The Small GTPase RAC1B: A Potent Negative Regulator of-and Useful Tool to Study-TGFβ Signaling

 ,
,  ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Basics of TGFβ Signaling
3. Deregulation of TGFβ Signaling in Cancer Cells
4. Interaction of RAC1 with TGFβ Signaling
4.1. Receptors
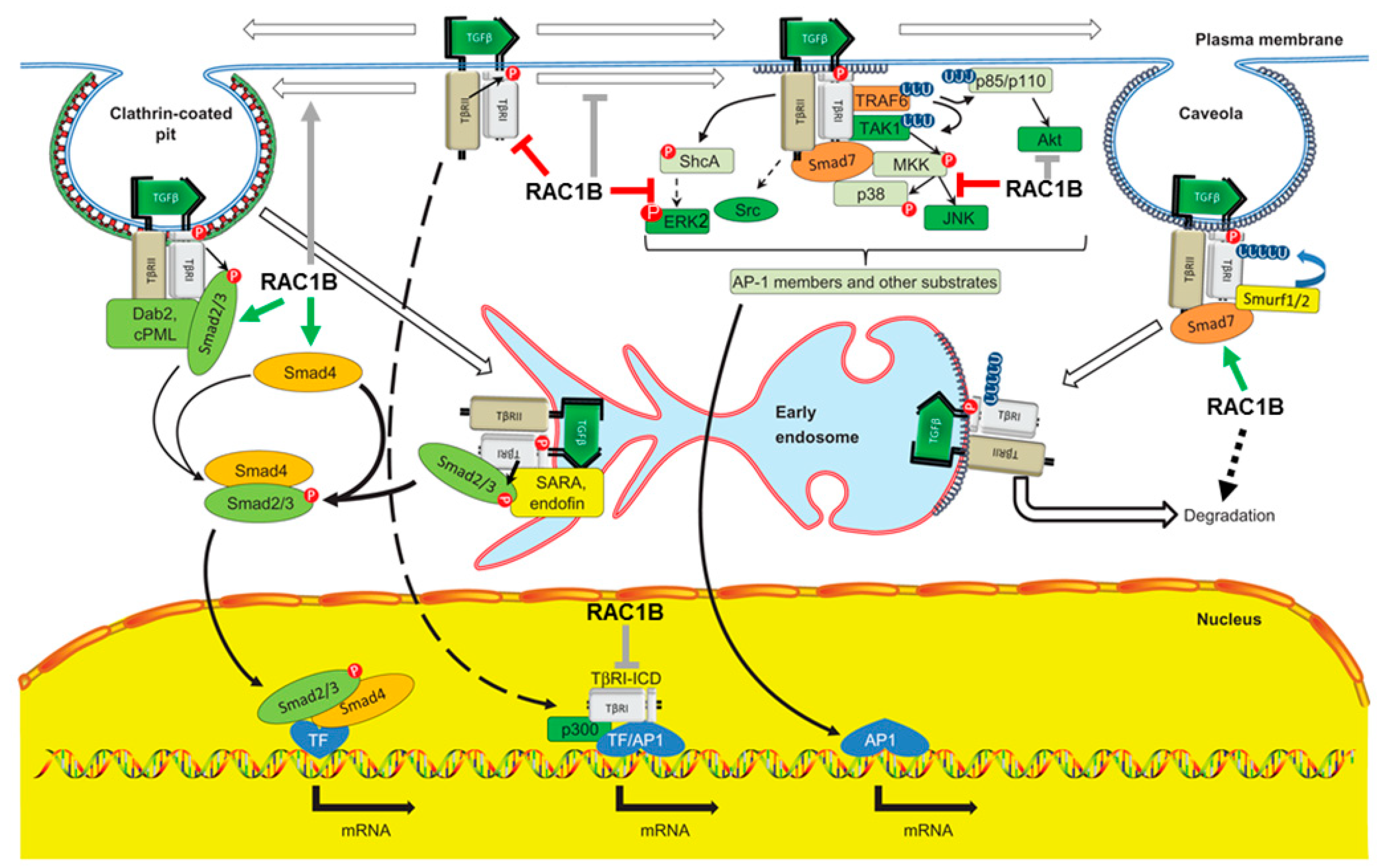
4.1.1. The TGFβ Type I Receptor ALK5
- (i)
- RAC1B inhibits autoinduction of TGFBR1 mRNA by TGFβ1. This became apparent only after knock-down or knock-out of RAC1B when ALK5 mRNA abundance increased in a time-dependent manner in response to stimulation with recombinant human TGFβ1 (rhTGFβ1) [73]. These data are compatible with TGFBR1 being a TFβ response gene [26,35].
- (ii)
- RAC1B inhibits proteins required to sustain ALK5 protein expression. We have previously shown that proteinase-activated receptor 2 (PAR2) encoded by F2RL1 was required for TGFβ1 signaling by its ability to sustain protein expression of ALK5 by an as-yet-unknown mechanism [78]. The combined knock-down in the PDAC cell line, Panc1, of RAC1B and PAR2 relieved the stimulatory effect of RAC1B single knock-down on the abundance of endogenous ALK5 mRNA and migratory activity, indicating that PAR2 is involved in mediating the suppressive effect of RAC1B on ALK5 and ALK5-dependent cell migration. Conversely, F2RL1 itself is a TGFβ target gene [79] and its mRNA expression was induced by treatment of cells with rhTGFβ. Consequently, the RAC1B knock-down-induced rise in PAR2 mRNA was likely the result of derepressed ALK5 levels activated by autocrine TGFβ, since PAR2 upregulation was relieved by co-transfection of RAC1B small interfering RNA (siRNA) with ALK5 siRNA [79]. This indicated that RAC1B disrupts an autoregulatory feed-forward loop between ALK5 and PAR2.
