Novel FGFR4-Targeting Single-Domain Antibodies for Multiple Targeted Therapies against Rhabdomyosarcoma

 ,
,  ,
, 
 , and
, and 
Simple Summary
Abstract

1. Introduction
2. Results
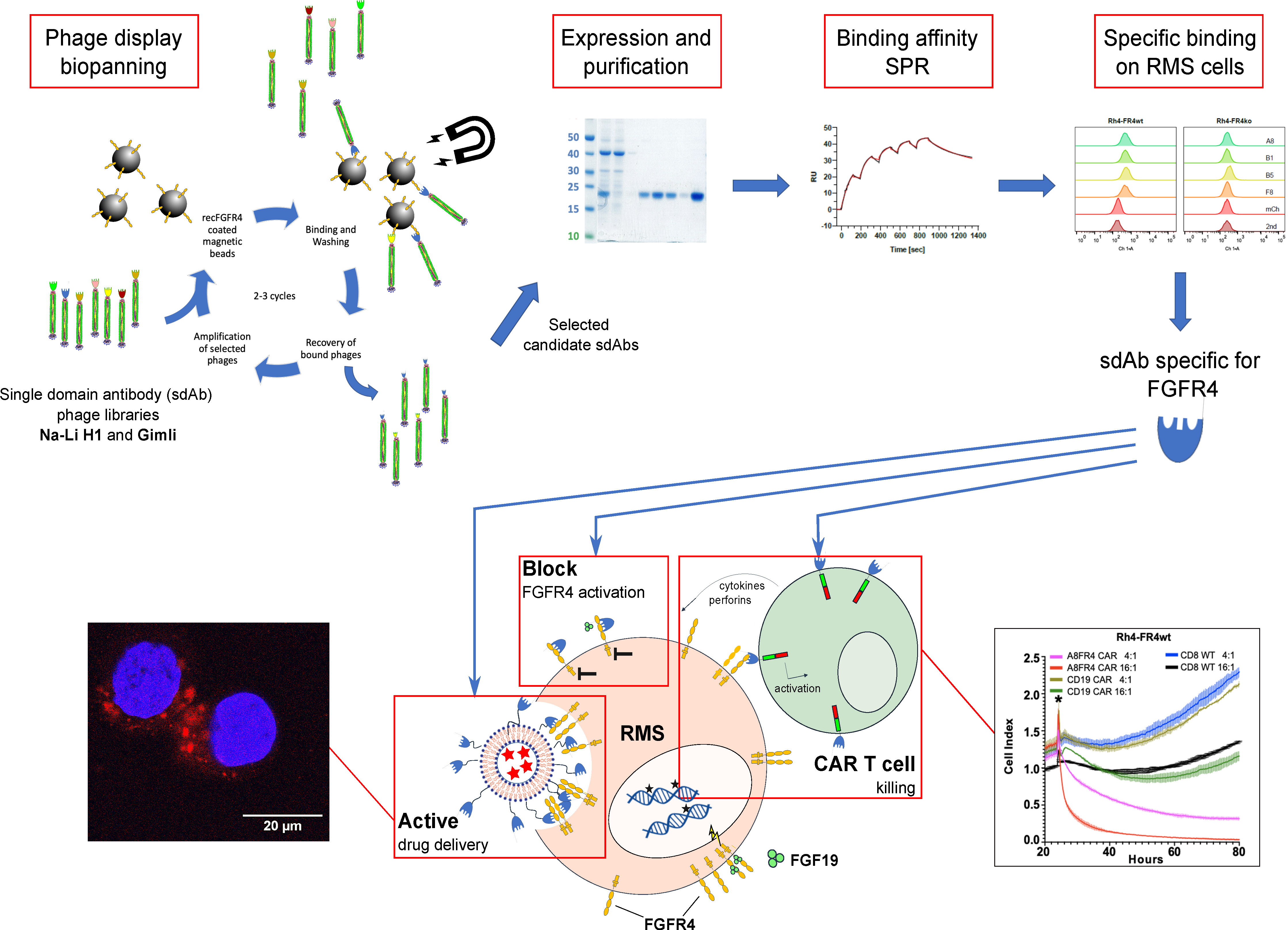
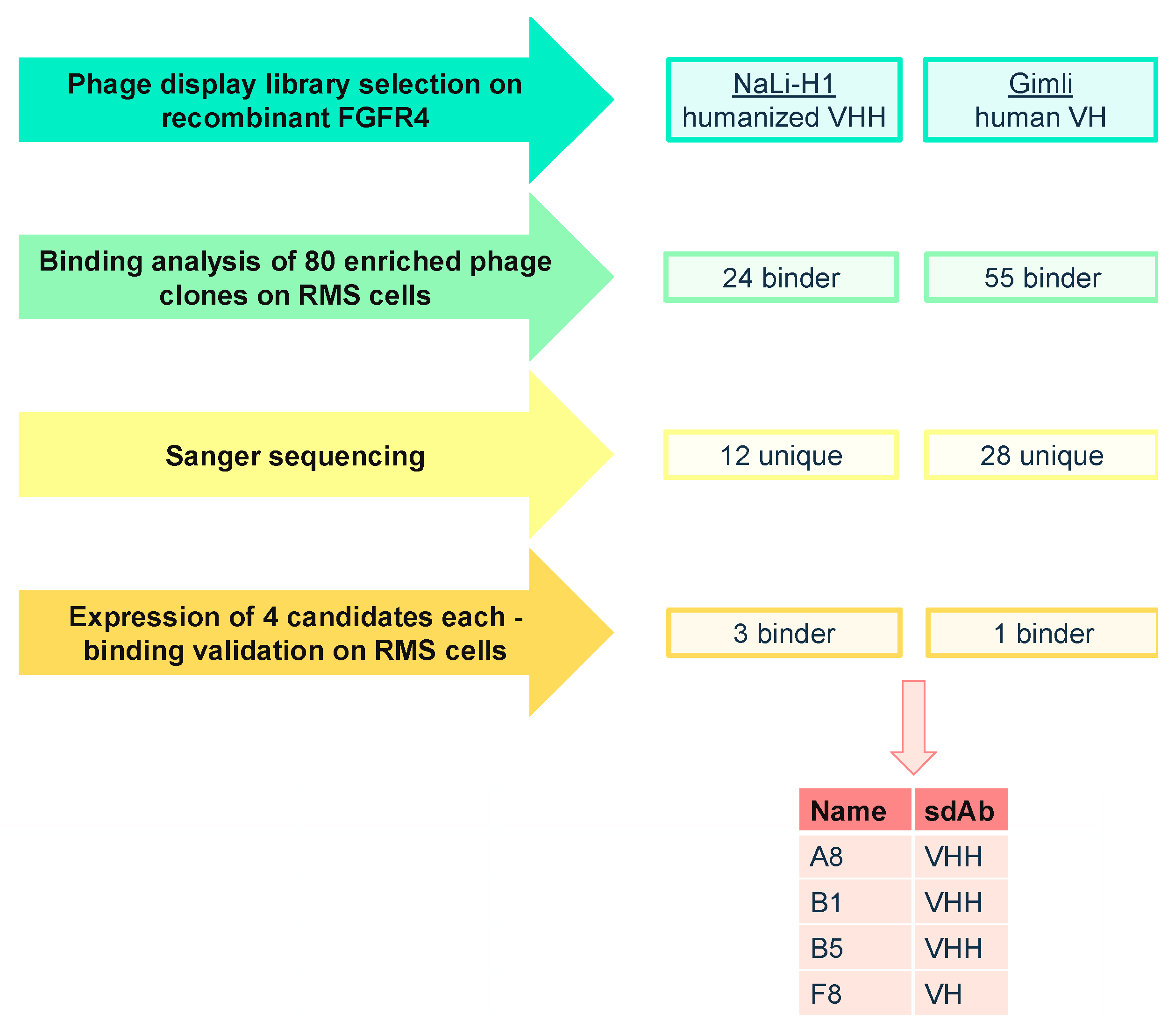
2.1. Phage Display Selection of FGFR4-Specific sdAb
2.2. Recombinant sdAb Bind to FR4wt RMS Cells But Not to FR4ko RMS Cells
2.3. Selected Recombinant Single-Domain Antibodies Inhibit FGFR4 Signaling
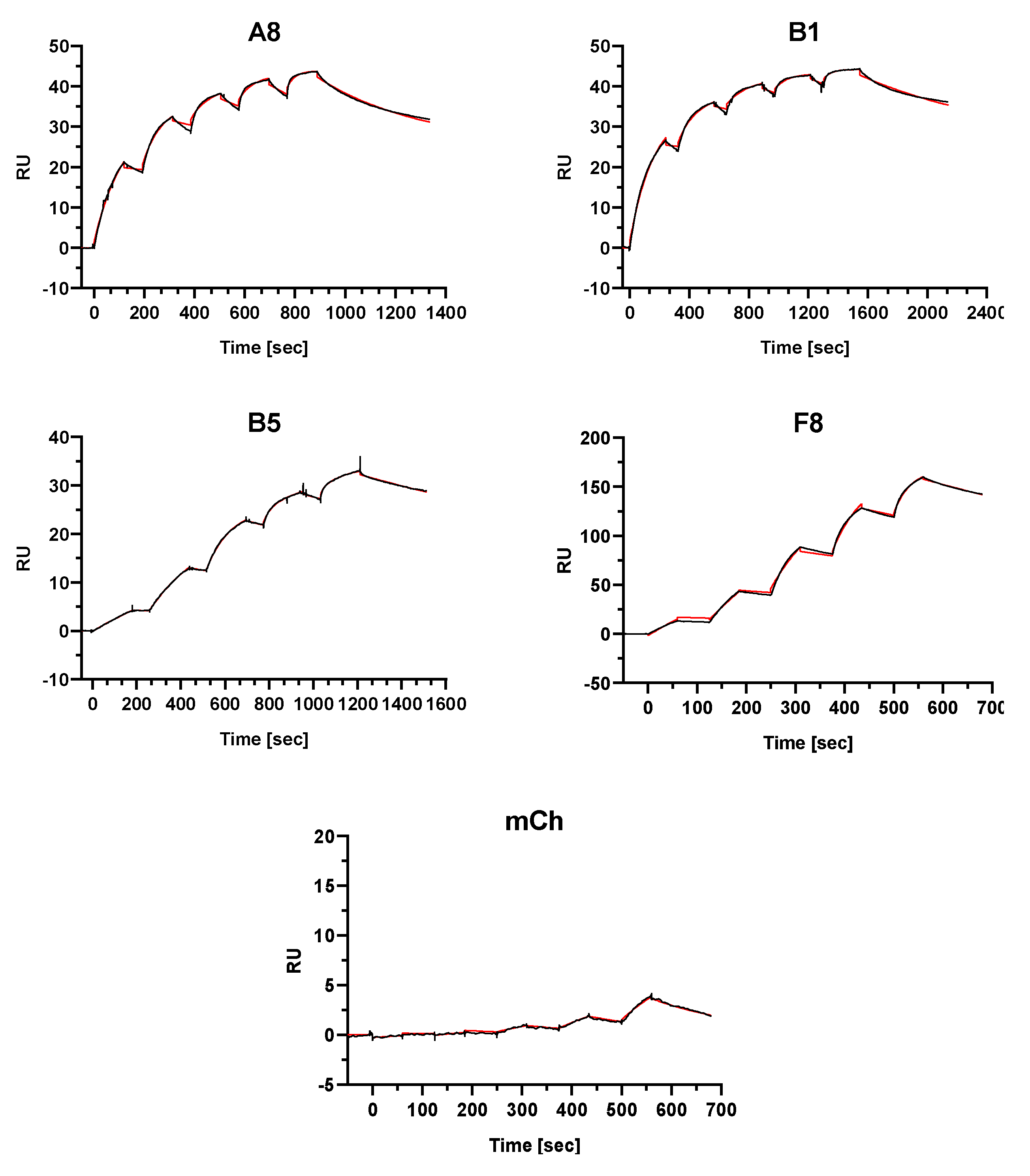
2.4. Single-Domain Antibodies Bind to FGFR4 with High Affinity
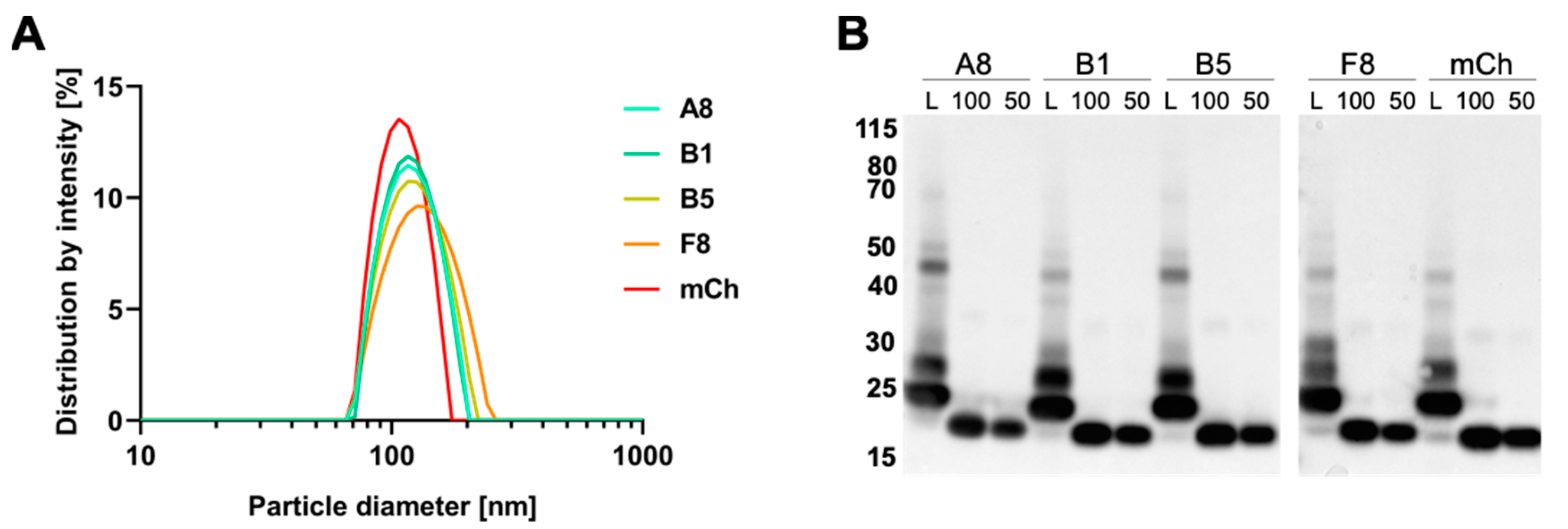
2.5. Preparation and Characterization of VCR-Loaded Targeting Liposomes
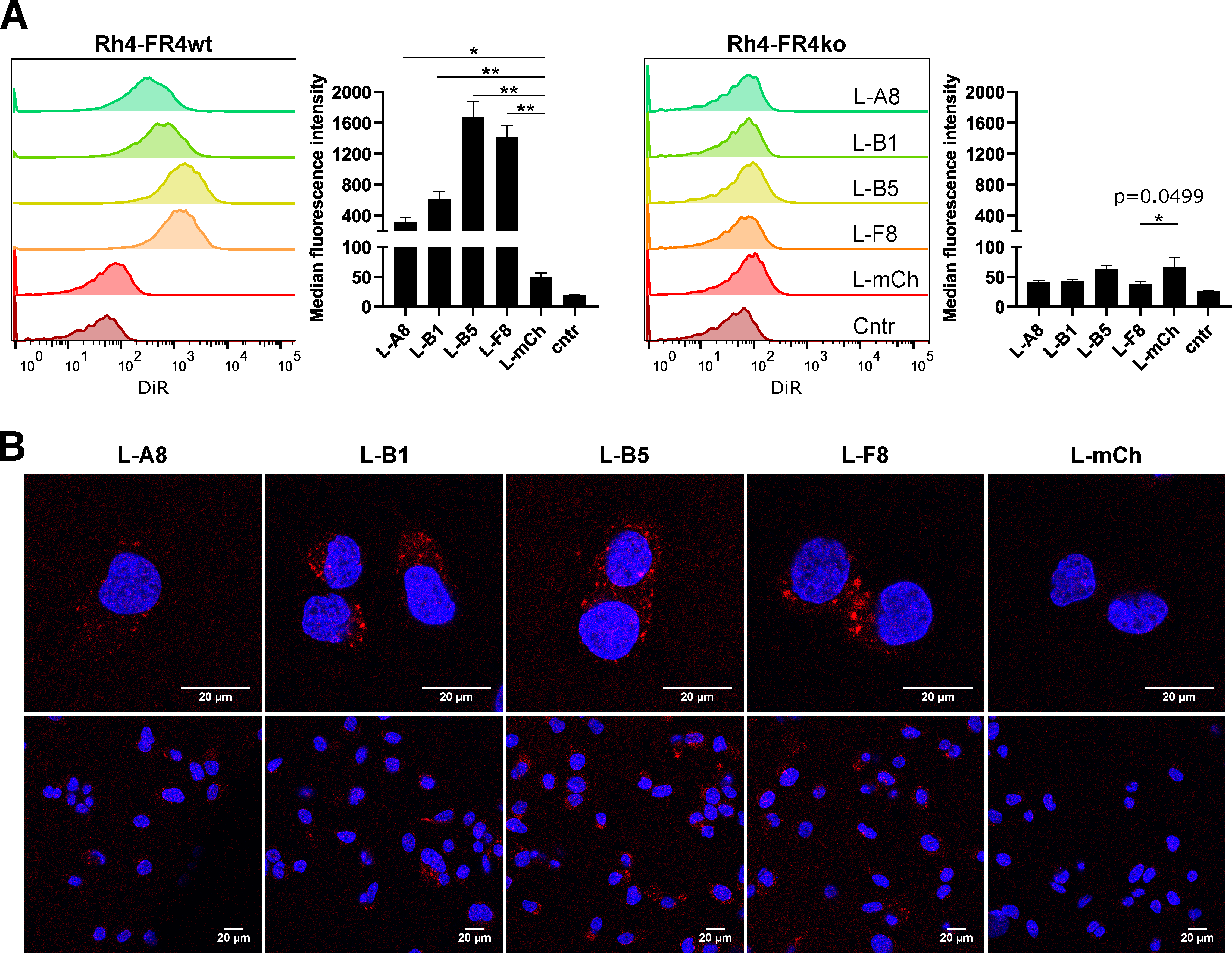
2.6. FGFR4-Targeted Liposomes Bind Specifically to FGFR4 Positive RMS Cells and Are Internalized
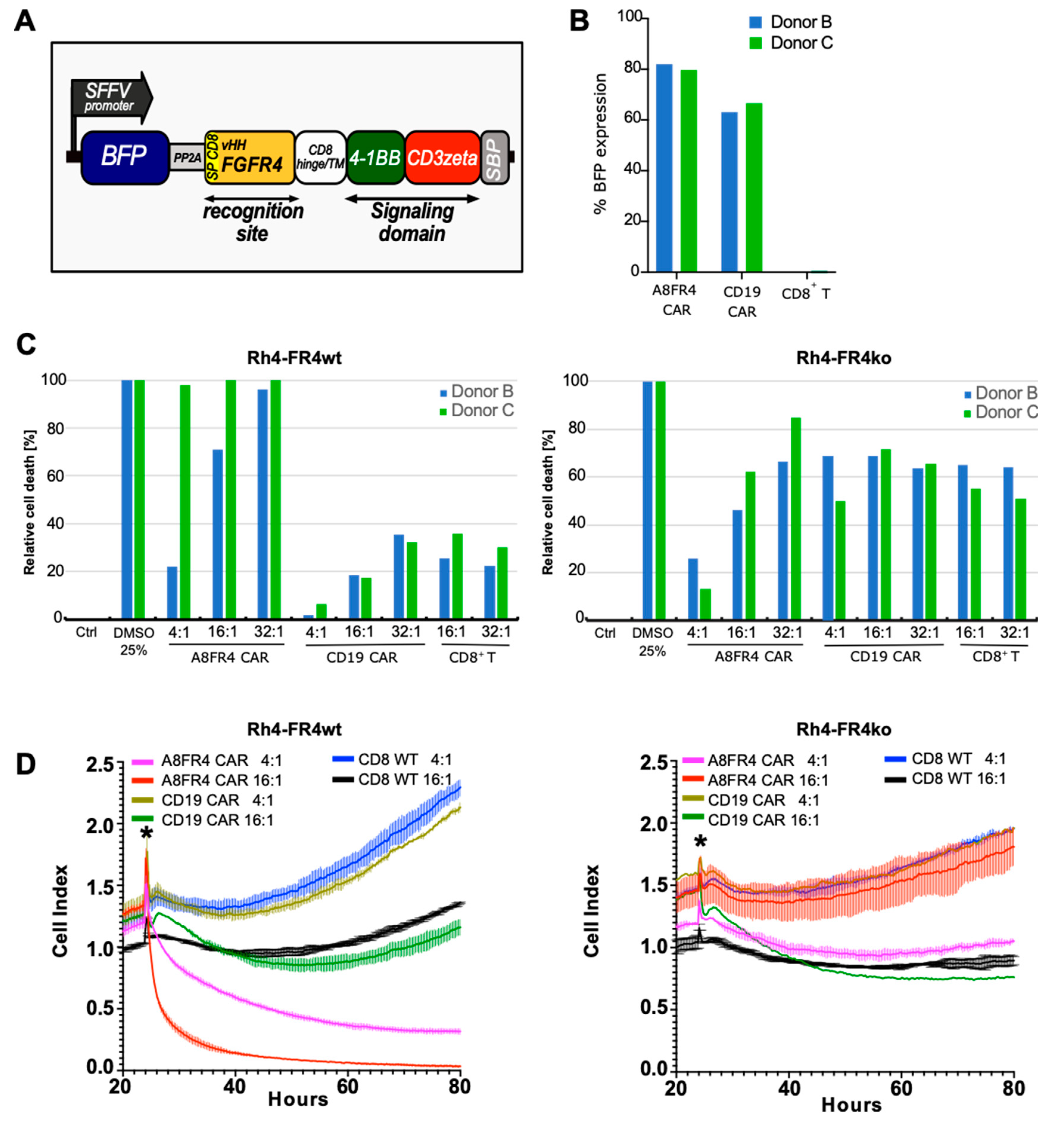
2.7. Cytotoxicity of FGFR4-CAR T Cells Targeting RMS Cells
3. Discussion
4. Materials and Methods
4.1. Plasmids and Cloning
4.2. Cell Lines
4.3. Generation of CRISPR/Cas9 FGFR4 Knockout Cells
4.4. Production of Lentiviral Vector for CAR T Cell Construction
4.5. T Cell Isolation and Transduction
4.6. Phage Display Selection
4.7. Protein Expression and Purification
4.8. Flow Cytometry
4.9. FGFR4 Activation Assay
4.10. Western Blotting
4.11. Surface Plasmon Resonance Spectroscopy
4.12. Preparation of Fluorescently Labelled VCR-Loaded Liposomes
4.13. Decoration of Liposomes with sdAb
4.14. VCR Quantification
4.15. Confocal Microscopy
4.16. CAR T Cell Cytotoxicity Assays
