BNIP3 in Lung Cancer: To Kill or Rescue?


{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
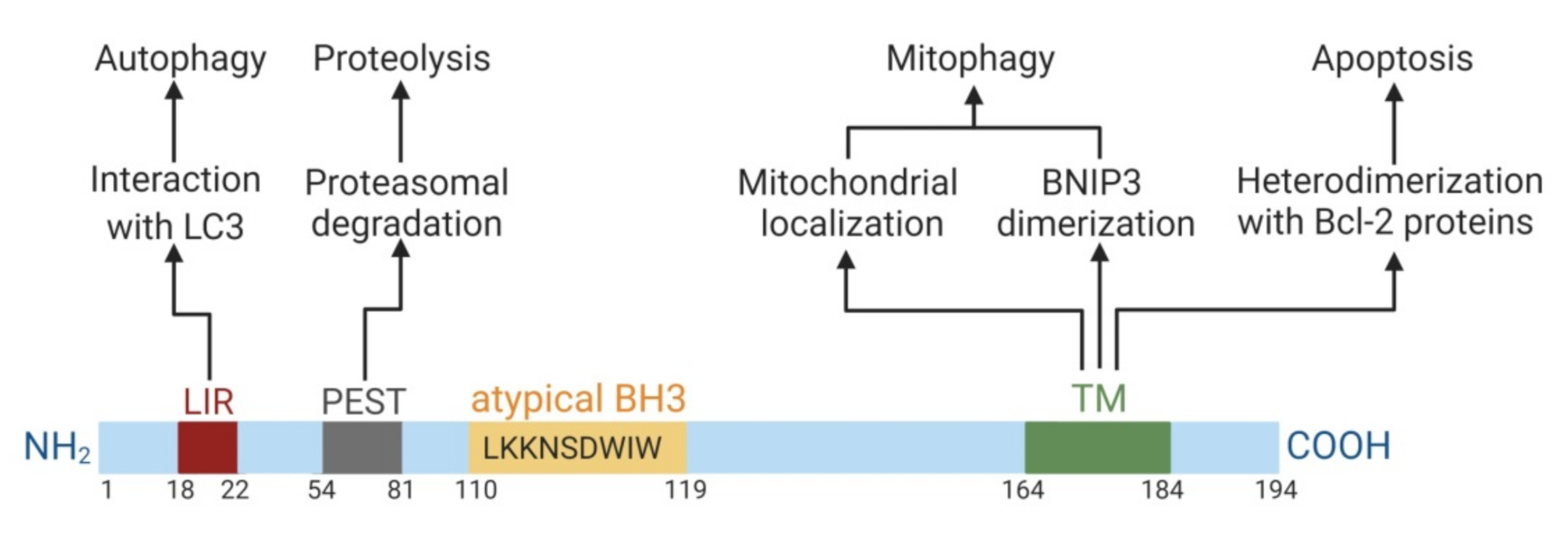
2. Characterization of BNIP3
3. Role of BNIP3 in Cancer
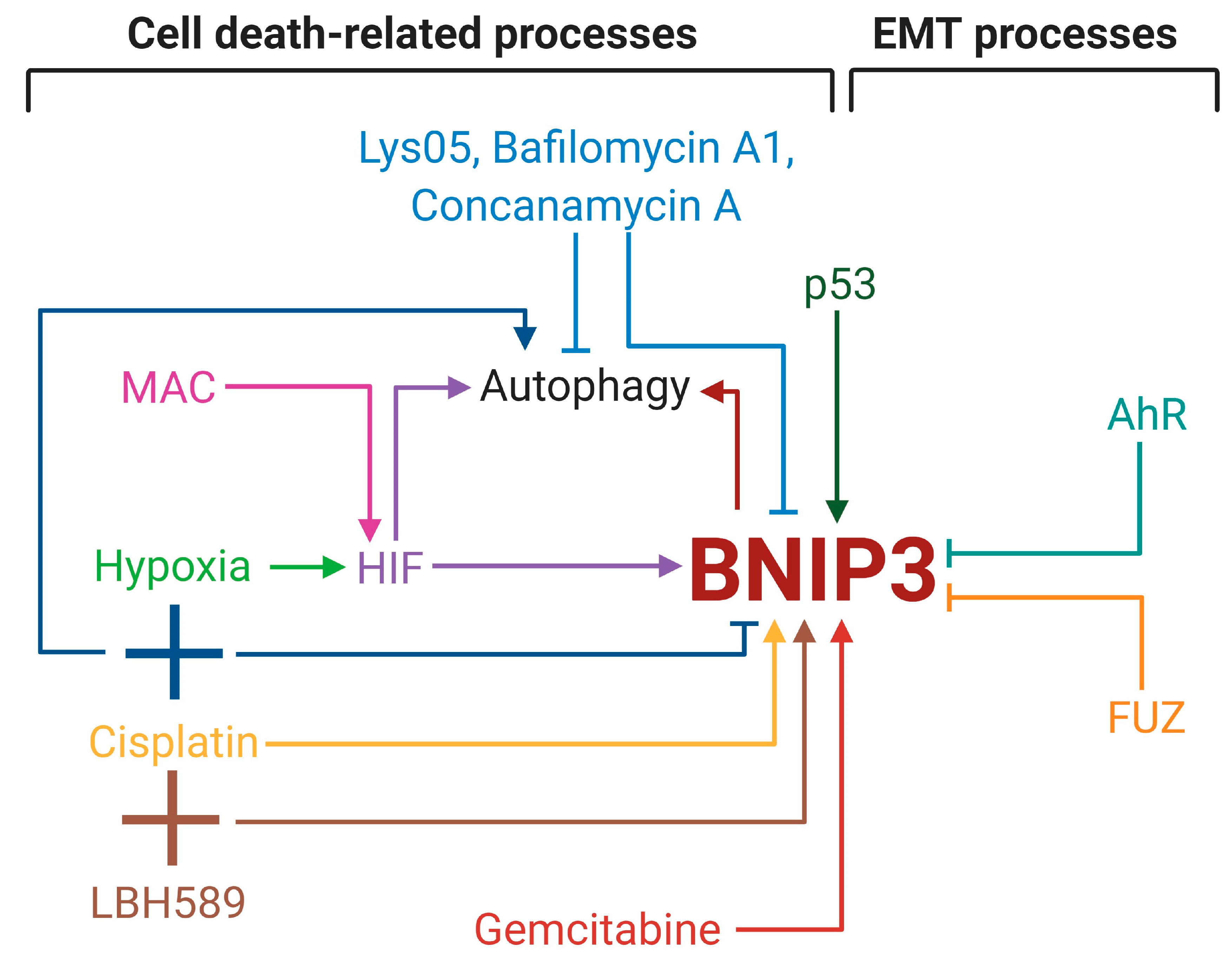
4. BNIP3 in Lung Cancer
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-Small Cell Lung Cancer: Epidemiology, Risk Factors, Treatment, and Survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Malhotra, J.; Malvezzi, M.; Negri, E.; La Vecchia, C.; Boffetta, P. Risk factors for lung cancer worldwide. Eur. Respir. J. 2016, 48, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Sholl, L.M.; Aisner, D.L.; Varella-Garcia, M.; Berry, L.D.; Dias-Santagata, D.; Wistuba, I.I.; Chen, H.; Fujimoto, J.; Kugler, K.; Franklin, W.A.; et al. Multi-institutional Oncogenic Driver Mutation Analysis in Lung Adenocarcinoma: The Lung Cancer Mutation Consortium Experience. J. Thorac. Oncol. 2015, 10, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Rurańska, B.; Stawicka, M.; Godlewski, D. The role of p53 gene in lung cancer with special emphasis on hereditary types. Rep. Pract. Oncol. 1997, 2, 56. [Google Scholar] [CrossRef]
- Canale, M.; Petracci, E.; Delmonte, A.; Bronte, G.; Chiadini, E.; Ludovini, V.; Dubini, A.; Papi, M.; Baglivo, S.; De Luigi, N.; et al. Concomitant TP53 Mutation Confers Worse Prognosis in EGFR-Mutated Non-Small Cell Lung Cancer Patients Treated with TKIs. J. Clin. Med. 2020, 9, 1047. [Google Scholar] [CrossRef] [PubMed]
- Vara-Perez, M.; Felipe-Abrio, B.; Agostinis, P. Mitophagy in Cancer: A Tale of Adaptation. Cells 2019, 8, 493. [Google Scholar] [CrossRef] [PubMed]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar] [PubMed]
- Colella, B.; Faienza, F.; Di Bartolomeo, S. EMT Regulation by Autophagy: A New Perspective in Glioblastoma Biology. Cancers (Basel) 2019, 11, 312. [Google Scholar] [CrossRef]
- Li, X.; Zhou, S.; Fan, T.; Feng, X. lncRNA DGCR 5/miR-27a-3p/BNIP3 promotes cell apoptosis in pancreatic cancer by regulating the p38 MAPK pathway. Int. J. Mol. Med. 2020, 46, 729–739. [Google Scholar] [CrossRef]
- Nollet, E.A.; Cardo-Vila, M.; Ganguly, S.S.; Tran, J.D.; Schulz, V.V.; Cress, A.; Corey, E.; Miranti, C.K. Androgen receptor-induced integrin α6β1 and Bnip3 promote survival and resistance to PI3K inhibitors in castration-resistant prostate cancer. Oncogene 2020, 39, 5390–5404. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Zhang, W.; Beaton, M.; Marsboom, G.; Gruber, M.; Simon, M.C.; Hart, J.; Dorn, G.W.; Brady, M.J.; Macleod, K.F. BNip3 Regulates Mitochondrial Function and Lipid Metabolism in the Liver. Mol. Cell. Biol. 2012, 32, 2570–2584. [Google Scholar] [CrossRef] [PubMed]