- (iii)
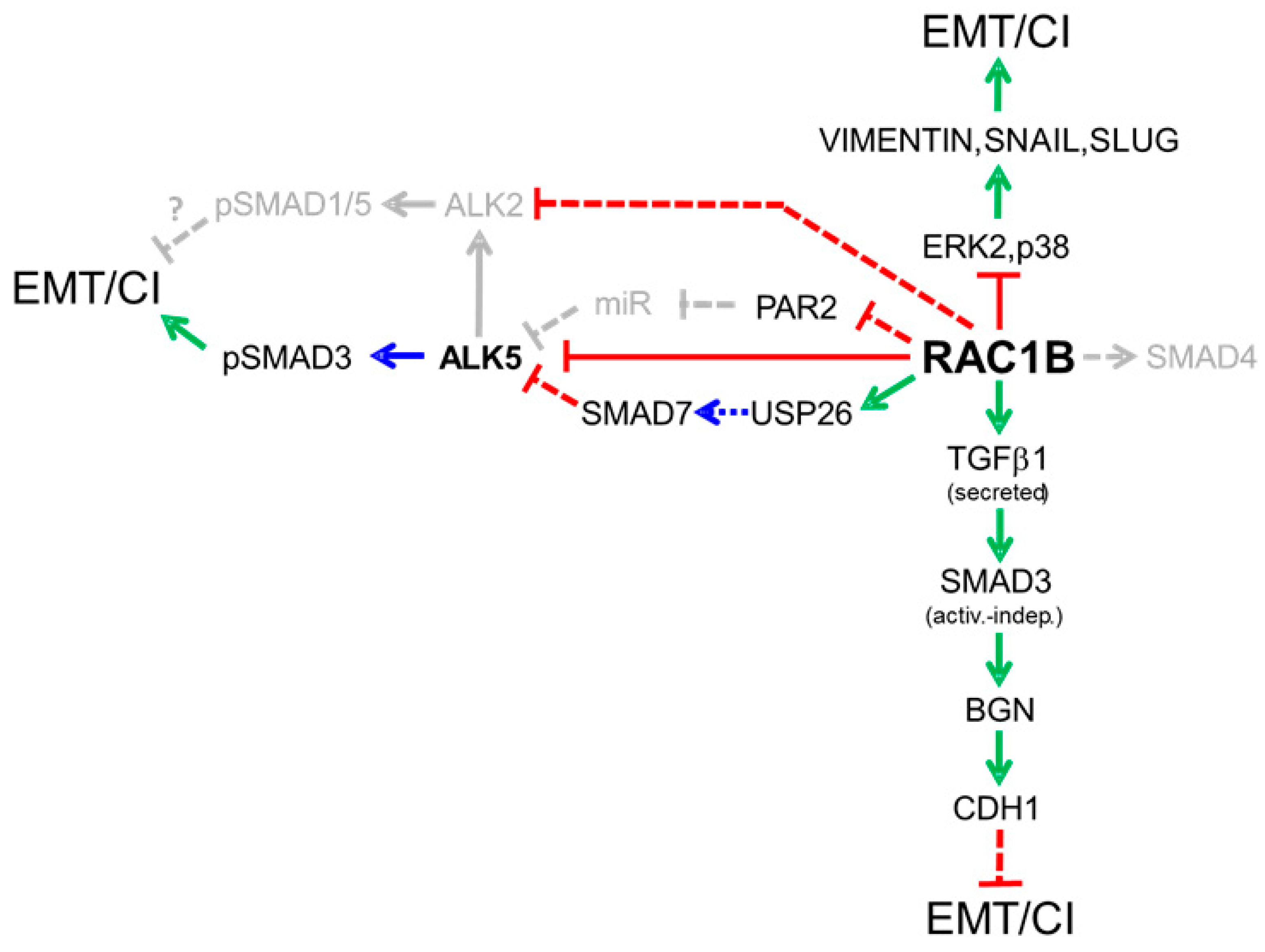
- RAC1B promotes ALK5 protein degradation via induction of SMAD7. Activated ALK5 is known to recruit SMAD7 in a complex with the E3 ligase Smurf2 to promote its internalization and eventual proteasomal degradation in order to terminate TGFβ signaling. Earlier, we demonstrated that the suppressive effect of RAC1B on ALK5 was dependent on SMAD7, and further that RAC1B upregulated protein expression of SMAD7 via intermittent induction of USP26 (see Section 4.2.1) [84].
4.1.2. Other Type I Receptors Involved in TGFβ Signaling
4.2. RAC1B Differentially Affects the Expression or Function of SMAD and MAPKs
4.2.1. Smad Proteins
- (i)
- SMAD2/3. Due to suppression of ALK5 and its kinase activity by RAC1B, C-terminal serine phosphorylation of SMAD2 and SMAD3 was concomitantly reduced [9]. However, in Panc1-RAC1B knock-out cells, we surprisingly detected lower levels of C-terminally phosphorylated SMAD3 (pSMAD3C) [73], although we expected the opposite as a result of derepression of ALK5. This was a puzzling observation before we realized that the abundance of total SMAD3 protein was dramatically decreased in RAC1B-depleted cells. Interestingly, in the same cells, SMAD2 expression remained unaffected, revealing that RAC1B selectively promoted SMAD3 expression.
- (ii)
- SMAD1/5. Preliminary data from Panc1 cells indicate that pSMAD1/5C levels in response to TGFβ1 stimulation increase much more strongly in cells in which RAC1 exon 3b has been deleted [85]. As outlined above, this likely reflects derepression of ALK5 and/or ALK2 and the associated increase in their kinase activities. In the murine cells, Smad1/5, in addition to Smad3 signaling, were also required for TGFβ-induced downregulation of the epithelial marker genes Cdh1 and Tjp1, and upregulation of the mesenchymal marker genes Acta2 and Fn1, together being indicative of EMT [29].
- (iii)
- SMAD4. In Panc1 cells we observed that following siRNA-mediated knock-down of RAC1B, SMAD4 protein expression was reduced [85]. This suggests that RAC1B coordinately drives the expression of both SMAD3 and SMAD4 in pancreatic epithelial cells. Mechanistically, SMAD4 upregulation may occur through transcriptional activation of DPC4 or a decrease in SMAD4 ubiquitination [88].
- (vi)
- SMAD7. In a recent study, we have shown that i) RAC1B promotes the expression of SMAD7 and ii) SMAD7 mediates the suppressive effect of RAC1B on ALK5 protein and its associated kinase activity [84]. We further revealed that upregulation of SMAD7 by RAC1B requires the rapid transcriptional induction of USP26 [84]. The involvement of USP26 strongly suggests that RAC1B increases SMAD7 protein stability by reducing the rate of proteasomal degradation; however, a direct demonstration of RAC1B-induced SMAD7 deubiquitination in pancreatic cells needs experimental verification. Of note, USP26 has been identified as a potent negative regulator of TGFβ signaling in breast cancer and glioma cells [28]. Our demonstration of RAC1B transcriptionally inducing USP26 in Panc1 cells, therefore, provides strong evidence in favor of RAC1B being a TGFβ antagonist in pancreatic epithelial cells. Moreover, since SMAD7 has been implicated in the inhibition of EMT and maintenance of the epithelial phenotype [89], its identification as a RAC1B target gene is a significant observation in light of the strong expression of RAC1B in PDAC cell lines of the epithelial/classical subtype [12,13]. RAC1B’s ability to promote USP26 and SMAD7 expression may thus have a crucial role in inhibiting EMT and promoting mesenchymal–epithelial transition (MET).