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; Barr, F.G.; Hawkins, D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Prim. 2019, 5, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hern, J.F.; Chen, L.; Chmielecki, J.; Wei, J.S.; Patidar, R.; Rosenberg, M.; Ambrogio, L.; Auclair, D.; Wang, J.; Song, Y.K.; et al. Comprehensive Genomic Analysis of Rhabdomyosarcoma Reveals a Landscape of Alterations Affecting a Common Genetic Axis in Fusion-Positive and Fusion-Negative Tumors. Cancer Discov. 2014, 4, 216–231. [Google Scholar] [CrossRef]
- Van Gaal, J.C.; Van Der Graaf, W.T.A.; Rikhof, B.; Van Hoesel, Q.G.C.M.; Teerenstra, S.; Suurmeijer, A.J.H.; Flucke, U.E.; Loeffen, J.L.C.M.; Sleijfer, S.; De Bont, E.S.J.M. The impact of age on outcome of embryonal and alveolar rhabdomyosarcoma patients. A multicenter study. Anticancer Res. 2012, 32, 4485–4497. [Google Scholar] [PubMed]
- Sultan, I.; Qaddoumi, I.; Yaser, S.; Rodriguez-Galindo, C.; Ferrari, A. Comparing Adult and Pediatric Rhabdomyosarcoma in the Surveillance, Epidemiology and End Results Program, 1973 to 2005: An Analysis of 2600 Patients. J. Clin. Oncol. 2009, 27, 3391–3397. [Google Scholar] [CrossRef] [PubMed]
- Punyko, J.A.; Mertens, A.C.; Gurney, J.G.; Yasui, Y.; Donaldson, S.S.; Rodeberg, D.A.; Raney, R.B.; Stovall, M.; Sklar, C.A.; Robison, L.L.; et al. Long-term medical effects of childhood and adolescent rhabdomyosarcoma: A report from the childhood cancer survivor study. Pediatr. Blood Cancer 2005, 44, 643–653. [Google Scholar] [CrossRef]
- Rodríguez-Nogales, C.; González-Fernández, Y.; Aldaz, A.; Couvreur, P.; Blanco-Prieto, M.J. Nanomedicines for Pediatric Cancers. ACS Nano 2018, 12, 7482–7496. [Google Scholar] [CrossRef]
