarchical clustering method: Clustering criterion and agglomerative algorithm. arXiv 2011, arXiv:1111.6285. [Google Scholar]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Nerurkar, S.N.; Goh, D.; Cheung, C.C.L.; Nga, P.Q.; Lim, J.C.; Yeong, J.P. Transcriptional Spatial Profiling of Cancer Tissues in the Era of Immunotherapy: The Potential and Promise. Cancers 2020, 12, 2572. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.L.; Matynia, A.P.; Factor, R.E.; Varley, K.E. Spatially-resolved quantification of proteins in triple negative breast cancers reveals differences in the immune microenvironment associated with prognosis. Sci. Rep. 2020, 10, 6598. [Google Scholar] [CrossRef] [Green Version]
- Zugazagoitia, J.; Gupta, S.; Liu, Y.; Fuhrman, K.; Gettinger, S.; Herbst, R.S.; Schalper, K.A.; Rimm, D.L. Biomarkers Associated with Beneficial PD-1 Checkpoint Blockade in Non-Small Cell Lung Cancer (NSCLC) Identified Using High-Plex Digital Spatial Profiling. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef] [Green Version]
- Toki, M.I.; Merritt, C.R.; Wong, P.F.; Smithy, J.W.; Kluger, H.M.; Syrigos, K.N.; Ong, G.T.; Warren, S.E.; Beechem, J.M.; Rimm, D.L. High-Plex Predictive Marker Discovery for Melanoma Immunotherapy–Treated Patients Using Digital Spatial Profiling. Clin. Cancer Res. 2019, 25, 5503. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Liu, J.; Liu, D.; Liu, B.; Wang, M.; Hu, Z.; Du, X.; Tang, L.; He, F. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. 2014, 74, 3418–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Koblish, H.K.; Horton, B.; Scherle, P.A.; Newton, R.; Gajewski, T.F. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J. Immunother. Cancer 2014, 2, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Controls | Immune Cell Profiling | IO Drug Target | Immune Activation Status | Immune Cell Typing | Pan-Tumour Module |
|---|---|---|---|---|---|
| Rb IgG | PD-1 | 4-1BB | CD127 | CD45RO | MART1 |
| Ms IgG1 | CD68 | LAG3 | CD25 | FOXP3 | NY-ESO-1 |
| Ms IgG2a | HLA-DR | OX40L | CD80 | CD34 | S100B |
| Histone H3 | Ki-67 | Tim-3 | ICOS | CD66b | Bcl-2 |
| S6 | Beta-2M | VISTA | PD-L2 | FAP-alpha | EpCAM |
| GAPDH | CD11c | ARG1 | CD40 | CD14 | Her2 |
| CD20 | B7-H3 | CD44 | CD163 | PTEN | |
| CD3 | IDO1 | CD27 | ER-alpha | ||
| CD4 | STING | PR | |||
| CD45 | GITR | ||||
| CD56 | |||||
| CD8 | |||||
| CTLA4 | |||||
| GZMB | |||||
| PD-L1 | |||||
| PanCk | |||||
| SMA | |||||
| Fibronectin |
| Characteristic | Overall, n = 96 | NAT, n = 19 1 | TME, n = 32 1 | Tumour, n = 45 1 | p-Value 2 |
|---|---|---|---|---|---|
| Age | 62 (54, 69) | 66 (56, 69) | 60 (54, 67) | 62 (54, 71) | 0.6 |
| Sex | 0.7 | ||||
| F | 42 (44%) | 9 (47%) | 12 (38%) | 21 (47%) | |
| M | 54 (56%) | 10 (53%) | 20 (62%) | 24 (53%) | |
| t | |||||
| 0 | 19 (20%) | 19 (100%) | 0 (0%) | 0 (0%) | |
| 1 | 24 (25%) | 0 (0%) | 11 (34%) | 13 (29%) | |
| 2 | 34 (35%) | 0 (0%) | 15 (47%) | 19 (42%) | |
| 3 | 12 (12%) | 0 (0%) | 4 (12%) | 8 (18%) | |
| 4 | 7 (7.3%) | 0 (0%) | 2 (6.2%) | 5 (11%) | |
| n | 0.2 | ||||
| 0 | 78 (81%) | 19 (100%) | 23 (72%) | 36 (80%) | |
| 1 | 13 (14%) | 0 (0%) | 6 (19%) | 7 (16%) | |
| 2 | 3 (3.1%) | 0 (0%) | 2 (6.2%) | 1 (2.2%) | |
| 3 | 2 (2.1%) | 0 (0%) | 1 (3.1%) | 1 (2.2%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monkman, J.; Taheri, T.; Ebrahimi Warkiani, M.; O’Leary, C.; Ladwa, R.; Richard, D.; O’Byrne, K.; Kulasinghe, A. High-Plex and High-Throughput Digital Spatial Profiling of Non-Small-Cell Lung Cancer (NSCLC). Cancers 2020, 12, 3551. https://doi.org/10.3390/cancers12123551
Monkman J, Taheri T, Ebrahimi Warkiani M, O’Leary C, Ladwa R, Richard D, O’Byrne K, Kulasinghe A. High-Plex and High-Throughput Digital Spatial Profiling of Non-Small-Cell Lung Cancer (NSCLC). Cancers. 2020; 12(12):3551. https://doi.org/10.3390/cancers12123551
Chicago/Turabian StyleMonkman, James, Touraj Taheri, Majid Ebrahimi Warkiani, Connor O’Leary, Rahul Ladwa, Derek Richard, Ken O’Byrne, and Arutha Kulasinghe. 2020. "High-Plex and High-Throughput Digital Spatial Profiling of Non-Small-Cell Lung Cancer (NSCLC)" Cancers 12, no. 12: 3551. https://doi.org/10.3390/cancers12123551
APA StyleMonkman, J., Taheri, T., Ebrahimi Warkiani, M., O’Leary, C., Ladwa, R., Richard, D., O’Byrne, K., & Kulasinghe, A. (2020). High-Plex and High-Throughput Digital Spatial Profiling of Non-Small-Cell Lung Cancer (NSCLC). Cancers, 12(12), 3551. https://doi.org/10.3390/cancers12123551