4.2.2. MAPKs
4.3. RAC1B Enhances Expression and Secretion of Autocrine TGFβ1: A Possible Role in Tumor Suppression
4.4. RAC1B Favors the Expression of Extracellular TGFβ Inhibitors
4.5. Other Possible but Still Hypothetical Targets
5. How Can the Differential Interactions of RAC1B with TGFβ Signaling Be Integrated with the Proposed Role of RAC1B as a Tumor Suppressor?
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Svensmark, J.H.; Brakebusch, C. Rho GTPases in cancer: Friend or foe? Oncogene 2019, 38, 7447–7456. [Google Scholar] [CrossRef]
- Bokoch, G.M. Regulation of innate immunity by Rho GTPases. Trends Cell Biol. 2005, 15, 163–171. [Google Scholar] [CrossRef]
- Kotelevets, L.; Chastre, E. Rac1 Signaling: From Intestinal Homeostasis to Colorectal Cancer Metastasis. Cancers 2020, 12, 665. [Google Scholar] [CrossRef]
- De, P.; Aske, J.C.; Dey, N. RAC1 Takes the Lead in Solid Tumors. Cells 2019, 8, 382. [Google Scholar] [CrossRef] [PubMed]
- Casado-Medrano, V.; Baker, M.J.; Lopez-Haber, C.; Cooke, M.; Wang, S.; Caloca, M.J.; Kazanietz, M.G. The role of Rac in tumor susceptibility and disease progression: From biochemistry to the clinic. Biochem. Soc. Trans. 2018, 46, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Moreno, V.; Gadea, G.; Ahn, J.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac Activation and Inactivation Control Plasticity of Tumor Cell Movement. Cell 2008, 135, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Alexander, S. Cancer Invasion and the Microenvironment: Plasticity and Reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Navarro-Serer, B.; Jeong, Y.J.; Chianchiano, P.; Xia, L.; Luchini, C.; Veronese, N.; Dowiak, C.; Ng, T.; Trujillo, M.A.; et al. Pattern of Invasion in Human Pancreatic Cancer Organoids Is Associated with Loss of SMAD4 and Clinical Outcome. Cancer Res. 2020, 80, 2804–2817. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Sebens, S.; Giehl, K.; Helm, O.; Groth, S.; Fandrich, F.; Rocken, C.; Sipos, B.; Lehnert, H.; Gieseler, F. Rac1b negatively regulates TGF-beta1-induced cell motility in pancreatic ductal epithelial cells by suppressing Smad signalling. Oncotarget 2014, 5, 277–290. [Google Scholar] [CrossRef]
- Matos, P.; Kotelevets, L.; Jordan, P.; Gonçalves, V.; Henriques, A.F.A.; Zerbib, P.; Moyer, M.P.; Chastre, E. Ibuprofen Inhibits Colitis-Induced Overexpression of TumorRelated Rac1b. Neoplasia 2013, 15, 102–111. [Google Scholar] [CrossRef]
- Kotelevets, L.; Walker, F.; Mamadou, G.; Lehy, T.; Jordan, P.; Chastre, E. The Rac1 splice form Rac1b favors mouse colonic mucosa regeneration and contributes to intestinal cancer progression. Oncogene 2018, 37, 6054–6068. [Google Scholar] [CrossRef] [PubMed]
- Zinn, R.; Otterbein, H.; Lehnert, H.; Ungefroren, H. RAC1B: A Guardian of the Epithelial Phenotype and Protector Against Epithelial-Mesenchymal Transition. Cells 2019, 8, 1569. [Google Scholar] [CrossRef] [PubMed]
- Otterbein, H.; Lehnert, H.; Ungefroren, H. Negative Control of Cell Migration by Rac1b in Highly Metastatic Pancreatic Cancer Cells Is Mediated by Sequential Induction of Nonactivated Smad3 and Biglycan. Cancers 2019, 11, 1959. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4. [Google Scholar] [CrossRef]
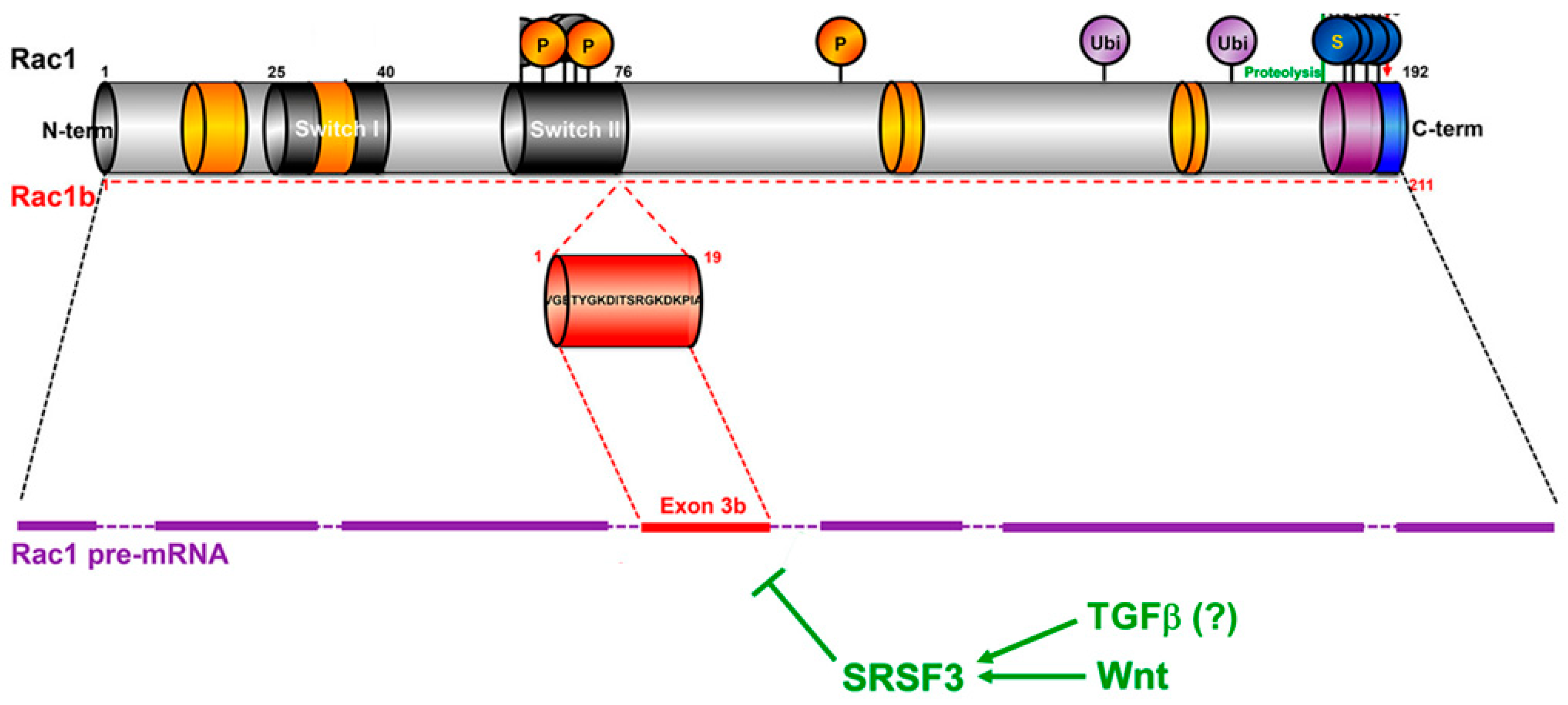
- Gonçalves, V.; Matos, P.; Jordan, P. Antagonistic SR proteins regulate alternative splicing of tumor-related Rac1b downstream of the PI3-kinase and Wnt pathways. Hum. Mol. Genet. 2009, 18, 3696–3707. [Google Scholar] [CrossRef]
- Gonçalves, V.; Henriques, A.F.A.; Pereira, J.F.S.; Costa, A.N.; Moyer, M.P.; Moita, L.F.; Gama-Carvalho, M.; Matos, P.; Jordan, P. Phosphorylation of SRSF1 by SRPK1 regulates alternative splicing of tumor-related Rac1b in colorectal cells. RNA 2014, 20, 474–482. [Google Scholar] [CrossRef]
- Matos, P.; Collard, J.G.; Jordan, P. Tumor-related Alternatively Spliced Rac1b Is Not Regulated by Rho-GDP Dissociation Inhibitors and Exhibits Selective Downstream Signaling. J. Biol. Chem. 2003, 278, 50442–50448. [Google Scholar] [CrossRef]
- Melzer, C.; Hass, R.; Lehnert, H.; Ungefroren, H. RAC1B: A Rho GTPase with Versatile Functions in Malignant Transformation and Tumor Progression. Cells 2019, 8, 21. [Google Scholar] [CrossRef]
- Matos, P.; Jordan, P. Increased Rac1b Expression Sustains Colorectal Tumor Cell Survival. Mol. Cancer Res. 2008, 6, 1178–1184. [Google Scholar] [CrossRef]
- Matos, P.; Jordan, P. Rac1, but Not Rac1B, Stimulates RelB-mediated Gene Transcription in Colorectal Cancer Cells. J. Biol. Chem. 2006, 281, 13724–13732. [Google Scholar] [CrossRef]