- Li, Z.; Tan, S.; Li, S.; Shen, Q.; Wang, K. Cancer drug delivery in the nano era: An overview and perspectives. Oncol. Rep. 2017, 38, 611–624. [Google Scholar] [CrossRef]
- Kumari, P.; Ghosh, B.; Biswas, S. Nanocarriers for cancer-targeted drug delivery. J. Drug Target. 2015, 24, 179–191. [Google Scholar] [CrossRef]
- Ferrari, M. Cancer nanotechnology: Opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Jain, R.K. Transport of Molecules, Particles, and Cells in Solid Tumors. Annu. Rev. Biomed. Eng. 1999, 1, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Sindhwani, S.; Syed, A.M.; Ngai, J.; Kingston, B.R.; Maiorino, L.; Rothschild, J.; Macmillan, P.; Zhang, Y.; Rajesh, N.U.; Hoang, T.; et al. The entry of nanoparticles into solid tumours. Nat. Mater. 2020, 19, 566–575. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Merchant, M.S.; Cole, D.E.; Jayaprakash, N.; Bernstein, D.; Delbrook, C.; Richards, K.; Widemann, B.C.; Wayne, A.S. Vincristine Sulfate Liposomes Injection (VSLI, Marqibo®): Results From a Phase I Study in Children, Adolescents, and Young Adults With Refractory Solid Tumors or Leukemias. Pediatr. Blood Cancer 2016, 63, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.S.; Wernz, J.; Scadden, D.T.; Cohen, P.; Mukwaya, G.M.; Von Roenn, J.H.; Jacobs, M.; Kempin, S.; Silverberg, I.; Gonzales, G.; et al. Randomized phase III trial of liposomal daunorubicin versus doxorubicin, bleomycin, and vincristine in AIDS-related Kaposi’s sarcoma. J. Clin. Oncol. 1996, 14, 2353–2364. [Google Scholar] [CrossRef]