- Gang, H.; Dhingra, R.; Lin, J.; Hai, Y.; Aviv, Y.; Margulets, V.; Hamedani, M.; Thanasupawat, T.; Leygue, E.; Klonisch, T.; et al. PDK2-mediated alternative splicing switches Bnip3 from cell death to cell survival. J. Cell Biol. 2015, 210, 1101–1115. [Google Scholar] [CrossRef] [PubMed]
- Rikka, S.; Quinsay, M.N.; Thomas, R.L.; Kubli, D.A.; Zhang, X.; Murphy, A.N.; Gustafsson, Å.B. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ. 2011, 18, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef]
- Guo, K.; Searfoss, G.; Krolikowski, D.; Pagnoni, M.; Franks, C.; Clark, K.; Yu, K.T.; Jaye, M.; Ivashchenko, Y. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001, 8, 367–376. [Google Scholar] [CrossRef]
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 9082–9087. [Google Scholar] [CrossRef]
- Kamino, H.; Nakamura, Y.; Tsuneki, M.; Sano, H.; Miyamoto, Y.; Kitamura, N.; Futamura, M.; Kanai, Y.; Taniguchi, H.; Shida, D.; et al. Mieap-regulated mitochondrial quality control is frequently inactivated in human colorectal cancer. Oncogenesis 2016, 5, e181. [Google Scholar] [CrossRef]
- Okami, J.; Simeone, D.M.; Logsdon, C.D. Silencing of the Hypoxia-Inducible Cell Death Protein BNIP3 in Pancreatic Cancer. Cancer Res. 2004, 64, 5338–5346. [Google Scholar] [CrossRef]
- Murai, M.; Toyota, M.; Satoh, A.; Suzuki, H.; Akino, K.; Mita, H.; Sasaki, Y.; Ishida, T.; Shen, L.; Garcia-Manero, G.; et al. Aberrant DNA methylation associated with silencing BNIP3 gene expression in haematopoietic tumours. Br. J. Cancer 2005, 92, 1165–1172. [Google Scholar] [CrossRef]
- Shao, Y.; Liu, Z.; Liu, J.; Wang, H.; Huang, L.; Lin, T.; Liu, J.; Wei, Q.; Zeng, H.; He, G.; et al. Expression and epigenetic regulatory mechanism of BNIP3 in clear cell renal cell carcinoma. Int. J. Oncol. 2018, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, Å.B. Microtubule-associated Protein 1 Light Chain 3 (LC3) Interacts with Bnip3 Protein to Selectively Remove Endoplasmic Reticulum and Mitochondria via Autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.; Wells, R.; Rechsteiner, M. Amino acid sequences common to rapidly degraded proteins: The PEST hypothesis. Science 1986, 234, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Aouacheria, A.; Brunet, F.; Gouy, M. Phylogenomics of Life-Or-Death Switches in Multicellular Animals: Bcl-2, BH3-Only, and BNip Families of Apoptotic Regulators. Mol. Biol. Evol. 2005, 22, 2395–2416. [Google Scholar] [CrossRef]