- Lozano, E.; Frasa, M.A.M.; Smolarczyk, K.; Knaus, U.G.; Braga, V.M. PAK is required for the disruption of E-cadherin adhesion by the small GTPase Rac. J. Cell Sci. 2008, 121, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Orlichenko, L.; Geyer, R.; Yanagisawa, M.; Khauv, D.; Radisky, E.S.; Anastasiadis, P.Z.; Radisky, D.C. The 19-Amino Acid Insertion in the Tumor-associated Splice Isoform Rac1b Confers Specific Binding to p120 Catenin. J. Biol. Chem. 2010, 285, 19153–19161. [Google Scholar] [CrossRef] [PubMed]
- Esufali, S.; Charames, G.S.; Pethe, V.V.; Buongiorno, P.; Bapat, B. Activation of Tumor-Specific Splice Variant Rac1b by Dishevelled Promotes Canonical Wnt Signaling and Decreased Adhesion of Colorectal Cancer Cells. Cancer Res. 2007, 67, 2469–2479. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2020, 1–26. [Google Scholar] [CrossRef]
- Tang, J.; Gifford, C.C.; Samarakoon, R.; Higgins, P.J. Deregulation of Negative Controls on TGF-β1 Signaling in Tumor Progression. Cancers 2018, 10, 159. [Google Scholar] [CrossRef]
- Kit Leng Lui, S.; Iyengar, P.V.; Jaynes, P.; Isa, Z.; Pang, B.; Tan, T.Z.; Eichhorn, P.J.A. USP 26 regulates TGF-beta signaling by deubiquitinating and stabilizing SMAD 7. EMBO Rep. 2017, 18, 797–808. [Google Scholar] [CrossRef]
- Ramachandran, A.; Vizán, P.; Das, D.; Chakravarty, P.; Vogt, J.; Rogers, K.W.; Müller, P.; Hinck, A.P.; Sapkota, G.P.; Hill, C.S. TGF-β uses a novel mode of receptor activation to phosphorylate SMAD1/5 and induce epithelial-to-mesenchymal transition. Elife 2018, 7, e31756. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef]
- Yakymovych, I.; Yakymovych, M.; Heldin, C.-H. Intracellular trafficking of transforming growth factor β receptors. Acta Biochim. Biophys. Sin. 2018, 50, 3–11. [Google Scholar] [CrossRef] [PubMed]
- McLean, S.; Di Guglielmo, G.M. TGF beta (transforming growth factor beta) receptor type III directs clathrin-mediated endocytosis of TGF beta receptor types I and II. Biochem. J. 2010, 429, 137–145. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Yan, X.; Li, N.; Dang, S.; Xu, L.; Zhao, B.; Li, Z.; Lv, Z.; Fang, X.; Zhang, Y.; et al. Internalization of the TGF-β type I receptor into caveolin-1 and EEA1 double-positive early endosomes. Cell Res. 2015, 25, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Margiotta, A.; Bucci, C. Coordination between Rac1 and Rab Proteins: Functional Implications in Health and Disease. Cells 2019, 8, 396. [Google Scholar] [CrossRef]
- Duan, D.; Derynck, R. Transforming growth factor–β (TGF-β)–induced up-regulation of TGF-β receptors at the cell surface amplifies the TGF-β response. J. Biol. Chem. 2019, 294, 8490–8504. [Google Scholar] [CrossRef]
- Vizán, P.; Miller, D.; Gori, I.; Das, D.; Schmierer, B.; Hill, C.S. Controlling Long-Term Signaling: Receptor Dynamics Determine Attenuation and Refractory Behavior of the TGF- Pathway. Sci. Signal. 2013, 6, ra106. [Google Scholar] [CrossRef]
- Horiguchi, K.; Sakamoto, K.; Koinuma, D.; Semba, K.; Inoue, A.; Inoue, S.; Fujii, H.; Yamaguchi, A.; Miyazawa, K.; Miyazono, K.; et al. TGF-β drives epithelial-mesenchymal transition through δEF1-mediated downregulation of ESRP. Oncogene 2012, 31, 3190–3201. [Google Scholar] [CrossRef]
- Hallgren, O.; Malmström, J.; Malmström, L.; Andersson-Sjöland, A.; Wildt, M.; Tufvesson, E.; Juhasz, P.; Marko-Varga, G.; Westergren-Thorsson, G. Splicosomal and serine and arginine-rich splicing factors as targets for TGF-β. Fibrogenesis Tissue Repair 2012, 5, 6. [Google Scholar] [CrossRef]
- Ritterhouse, L.L.; Wu, E.Y.; Kim, W.G.; Dillon, D.A.; Hirsch, M.S.; Sholl, L.M.; Agoston, A.T.; Setia, N.; Lauwers, G.Y.; Park, D.Y.; et al. Loss of SMAD4 protein expression in gastrointestinal and extra-gastrointestinal carcinomas. Histopathology 2019, 75, 546–551. [Google Scholar] [CrossRef]
- Descargues, P.; Sil, A.K.; Sano, Y.; Korchynskyi, O.; Han, G.; Owens, P.; Wang, X.J.; Karin, M. IKKalpha is a critical coregulator of a Smad4-independent TGFbeta-Smad2/3 signaling pathway that controls keratinocyte differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 2487–2492. [Google Scholar] [CrossRef]
- Levy, L.; Hill, C.S. Smad4 Dependency Defines Two Classes of Transforming Growth Factor β (TGF-β) Target Genes and Distinguishes TGF-β-Induced Epithelial-Mesenchymal Transition from Its Antiproliferative and Migratory Responses. Mol. Cell. Biol. 2005, 25, 8108–8125. [Google Scholar] [CrossRef] [PubMed]
- Whittle, M.C.; Izeradjene, K.; Rani, P.G.; Feng, L.; Carlson, M.A.; DelGiorno, K.E.; Wood, L.D.; Goggins, M.; Hruban, R.H.; Chang, A.E.; et al. RUNX3 Controls a Metastatic Switch in Pancreatic Ductal Adenocarcinoma. Cell 2015, 161, 1345–1360. [Google Scholar] [CrossRef] [PubMed]
- Suriyamurthy, S.; Baker, D.; Dijke, P.T.; Iyengar, P.V. Epigenetic Reprogramming of TGF-β Signaling in Breast Cancer. Cancers 2019, 11, 726. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, M.; Doody, J.; Timokhina, I.; Massagué, J. A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev. 1999, 13, 804–816. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, N.; Lee, C. Vicious cycle of TGF-β signaling in tumor progression and metastasis. Am. J. Clin. Exp. Urol. 2014, 2, 149–155. [Google Scholar]