- O’Brien, M.E.R.; Wigler, N.; Inbar, M.; Rosso, R.; Grischke, E.; Santoro, A.; Catane, R.; Kieback, D.G.; Tomczak, P.; Ackland, S.P.; et al. Reduced cardiotoxicity and comparable efficacy in a phase IIItrial of pegylated liposomal doxorubicin HCl(CAELYX™/Doxil®) versus conventional doxorubicin forfirst-line treatment of metastatic breast cancer. Ann. Oncol. 2004, 15, 440–449. [Google Scholar] [CrossRef]
- Roveri, M.; Bernasconi, M.; Leroux, J.-C.; Luciani, P. Peptides for tumor-specific drug targeting: State of the art and beyond. J. Mater. Chem. B 2017, 5, 4348–4364. [Google Scholar] [CrossRef]
- Kirpotin, D.B.; Drummond, D.C.; Shao, Y.; Shalaby, M.R.; Hong, K.; Nielsen, U.B.; Marks, J.D.; Benz, C.; Park, J.W. Antibody Targeting of Long-Circulating Lipidic Nanoparticles Does Not Increase Tumor Localization but Does Increase Internalization in Animal Models. Cancer Res. 2006, 66, 6732–6740. [Google Scholar] [CrossRef]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Oliveira, S.; Heukers, R.; Sornkom, J.; Kok, R.J.; van Bergen En Henegouwen, P.M.P. Targeting tumors with nanobodies for cancer imaging and therapy. J. Control. Release 2013, 172, 607–617. [Google Scholar] [CrossRef]
- Roveri, M.; Pfohl, A.; Jaaks, P.; Alijaj, N.; Leroux, J.-C.; Luciani, P.; Bernasconi, M. Prolonged circulation and increased tumor accumulation of liposomal vincristine in a mouse model of rhabdomyosarcoma. Nanomedicine 2017, 12, 1135–1151. [Google Scholar] [CrossRef]