- Yasuda, M.; Theodorakis, P.; Subramanian, T.; Chinnadurai, G. Adenovirus E1B-19K/BCL-2 Interacting Protein BNIP3 Contains a BH3 Domain and a Mitochondrial Targeting Sequence. J. Biol. Chem. 1998, 273, 12415–12421. [Google Scholar] [CrossRef]
- Ray, R.; Chen, G.; Velde, C.V.; Cizeau, J.; Park, J.H.; Reed, J.C.; Gietz, R.D.; Greenberg, A.H. BNIP3 Heterodimerizes with Bcl-2/Bcl-XL and Induces Cell Death Independent of a Bcl-2 Homology 3 (BH3) Domain at Both Mitochondrial and Nonmitochondrial Sites. J. Biol. Chem. 2000, 275, 1439–1448. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Cho, J.-J.; Ha, J.; Park, J.-H. The Carboxy Terminal C-Tail of BNip3 Is Crucial in Induction of Mitochondrial Permeability Transition in Isolated Mitochondria. Arch. Biochem. Biophys. 2002, 398, 147–152. [Google Scholar] [CrossRef]
- Vasagiri, N.; Kutala, V.K. Structure, function, and epigenetic regulation of BNIP3: A pathophysiological relevance. Mol. Biol. Rep. 2014, 41, 7705–7714. [Google Scholar] [CrossRef]
- Chen, G.; Ray, R.; Dubik, D.; Shi, L.; Cizeau, J.; Bleackley, R.C.; Saxena, S.; Gietz, R.D.; Greenberg, A.H. The E1B 19K/Bcl-2–binding Protein Nip3 is a Dimeric Mitochondrial Protein that Activates Apoptosis. J. Exp. Med. 1997, 186, 1975–1983. [Google Scholar] [CrossRef]
- Liu, K.E.; Frazier, W.A. Phosphorylation of the BNIP3 C-terminus inhibits mitochondrial damage and cell death without blocking autophagy. PLoS ONE 2015, 10, e0129667. [Google Scholar] [CrossRef]
- Frazier, D.P.; Wilson, A.; Graham, R.M.; Thompson, J.W.; Bishopric, N.H.; Webster, K.A. Acidosis Regulates the Stability, Hydrophobicity, and Activity of the BH3-Only Protein Bnip3. Antioxid. Redox Signal. 2006, 8, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Chinnadurai, G.; Vijayalingam, S.; Gibson, S.B. BNIP3 subfamily BH3-only proteins: Mitochondrial stress sensors in normal and pathological functions. Oncogene 2008, 27, S114–S127. [Google Scholar] [CrossRef] [PubMed]
- Kubasiak, L.A.; Hernandez, O.M.; Bishopric, N.H.; Webster, K.A. Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc. Natl. Acad. Sci. USA 2002, 99, 12825–12830. [Google Scholar] [CrossRef] [PubMed]
- Regula, K.M.; Ens, K.; Kirshenbaum, L.A. Inducible Expression of BNIP3 Provokes Mitochondrial Defects and Hypoxia-Mediated Cell Death of Ventricular Myocytes. Circ. Res. 2002, 91, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Vijayalingam, S.; Pillai, S.G.; Rashmi, R.; Subramanian, T.; Sagartz, J.E.; Chinnadurai, G. Overexpression of BH3-Only Protein BNIP3 Leads to Enhanced Tumor Growth. Genes Cancer 2010, 1, 964–971. [Google Scholar] [CrossRef]