- Meriane, M.; Charrasse, S.; Comunale, F.; Gauthier-Rouvière, C. Transforming growth factor beta activates Rac1 and Cdc42Hs GTPases and the JNK pathway in skeletal muscle cells. Biol. Cell 2002, 94, 535–543. [Google Scholar] [CrossRef]
- Kim, H.P.; Kim, T.Y.; Lee, M.S.; Jong, H.S.; Kim, T.Y.; Lee, J.W.; Bang, Y.J. TGF-beta1-mediated activations of c-Src and Rac1 modulate levels of cyclins and p27(Kip1) CDK inhibitor in hepatoma cells replated on fibronectin. Biochim. Biophys. Acta 2005, 1743, 151–161. [Google Scholar] [CrossRef]
- Groth, S.; Schulze, M.; Kalthoff, H.; Fändrich, F.; Ungefroren, H. Adhesion and Rac1-dependent regulation of biglycan gene expression by transforming growth factor-beta. Evidence for oxidative signaling through NADPH oxidase. J. Biol. Chem. 2005, 280, 33190–33199. [Google Scholar] [CrossRef]
- Woods, A.; Pala, D.; Kennedy, L.; McLean, S.; Rockel, J.S.; Wang, G.; Leask, A.; Beier, F. Rac1 signaling regulates CTGF/CCN2 gene expression via TGFbeta/Smad signaling in chondrocytes. Osteoarthritis Cartil. 2009, 17, 406–413. [Google Scholar] [CrossRef]
- Hubchak, S.C.; Sparks, E.E.; Hayashida, T.; Schnaper, H.W. Rac1 promotes TGF-beta-stimulated mesangial cell type I collagen expression through a PI3K/Akt-dependent mechanism. Am. J. Physiol. Ren. Physiol. 2009, 297, F1316–F1323. [Google Scholar] [CrossRef]
- Santibanez, J.F.; Kocić, J.; Fabra, A.; Cano, A.; Quintanilla, M. Rac1 modulates TGF-β1-mediated epithelial cell plasticity and MMP9 production in transformed keratinocytes. FEBS Lett. 2010, 584, 2305–2310. [Google Scholar] [CrossRef] [PubMed]
- Varon, C.; Basoni, C.; Reuzeau, E.; Moreau, V.; Kramer, I.J.; Génot, E. TGFbeta1-induced aortic endothelial morphogenesis requires signaling by small GTPases Rac1 and RhoA. Exp. Cell Res. 2006, 312, 3604–3619. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Wang, S.; Dumont, N.; Yi, J.Y.; Koh, Y.; Arteaga, C.L. Overexpression of HER2 (erbB2) in Human Breast Epithelial Cells Unmasks Transforming Growth Factor β-induced Cell Motility. J. Biol. Chem. 2004, 279, 24505–24513. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.E.; Yu, Y.; Criswell, T.L.; DeBusk, L.M.; Lin, P.C.; Zent, R.; Johnson, D.H.; Ren, X.; Arteaga, C.L. Oncogenic mutations regulate tumor microenvironment through induction of growth factors and angiogenic mediators. Oncogene 2010, 29, 3335–3348. [Google Scholar] [CrossRef]
- Shen, H.J.; Sun, Y.H.; Zhang, S.J.; Jiang, J.X.; Dong, X.W.; Jia, Y.L.; Shen, J.; Guan, Y.; Zhang, L.H.; Li, F.F.; et al. Cigarette smoke-induced alveolar epithelial–mesenchymal transition is mediated by Rac1 activation. Biochim. Biophys. Acta 2014, 1840, 1838–1849. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, B.; You, W.; Li, P.; Kuang, Y. Rab23 Promotes Hepatocellular Carcinoma Cell Migration Via Rac1/TGF-β Signaling. Pathol. Oncol. Res. 2020, 26, 301–306. [Google Scholar] [CrossRef]
- Ungefroren, H.; Groth, S.; Sebens, S.; Lehnert, H.; Gieseler, F.; Fändrich, F. Differential roles of Smad2 and Smad3 in the regulation of TGF-β1-mediated growth inhibition and cell migration in pancreatic ductal adenocarcinoma cells: Control by Rac1. Mol. Cancer 2011, 10, 67. [Google Scholar] [CrossRef]
- Chiu, C.; Maddock, D.A.; Zhang, Q.; Souza, K.P.; Townsend, A.R.; Wan, Y. TGF-beta-induced p38 activation is mediated by Rac1-regulated generation of reactive oxygen species in cultured human keratinocytes. Int. J. Mol. Med. 2001, 8, 251–255. [Google Scholar]
- Ungefroren, H.; Lenschow, W.; Chen, W.B.; Faendrich, F.; Kalthoff, H. Regulation of Biglycan Gene Expression by Transforming Growth Factor-β Requires MKK6-p38 Mitogen-activated Protein Kinase Signaling Downstream of Smad Signaling. J. Biol. Chem. 2003, 278, 11041–11049. [Google Scholar] [CrossRef]
- Patel, S.; Tang, J.; Overstreet, J.M.; Anorga, S.; Lian, F.; Arnouk, A.; Goldschmeding, R.; Higgins, P.J.; Samarakoon, R. Rac-GTPase promotes fibrotic TGF-β1 signaling and chronic kidney disease via EGFR, p53, and Hippo/YAP/TAZ pathways. FASEB J. 2019, 33, 9797–9810. [Google Scholar] [CrossRef]