- Zhao, P.; Caretti, G.; Mitchell, S.; McKeehan, W.L.; Boskey, A.L.; Pachman, L.M.; Sartorelli, V.; Hoffman, E.P. Fgfr4 Is Required for Effective Muscle Regenerationin Vivo. J. Biol. Chem. 2006, 281, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Marics, I.; Padilla, F.; Guillemot, J.-F.; Scaal, M.; Marcelle, C. FGFR4 signaling is a necessary step in limb muscle differentiation. Development 2002, 129, 4559–4569. [Google Scholar]
- Khan, J.; Wei, J.S.; Ringnér, M.; Saal, L.H.; Ladanyi, M.; Westermann, F.; Berthold, F.; Schwab, M.; Antonescu, C.R.; Peterson, C.; et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat. Med. 2001, 7, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Tiong, K.H.; Tan, B.S.; Choo, H.L.; Chung, F.F.-L.; Hii, L.-W.; Tan, S.H.; Khor, N.T.W.; Wong, S.F.; See, S.-J.; Tan, Y.-F.; et al. Fibroblast growth factor receptor 4 (FGFR4) and fibroblast growth factor 19 (FGF19) autocrine enhance breast cancer cells survival. Oncotarget 2016, 7, 57633–57650. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Lang, L.; Zhao, X.; Shay, C.; Shull, A.Y.; Teng, Y. FGF19 amplification reveals an oncogenic dependency upon autocrine FGF19/FGFR4 signaling in head and neck squamous cell carcinoma. Oncogene 2018, 38, 2394–2404. [Google Scholar] [CrossRef]
- Sawey, E.T.; Chanrion, M.; Cai, C.; Wu, G.; Zhang, J.; Zender, L.; Zhao, A.; Busuttil, R.W.; Yee, H.; Stein, L.; et al. Identification of a Therapeutic Strategy Targeting Amplified FGF19 in Liver Cancer by Oncogenomic Screening. Cancer Cell 2011, 19, 347–358. [Google Scholar] [CrossRef]