- Shaida, N.; Launchbury, R.; Boddy, J.L.; Jones, C.; Campo, L.; Turley, H.; Kanga, S.; Banham, A.H.; Malone, P.R.; Harris, A.L.; et al. Expression of BNIP3 correlates with hypoxia-inducible factor (HIF)-1α, HIF-2α and the androgen receptor in prostate cancer and is regulated directly by hypoxia but not androgens in cell lines. Prostate 2008, 68, 336–343. [Google Scholar] [CrossRef]
- Giatromanolaki, A. BNIP3 Expression Is Linked with Hypoxia-Regulated Protein Expression and with Poor Prognosis in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2004, 10, 5566–5571. [Google Scholar] [CrossRef]
- Burton, T.R.; Gibson, S.B. The role of Bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ. 2009, 16, 515–523. [Google Scholar] [CrossRef]
- Leo, C.; Horn, L.-C.; Hockel, M. Hypoxia and expression of the proapoptotic regulator BNIP3 in cervical cancer. Int. J. Gynecol. Cancer 2006, 16, 1314–1320. [Google Scholar] [CrossRef]
- Sowter, H.M.; Ferguson, M.; Pym, C.; Watson, P.; Fox, S.B.; Han, C.; Harris, A.L. Expression of the cell death genes BNip3 and NIX in ductal carcinomain situ of the breast; correlation of BNip3 levels with necrosis and grade. J. Pathol. 2003, 201, 573–580. [Google Scholar] [CrossRef]
- Chen, X.; Gong, J.; Zeng, H.; Chen, N.; Huang, R.; Huang, Y.; Nie, L.; Xu, M.; Xia, J.; Zhao, F.; et al. MicroRNA145 Targets BNIP3 and Suppresses Prostate Cancer Progression. Cancer Res. 2010, 70, 2728–2738. [Google Scholar] [CrossRef] [PubMed]
- Koop, E.A.; van Laar, T.; van Wichen, D.F.; de Weger, R.A.; van der Wall, E.; van Diest, P.J. Expression of BNIP3 in invasive breast cancer: Correlations with the hypoxic response and clinicopathological features. BMC Cancer 2009, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.Y.; Campo, L.; Han, C.; Turley, H.; Pezzella, F.; Gatter, K.C.; Harris, A.L.; Fox, S.B. BNIP3 as a Progression Marker in Primary Human Breast Cancer; Opposing Functions in In situ Versus Invasive Cancer. Clin. Cancer Res. 2007, 13, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Toyota, M.; Suzuki, H.; Murai, M.; Akino, K.; Ueno, M.; Nojima, M.; Yawata, A.; Miyakawa, H.; Suga, T.; et al. Upregulation of BNIP3 by 5-aza-2′-deoxycytidine sensitizes pancreatic cancer cells to hypoxia-mediated cell death. J. Gastroenterol. 2005, 40, 504–510. [Google Scholar] [CrossRef]
- Murai, M.; Toyota, M.; Suzuki, H.; Satoh, A.; Sasaki, Y.; Akino, K.; Ueno, M.; Takahashi, F.; Kusano, M.; Mita, H.; et al. Aberrant methylation and silencing of the BNIP3 gene in colorectal and gastric cancer. Clin. Cancer Res. 2005, 11, 1021–1027. [Google Scholar]