- Varon, C.; Rottiers, P.; Ezan, J.; Reuzeau, E.; Basoni, C.; Kramer, I.; Génot, E. TGFbeta1 regulates endothelial cell spreading and hypertrophy through a Rac-p38-mediated pathway. Biol. Cell. 2008, 100, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Al-Azayzih, A.; Gao, F.; Somanath, P.R. P21 activated kinase-1 mediates transforming growth factor β1-induced prostate cancer cell epithelial to mesenchymal transition. Biochim. Biophys. Acta 2015, 1853, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Su, J.; Zeng, Y.; Ding, E.; Liu, F.; Tan, T.; Xia, H.; Wu, Y.; Zeng, X.; Ling, H.; et al. Diallyl disulfide inhibits TGF-β1-induced upregulation of Rac1 and β-catenin in epithelial-mesenchymal transition and tumor growth of gastric cancer. Oncol. Rep. 2018, 39, 2797–2806. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Li, Y.; Zhao, W.; Hou, F.; Li, X.; Sun, L.; Chen, W.; Yang, A.; Wu, S.; Zhang, B.; et al. PHP14 regulates hepatic stellate cells migration in liver fibrosis via mediating TGF-β1 signaling to PI3Kγ/AKT/Rac1 pathway. J. Mol. Med. 2017, 96, 119–133. [Google Scholar] [CrossRef]
- Witte, D.; Bartscht, T.; Kaufmann, R.; Pries, R.; Settmacher, U.; Lehnert, H.; Ungefroren, H. TGF-β1-induced cell migration in pancreatic carcinoma cells is RAC1 and NOX4-dependent and requires RAC1 and NOX4-dependent activation of p38MAPK. Oncol. Rep. 2017, 38, 3693–3701. [Google Scholar] [CrossRef]
- Binker, M.G.; Binker-Cosen, A.A.; Gaisano, H.Y.; de Cosen, R.H.; Cosen-Binker, L.I. TGF-β1 increases invasiveness of SW1990 cells through Rac1/ROS/NF-κB/IL-6/MMP-2. Biochem. Biophys. Res. Commun. 2011, 405, 140–145. [Google Scholar] [CrossRef]
- Yu, J.R.; Tai, Y.; Jin, Y.; Hammell, M.G.; Wilkinson, J.E.; Roe, J.S.; Vakoc, C.R.; Van Aelst, L. TGF-β/Smad signaling through DOCK4 facilitates lung adenocarcinoma metastasis. Genes Dev. 2015, 29, 250–261. [Google Scholar] [CrossRef]
- Oltean, S.; Bates, D.O. Hallmarks of alternative splicing in cancer. Oncogene 2014, 33, 5311–5318. [Google Scholar] [CrossRef]
- Wang, B.-D.; Lee, N.H. Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef]
- Abdel-Samad, R.; Zalzali, H.; Rammah, C.; Giraud, J.; Naudin, C.; Dupasquier, S.; Poulat, F.; Boizet-Bonhoure, B.; Lumbroso, S.; Mouzat, K.; et al. MiniSOX9, a dominant-negative variant in colon cancer cells. Oncogene 2011, 30, 2493–2503. [Google Scholar] [CrossRef][Green Version]
- Boise, L.H.; González-García, M.; Postema, C.E.; Ding, L.; Lindsten, T.; Turka, L.A.; Mao, X.; Nuñez, G.; Thompson, C.B. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993, 74, 597–608. [Google Scholar] [CrossRef]
- Logotheti, S.; Pavlopoulou, A.; Galtsidis, S.; Vojtesek, B.; Zoumpourlis, V. Functions, divergence and clinical value of TAp73 isoforms in cancer. Cancer Metastasis Rev. 2013, 32, 511–534. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Otterbein, H.; Fiedler, C.; Mihara, K.; Hollenberg, M.D.; Gieseler, F.; Lehnert, H.; Witte, D. RAC1B Suppresses TGF-β1-Dependent Cell Migration in Pancreatic Carcinoma Cells through Inhibition of the TGF-β Type I Receptor ALK5. Cancers 2019, 11, 691. [Google Scholar] [CrossRef] [PubMed]
- Witte, D.; Otterbein, H.; Forster, M.; Giehl, K.; Zeiser, R.; Lehnert, H.; Ungefroren, H. Negative regulation of TGF-beta1-induced MKK6-p38 and MEK-ERK signalling and epithelial-mesenchymal transition by Rac1b. Sci. Rep. 2017, 7, 17313. [Google Scholar] [CrossRef]
- Fu, G.; Ye, G.; Nadeem, L.; Ji, L.; Manchanda, T.; Wang, Y.; Zhao, Y.; Qiao, J.; Wang, Y.L.; Lye, S.; et al. MicroRNA-376c Impairs Transforming Growth Factor-β and Nodal Signaling to Promote Trophoblast Cell Proliferation and Invasion. Hypertension 2013, 61, 864–872. [Google Scholar] [CrossRef]
- Shen, N.; Lin, H.; Wu, T.; Wang, D.; Wang, W.; Xie, H.; Zhang, J.; Feng, Z. Inhibition of TGF-β1-receptor posttranslational core fucosylation attenuates rat renal interstitial fibrosis. Kidney Int. 2013, 84, 64–77. [Google Scholar] [CrossRef]
- Kim, S.Y.; Baek, K.H. TGF-β signaling pathway mediated by deubiquitinating enzymes. Cell. Mol. Life Sci. 2019, 76, 653–665. [Google Scholar] [CrossRef]
- Zeeh, F.; Witte, D.; Gädeken, T.; Rauch, B.H.; Grage-Griebenow, E.; Leinung, N.; Fromm, S.J.; Stölting, S.; Mihara, K.; Kaufmann, R.; et al. Proteinase-activated receptor 2 promotes TGF-β-dependent cell motility in pancreatic cancer cells by sustaining expression of the TGF-β type I receptor ALK5. Oncotarget 2016, 7, 41095–41109. [Google Scholar] [CrossRef]
- Otterbein, H.; Mihara, K.; Hollenberg, M.D.; Lehnert, H.; Witte, D.; Ungefroren, H. RAC1B Suppresses TGF-β-Dependent Chemokinesis and Growth Inhibition through an Autoregulatory Feed-Forward Loop Involving PAR2 and ALK5. Cancers 2019, 11, 1211. [Google Scholar] [CrossRef]
- Cunningham, M.R.; McIntosh, K.A.; Pediani, J.D.; Robben, J.; Cooke, A.E.; Nilsson, M.F.; Gould, G.W.; Mundell, S.; Milligan, G.; Plevin, R. Novel Role for Proteinase-activated Receptor 2 (PAR2) in Membrane Trafficking of Proteinase-activated Receptor 4 (PAR4). J. Biol. Chem. 2012, 287, 16656–16669. [Google Scholar] [CrossRef]