- French, D.M.; Lin, B.C.; Wang, M.; Adams, C.; Shek, T.; Hötzel, K.; Bolon, B.; Ferrando, R.; Blackmore, C.; Schroeder, K.; et al. Targeting FGFR4 Inhibits Hepatocellular Carcinoma in Preclinical Mouse Models. PLoS ONE 2012, 7, e36713. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Gill, S.; Maus, M.V.; Porter, D.L. Chimeric antigen receptor T cell therapy: 25years in the making. Blood Rev. 2016, 30, 157–167. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor–transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef] [PubMed]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T cells expressing an anti–B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Hegde, M.; Joseph, S.K.; Pashankar, F.; Derenzo, C.; Sanber, K.; Navai, S.; Byrd, T.T.; Hicks, J.; Xu, M.L.; Gerken, C.; et al. Tumor response and endogenous immune reactivity after administration of HER2 CAR T cells in a child with metastatic rhabdomyosarcoma. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Moutel, S.; Bery, N.; Bernard, V.; Keller, L.; Lemesre, E.; De Marco, A.; Ligat, L.; Rain, J.-C.; Favre, G.; Olichon, A.; et al. NaLi-H1: A universal synthetic library of humanized nanobodies providing highly functional antibodies and intrabodies. eLife 2016, 5, e16228. [Google Scholar] [CrossRef]
- Moutel, S.; Perez, F. Synthetic Single Domain Antibody. European Patent Application Number EP20305536, 20 May 2020. [Google Scholar]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Sapra, P.; Allen, T.M. Internalizing antibodies are necessary for improved therapeutic efficacy of antibody-targeted liposomal drugs. Cancer Res. 2002, 62, 7190–7194. [Google Scholar]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Cao, L.; Yu, Y.; Bilke, S.; Walker, R.L.; Mayeenuddin, L.H.; Azorsa, D.O.; Yang, F.; Pineda, M.; Helman, L.J.; Meltzer, P.S. Genome-Wide Identification of PAX3-FKHR Binding Sites in Rhabdomyosarcoma Reveals Candidate Target Genes Important for Development and Cancer. Cancer Res. 2010, 70, 6497–6508. [Google Scholar] [CrossRef]