- Erkan, M.; Kleeff, J.; Esposito, I.; Giese, T.; Ketterer, K.; Büchler, M.W.; Giese, N.A.; Friess, H. Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene 2005, 24, 4421–4432. [Google Scholar] [CrossRef]
- He, J.; Pei, L.; Jiang, H.; Yang, W.; Chen, J.; Liang, H. Chemoresistance of colorectal cancer to 5-fluorouracil is associated with silencing of the BNIP3 gene through aberrant methylation. J. Cancer 2017, 8, 1187–1196. [Google Scholar] [CrossRef]
- Méndez-Blanco, C.; Fondevila, F.; Fernández-Palanca, P.; García-Palomo, A.; van Pelt, J.; Verslype, C.; González-Gallego, J.; Mauriz, J.L. Stabilization of Hypoxia-Inducible Factors and BNIP3 Promoter Methylation Contribute to Acquired Sorafenib Resistance in Human Hepatocarcinoma Cells. Cancers (Basel) 2019, 11, 1984. [Google Scholar] [CrossRef]
- Velde, C.V.; Cizeau, J.; Dubik, D.; Alimonti, J.; Brown, T.; Israels, S.; Hakem, R.; Greenberg, A.H. BNIP3 and Genetic Control of Necrosis-Like Cell Death through the Mitochondrial Permeability Transition Pore. Mol. Cell. Biol. 2000, 20, 5454–5468. [Google Scholar] [CrossRef]
- Kothari, S.; Cizeau, J.; McMillan-Ward, E.; Israels, S.J.; Bailes, M.; Ens, K.; Kirshenbaum, L.A.; Gibson, S.B. BNIP3 plays a role in hypoxic cell death in human epithelial cells that is inhibited by growth factors EGF and IGF. Oncogene 2003, 22, 4734–4744. [Google Scholar] [CrossRef]
- Burton, T.R.; Henson, E.S.; Baijal, P.; Eisenstat, D.D.; Gibson, S.B. The pro-cell death Bcl-2 family member, BNIP3, is localized to the nucleus of human glial cells: Implications for glioblastoma multiforme tumor cell survival under hypoxia. Int. J. Cancer 2006, 118, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Paik, S.-G. Regulation of BNIP3 in normal and cancer cells. Mol. Cells 2006, 21, 1–6. [Google Scholar] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef]
- Shintani, T. Autophagy in Health and Disease: A Double-Edged Sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef]
- Denisenko, T.; Pivnyuk, A.; Zhivotovsky, B. p53-Autophagy-Metastasis Link. Cancers (Basel) 2018, 10, 148. [Google Scholar] [CrossRef]
- Zhang, L.; Li, L.; Liu, H.; Borowitz, J.L.; Isom, G.E. BNIP3 mediates cell death by different pathways following localization to endoplasmic reticulum and mitochondrion. FASEB J. 2009, 23, 3405–3414. [Google Scholar] [CrossRef]