- Zhang, J.; Dijke, P.T.; Wuhrer, M.; Zhang, T. Role of glycosylation in TGF-β signaling and epithelial-to-mesenchymal transition in cancer. Protein Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Witte, D.; Mihara, K.; Rauch, B.H.; Henklein, P.; Jöhren, O.; Bonni, S.; Settmacher, U.; Lehnert, H.; Hollenberg, M.D.; et al. Transforming Growth Factor-β1/Activin Receptor-like Kinase 5-Mediated Cell Migration is Dependent on the Protein Proteinase-Activated Receptor 2 but not on Proteinase-Activated Receptor 2-Stimulated Gq-Calcium Signaling. Mol. Pharmacol. 2017, 92, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Mußbach, F.; Ungefroren, H.; Günther, B.; Katenkamp, K.; Henklein, P.; Westermann, M.; Settmacher, U.; Lenk, L.; Sebens, S.; Müller, J.P.; et al. Proteinase-activated receptor 2 (PAR2) in hepatic stellate cells–evidence for a role in hepatocellular carcinoma growth in vivo. Mol. Cancer 2016, 15, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Kumarasinghe, A.; Musfeldt, M.; Fiedler, C.; Lehnert, H.; Marquardt, J.U. RAC1B Induces SMAD7 via USP26 to Suppress TGFβ1-Dependent Cell Migration in Mesenchymal-Subtype Carcinoma Cells. Cancers 2020, 12, 1545. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H. The effect of RAC1B on the Smad1/5 arm of TGFβ signaling. (Manuscript in preparation).
- Holtzhausen, A.; Golzio, C.; How, T.; Lee, Y.; Schiemann, W.P.; Katsanis, N.; Blobe, G.C. Novel bone morphogenetic protein signaling through Smad2 and Smad3 to regulate cancer progression and development. FASEB J. 2014, 28, 1248–1267. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.-M.; Cha, J.Y.; Kim, J.; Kim, D.; Trang, H.T.H.; Kim, Y.M.; Cho, Y.H.; Park, D.; Hong, S. Smad3 regulates E-cadherin via miRNA-200 pathway. Oncogene 2012, 31, 3051–3059. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Inui, M.; Newfeld, S.J. Regulation of TGF-β signal transduction by mono- and deubiquitylation of Smads. FEBS Lett. 2012, 586, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gong, W.; Ma, X.; Sun, X.; Jiang, H.; Chen, T. Smad7 maintains epithelial phenotype of ovarian cancer stem-like cells and supports tumor colonization by mesenchymal-epithelial transition. Mol. Med. Rep. 2014, 11, 309–316. [Google Scholar] [CrossRef]
- Melzer, C.; Von Der Ohe, J.; Hass, R.; Ungefroren, H. TGF-β-Dependent Growth Arrest and Cell Migration in Benign and Malignant Breast Epithelial Cells Are Antagonistically Controlled by Rac1 and Rac1b. Int. J. Mol. Sci. 2017, 18, 1574. [Google Scholar] [CrossRef]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef]
- Cisowski, J.; Sayin, V.I.; Liu, M.; Karlsson, C.; Bergo, M.O.; Liu, M. Oncogene-induced senescence underlies the mutual exclusive nature of oncogenic KRAS and BRAF. Oncogene 2015, 35, 1328–1333. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, Y.; Takahashi, M.; Van Hook, K.; Kampa-Schittenhelm, K.M.; Sheppard, B.C.; Sears, R.C.; Stork, P.J.S.; Lopez, C.D. N terminus of ASPP2 binds to Ras and enhances Ras/Raf/MEK/ERK activation to promote oncogene-induced senescence. Proc. Natl. Acad. Sci. USA 2013, 110, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Yang, J.; Lee, J.C.; Baek, K.-H. Depletion of ERK2 but not ERK1 abrogates oncogenic Ras-induced senescence. Cell. Signal. 2013, 25, 2540–2547. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.F.; Barros, P.; Moyer, M.P.; Matos, P.; Jordan, P. Expression of tumor-related Rac1b antagonizes B-Raf-induced senescence in colorectal cells. Cancer Lett. 2015, 369, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Cipriano, R.; Kan, C.E.; Graham, J.; Danielpour, D.; Stampfer, M.; Jackson, M.W. TGF-beta signaling engages an ATM-CHK2-p53-independent RAS-induced senescence and prevents malignant transformation in human mammary epithelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8668–8673. [Google Scholar] [CrossRef]
- Fang, D.; Chen, H.; Zhu, J.Y.; Wang, W.; Teng, Y.; Ding, H.F.; Jing, Q.; Su, S.B.; Huang, S. Epithelial-Mesenchymal Transition of Ovarian Cancer Cells Is Sustained by Rac1 through Simultaneous Activation of MEK1/2 and Src Signaling Pathways. Oncogene 2017, 36, 1546–1558. [Google Scholar] [CrossRef]
- Carl, C.; Flindt, A.; Hartmann, J.; Dahlke, M.; Rades, D.; Dunst, J.; Lehnert, H.; Gieseler, F.; Ungefroren, H. Ionizing radiation induces a motile phenotype in human carcinoma cells in vitro through hyperactivation of the TGF-beta signaling pathway. Cell. Mol. Life Sci. 2015, 73, 427–443. [Google Scholar] [CrossRef]
- Barnhouse, V.R.; Weist, J.L.; Shukla, V.C.; Ghadiali, S.N.; Kniss, D.A.; Leight, J.L. Myoferlin regulates epithelial cancer cell plasticity and migration through autocrine TGF-β1 signaling. Oncotarget 2018, 9, 19209–19222. [Google Scholar] [CrossRef]
- Ungefroren, H.; Otterbein, H.; Wellner, U.F.; Keck, T.; Lehnert, H.; Marquardt, J.U. RAC1B regulation of TGFB1 reveals an unexpected role of autocrine TGFβ1 in the suppression of cell motility. Cancers, in revision.