- Vi, J.G.T.; Cheuk, A.T.; Tsang, P.S.; Chung, J.-Y.; Song, Y.K.; Desai, K.; Yu, Y.; Chen, Q.-R.; Shah, K.; Youngblood, V.; et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Investig. 2009, 119, 3395–3407. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Guo, W.; Shen, J.K.; Mankin, H.J.; Hornicek, F.J.; Duan, Z. Rhabdomyosarcoma: Advances in Molecular and Cellular Biology. Sarcoma 2015, 2015, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Nishimura, R.; Yoshida, K.; Shimamura, T.; Shiraishi, Y.; Sato, Y.; Kato, M.; Chiba, K.; Tanaka, H.; Hoshino, N.; et al. Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nat. Commun. 2015, 6, 7557. [Google Scholar] [CrossRef] [PubMed]
- Crose, L.E.; Etheridge, K.T.; Chen, C.; Belyea, B.; Talbot, L.J.; Bentley, R.C.; Linardic, C.M. FGFR4 blockade exerts distinct antitumorigenic effects in human embryonal versus alveolar rhabdomyosarcoma. Clin. Cancer Res. 2012, 18, 3780–3790. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Q.; Cheuk, A.T.; Shern, J.F.; Song, Y.K.; Hurd, L.; Liao, H.; Wei, J.S.; Khan, J. Targeting Wild-Type and Mutationally Activated FGFR4 in Rhabdomyosarcoma with the Inhibitor Ponatinib (AP24534). PLoS ONE 2013, 8, e76551. [Google Scholar] [CrossRef]
- Helsten, T.; Schwaederle, M.; Kurzrock, R. Fibroblast growth factor receptor signaling in hereditary and neoplastic disease: Biologic and clinical implications. Cancer Metastasis Rev. 2015, 34, 479–496. [Google Scholar] [CrossRef]