- Ney, P.A. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 2775–2783. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Kim, E.; Beemiller, P.; Wang, C.-Y.; Swanson, J.; You, M.; Guan, K.-L. Bnip3 Mediates the Hypoxia-induced Inhibition on Mammalian Target of Rapamycin by Interacting with Rheb. J. Biol. Chem. 2007, 282, 35803–35813. [Google Scholar] [CrossRef]
- Azad, M.B.; Chen, Y.; Henson, E.S.; Cizeau, J.; McMillan-Ward, E.; Israels, S.J.; Gibson, S.B. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy 2008, 4, 195–204. [Google Scholar] [CrossRef]
- Kanzawa, T.; Zhang, L.; Xiao, L.; Germano, I.M.; Kondo, Y.; Kondo, S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene 2005, 24, 980–991. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.; Jiang, J.; Xie, F.; Chen, L. Bnip3 in mitophagy: Novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin. Chim. Acta 2020, 506, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, A.; Jayas, R.; Afshar, P.; Guberman, M.; Maddaford, G.; Gerstein, J.; Lieberman, B.; Nepon, H.; Margulets, V.; Dhingra, R.; et al. Ellagic acid antagonizes Bnip3-mediated mitochondrial injury and necrotic cell death of cardiac myocytes. Free Radic. Biol. Med. 2017, 112, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Chourasia, A.H.; Boland, M.L.; Macleod, K.F. Mitophagy and cancer. Cancer Metab. 2015, 3, 4. [Google Scholar] [CrossRef]
- Chang, J.Y.; Yi, H.-S.; Kim, H.-W.; Shong, M. Dysregulation of mitophagy in carcinogenesis and tumor progression. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Drake, L.E.; Springer, M.Z.; Poole, L.P.; Kim, C.J.; Macleod, K.F. Expanding perspectives on the significance of mitophagy in cancer. Semin. Cancer Biol. 2017, 47, 110–124. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Mancias, J.D.; Kimmelman, A.C. Mechanisms of Selective Autophagy in Normal Physiology and Cancer. J. Mol. Biol. 2016, 428, 1659–1680. [Google Scholar] [CrossRef]
- Chourasia, A.H.; Tracy, K.; Frankenberger, C.; Boland, M.L.; Sharifi, M.N.; Drake, L.E.; Sachleben, J.R.; Asara, J.M.; Locasale, J.W.; Karczmar, G.S.; et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 2015, 16, 1145–1163. [Google Scholar] [CrossRef]
- Lyons, A.; Coleman, M.; Riis, S.; Favre, C.; O’Flanagan, C.H.; Zhdanov, A.V.; Papkovsky, D.B.; Hursting, S.D.; O’Connor, R. Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J. Biol. Chem. 2017, 292, 16983–16998. [Google Scholar] [CrossRef]
- Chang, H.W.; Kim, M.R.; Lee, H.J.; Lee, H.M.; Kim, G.C.; Lee, Y.S.; Nam, H.Y.; Lee, M.; Jang, H.J.; Lee, K.E.; et al. p53/BNIP3-dependent mitophagy limits glycolytic shift in radioresistant cancer. Oncogene 2019, 38, 3729–3742. [Google Scholar] [CrossRef] [PubMed]