- Ungefroren, H.; Groth, S.; Ruhnke, M.; Kalthoff, H.; Fändrich, F. Transforming growth factor-beta (TGF-beta) type I receptor/ALK5-dependent activation of the GADD45beta gene mediates the induction of biglycan expression by TGF-beta. J. Biol. Chem. 2005, 280, 2644–2652. [Google Scholar] [CrossRef]
- Chen, W.B.; Lenschow, W.; Tiede, K.; Fischer, J.W.; Kalthoff, H.; Ungefroren, H. Smad4/DPC4-dependent regulation of biglycan gene expression by transforming growth factor-beta in pancreatic tumor cells. J. Biol. Chem. 2002, 277, 36118–36128. [Google Scholar] [CrossRef]
- Thakur, A.K.; Nigri, J.; Lac, S.; Leca, J.; Bressy, C.; Berthezene, P.; Bartholin, L.; Chan, P.; Calvo, E.; Iovanna, J.L.; et al. TAp73 loss favors Smad-independent TGF-β signaling that drives EMT in pancreatic ductal adenocarcinoma. Cell Death Differ. 2016, 23, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Brabletz, T.; Keck, T. ZEB1 in Pancreatic Cancer. Cancers 2010, 2, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Abdrabou, A.; Wang, Z. Post-Translational Modification and Subcellular Distribution of Rac1: An Update. Cells 2018, 7, 263. [Google Scholar] [CrossRef]
- Michaelson, D.; Abidi, W.; Guardavaccaro, D.; Zhou, M.; Ahearn, I.; Pagano, M.; Philips, M.R. Rac1 accumulates in the nucleus during the G2 phase of the cell cycle and promotes cell division. J. Cell Biol. 2008, 181, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Lérida, I.; Pellinen, T.; Sánchez, S.A.; Guadamillas, M.C.; Wang, Y.; Mirtti, T.; Calvo, E.; Del Pozo, M.A. Rac1 Nucleocytoplasmic Shuttling Drives Nuclear Shape Changes and Tumor Invasion. Dev. Cell 2015, 32, 318–334. [Google Scholar] [CrossRef]
- Mu, Y.; Sundar, R.; Thakur, N.; Ekman, M.; Gudey, S.K.; Yakymovych, M.; Hermansson, A.; Dimitriou, H.; Bengoechea-Alonso, M.T.; Ericsson, J.; et al. TRAF6 ubiquitinates TGFβtype I receptor to promote its cleavage and nuclear translocation in cancer. Nat. Commun. 2011, 2, 330. [Google Scholar] [CrossRef]
- Reinacher-Schick, A.; Baldus, S.E.; Romdhana, B.; Landsberg, S.; Zapatka, M.; Mönig, S.P.; Hölscher, A.H.; Dienes, H.P.; Schmiegel, W.; Schwarte-Waldhoff, I. Loss of Smad4 correlates with loss of the invasion suppressor E-cadherin in advanced colorectal carcinomas. J. Pathol. 2004, 202, 412–420. [Google Scholar] [CrossRef]
- Muller, N.; Reinacher-Schick, A.; Baldus, S.; Van Hengel, J.; Berx, G.; Baar, A.; Van Roy, F.; Schmiegel, W.; Schwarte-Waldhoff, I. Smad4 induces the tumor suppressor E-cadherin and P-cadherin in colon carcinoma cells. Oncogene 2002, 21, 6049–6058. [Google Scholar] [CrossRef]
- Pohl, M.; Radacz, Y.; Pawlik, N.; Schoeneck, A.; Baldus, S.E.; Munding, J.; Schmiegel, W.; Schwarte-Waldhoff, I.; Reinacher-Schick, A. SMAD4 mediates mesenchymal-epithelial reversion in SW480 colon carcinoma cells. Anticancer Res. 2010, 30, 2603–2613. [Google Scholar]
- Ou, H.; Hoffmann, R.; González-López, C.; Doherty, G.J.; Korkola, J.E.; Muñoz-Espín, D. Cellular senescence in cancer: From mechanisms to detection. Mol. Oncol. 2020. [Google Scholar] [CrossRef]
- Iacobuzio-Donahue, C.; Michael, C.; Baez, P.; Kappagantula, R.; Hooper, J.E.; Hollman, T.J. Cancer biology as revealed by the research autopsy. Nat. Rev. Cancer 2019, 19, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.D.; Parvani, J.G.; Schiemann, W.P. The relevance of the TGF-β Paradox to EMT-MET programs. Cancer Lett. 2013, 341, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, N.; Lee, C. Mysteries of TGF-β Paradox in Benign and Malignant Cells. Front. Oncol. 2014, 4, 94. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungefroren, H.; Wellner, U.F.; Keck, T.; Lehnert, H.; Marquardt, J.-U. The Small GTPase RAC1B: A Potent Negative Regulator of-and Useful Tool to Study-TGFβ Signaling. Cancers 2020, 12, 3475. https://doi.org/10.3390/cancers12113475
Ungefroren H, Wellner UF, Keck T, Lehnert H, Marquardt J-U. The Small GTPase RAC1B: A Potent Negative Regulator of-and Useful Tool to Study-TGFβ Signaling. Cancers. 2020; 12(11):3475. https://doi.org/10.3390/cancers12113475
Chicago/Turabian StyleUngefroren, Hendrik, Ulrich F. Wellner, Tobias Keck, Hendrik Lehnert, and Jens-Uwe Marquardt. 2020. "The Small GTPase RAC1B: A Potent Negative Regulator of-and Useful Tool to Study-TGFβ Signaling" Cancers 12, no. 11: 3475. https://doi.org/10.3390/cancers12113475
APA StyleUngefroren, H., Wellner, U. F., Keck, T., Lehnert, H., & Marquardt, J.-U. (2020). The Small GTPase RAC1B: A Potent Negative Regulator of-and Useful Tool to Study-TGFβ Signaling. Cancers, 12(11), 3475. https://doi.org/10.3390/cancers12113475