- Maes, H.; Van Eygen, S.; Krysko, D.V.; Vandenabeele, P.; Nys, K.; Rillaerts, K.; Garg, A.D.; Verfaillie, T.; Agostinis, P. BNIP3 supports melanoma cell migration and vasculogenic mimicry by orchestrating the actin cytoskeleton. Cell Death Dis. 2014, 5, e1127. [Google Scholar] [CrossRef] [PubMed]
- The Human Protein Atlas. Available online: https://web.archive.org/web/20201001145147/https://www.proteinatlas.org/ENSG00000176171-BNIP3/pathology/lung+cancer (accessed on 1 October 2020).
- Karpathiou, G.; Sivridis, E.; Koukourakis, M.; Mikroulis, D.; Bouros, D.; Froudarakis, M.; Bougioukas, G.; Maltezos, E.; Giatromanolaki, A. Autophagy and Bcl-2/BNIP3 death regulatory pathway in non-small cell lung carcinomas. APMIS 2013, 121, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Überall, I.; Kolek, V.; Klein, J.; Krejčí, V.; Šťastná, J.; Radová, L.; Škarda, J.; Fridman, E. The immunohistochemical expression of BNIP3 protein in non-small-cell lung cancer: A tissue microarray study. APMIS 2010. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, M.; Hu, D. Development of an autophagy-related gene prognostic signature in lung adenocarcinoma and lung squamous cell carcinoma. PeerJ 2020, 8, e8288. [Google Scholar] [CrossRef]
- Chaachouay, H.; Fehrenbacher, B.; Toulany, M.; Schaller, M.; Multhoff, G.; Rodemann, H.P. AMPK-independent autophagy promotes radioresistance of human tumor cells under clinical relevant hypoxia in vitro. Radiother. Oncol. 2015, 116, 409–416. [Google Scholar] [CrossRef]
- Wu, H.-M.; Jiang, Z.-F.; Ding, P.-S.; Shao, L.-J.; Liu, R.-Y. Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci. Rep. 2015, 5, 12291. [Google Scholar] [CrossRef]
- Munksgaard Persson, M.; Johansson, M.E.; Monsef, N.; Planck, M.; Beckman, S.; Seckl, M.J.; Rönnstrand, L.; Påhlman, S.; Pettersson, H.M. HIF-2α Expression Is Suppressed in SCLC Cells, Which Survive in Moderate and Severe Hypoxia When HIF-1α Is Repressed. Am. J. Pathol. 2012, 180, 494–504. [Google Scholar] [CrossRef]
- Seok, J.-Y.; Jeong, Y.-J.; Hwang, S.-K.; Kim, C.-H.; Magae, J.; Chang, Y.-C. Upregulation of AMPK by 4-O-methylascochlorin promotes autophagy via the HIF-1α expression. J. Cell. Mol. Med. 2018, 22, 6345–6356. [Google Scholar] [CrossRef]
- Jin, H.-O.; Hong, S.-E.; Kim, C.S.; Park, J.-A.; Kim, J.-H.; Kim, J.-Y.; Kim, B.; Chang, Y.H.; Hong, S.-I.; Hong, Y.J.; et al. Combined effects of EGFR tyrosine kinase inhibitors and vATPase inhibitors in NSCLC cells. Toxicol. Appl. Pharmacol. 2015, 287, 17–25. [Google Scholar] [CrossRef]
- Wu, H.-M.; Shao, L.-J.; Jiang, Z.-F.; Liu, R.-Y. Gemcitabine-Induced Autophagy Protects Human Lung Cancer Cells from Apoptotic Death. Lung 2016, 194, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yin, H.; Zhang, Y.; Li, X.; Tong, H.; Zeng, Y.; Wang, Q.; He, W. Hypoxia-induced autophagy promotes gemcitabine resistance in human bladder cancer cells through hypoxia-inducible factor 1α activation. Int. J. Oncol. 2018. [Google Scholar] [CrossRef]
- Cechakova, L.; Ondrej, M.; Pavlik, V.; Jost, P.; Cizkova, D.; Bezrouk, A.; Pejchal, J.; Amaravadi, R.K.; Winkler, J.D.; Tichy, A. A Potent Autophagy Inhibitor (Lys05) Enhances the Impact of Ionizing Radiation on Human Lung Cancer Cells H1299. Int. J. Mol. Sci. 2019, 20, 5881. [Google Scholar] [CrossRef]
- Chung, L.; Tang, S.; Wu, Y.; Yang, K.; Huang, H.; Sun, G.; Sun, K. Platinum-based combination chemotherapy triggers cancer cell death through induction of BNIP3 and ROS, but not autophagy. J. Cell. Mol. Med. 2020, 24, 1993–2003. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Yun, H.; Yang, Y.; Jing, B.; Feng, C.; Song-bin, F. Upregulation of BNIP3 promotes apoptosis of lung cancer cells that were induced by p53. Biochem. Biophys. Res. Commun. 2006, 346, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef]
- Tsai, C.-H.; Li, C.-H.; Cheng, Y.-W.; Lee, C.-C.; Liao, P.-L.; Lin, C.-H.; Huang, S.-H.; Kang, J.-J. The inhibition of lung cancer cell migration by AhR-regulated autophagy. Sci. Rep. 2017, 7, 41927. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Li, K.; Yu, C.; Lv, B.; Zhao, N.; Deng, J.; Cao, L.; Huang, H.; Yin, A.; Shi, T.; et al. In vitro study of FUZ as a novel potential therapeutic target in non-small-cell lung cancer. Life Sci. 2018, 197, 91–100. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorbunova, A.S.; Yapryntseva, M.A.; Denisenko, T.V.; Zhivotovsky, B. BNIP3 in Lung Cancer: To Kill or Rescue? Cancers 2020, 12, 3390. https://doi.org/10.3390/cancers12113390
Gorbunova AS, Yapryntseva MA, Denisenko TV, Zhivotovsky B. BNIP3 in Lung Cancer: To Kill or Rescue? Cancers. 2020; 12(11):3390. https://doi.org/10.3390/cancers12113390
Chicago/Turabian StyleGorbunova, Anna S., Maria A. Yapryntseva, Tatiana V. Denisenko, and Boris Zhivotovsky. 2020. "BNIP3 in Lung Cancer: To Kill or Rescue?" Cancers 12, no. 11: 3390. https://doi.org/10.3390/cancers12113390
APA StyleGorbunova, A. S., Yapryntseva, M. A., Denisenko, T. V., & Zhivotovsky, B. (2020). BNIP3 in Lung Cancer: To Kill or Rescue? Cancers, 12(11), 3390. https://doi.org/10.3390/cancers12113390