- Oliveira, S.; Schiffelers, R.M.; Van Der Veeken, J.; Van Der Meel, R.; Vongpromek, R.; van Bergen en Henegouwen, P.M.P.; Storm, G.; Roovers, R.C. Downregulation of EGFR by a novel multivalent nanobody-liposome platform. J. Control. Release 2010, 145, 165–175. [Google Scholar] [CrossRef]
- Caruso, H.G.; Hurton, L.V.; Najjar, A.; Rushworth, D.; Ang, S.; Olivares, S.; Mi, T.; Switzer, K.; Singh, H.; Huls, H.; et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res. 2015, 75, 3505–3518. [Google Scholar] [CrossRef]
- Walker, A.J.; Majzner, R.G.; Zhang, L.; Wanhainen, K.; Long, A.H.; Nguyen, S.M.; Lopomo, P.; Vigny, M.; Fry, T.J.; Orentas, R.J.; et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol. Ther. 2017, 25, 2189–2201. [Google Scholar] [CrossRef]
- Geertsma, E.R.; Dutzler, R. A Versatile and Efficient High-Throughput Cloning Tool for Structural Biology. Biochemistry 2011, 50, 3272–3278. [Google Scholar] [CrossRef]
- Gentili, M.; Kowal, J.; Tkach, M.; Satoh, T.; Lahaye, X.; Conrad, C.; Boyron, M.; Lombard, B.; Durand, S.; Kroemer, G.; et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science 2015, 349, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Hinson, A.R.P.; Ejones, R.; Crose, L.E.S.; Belyea, B.C.; Barr, F.G.; Linardic, C.M. Human Rhabdomyosarcoma Cell Lines for Rhabdomyosarcoma Research: Utility and Pitfalls. Front. Oncol. 2013, 3, 183. [Google Scholar] [CrossRef] [PubMed]
- Nizak, C.; Moutel, S.; Goud, B.; Perez, F. Selection and Application of Recombinant Antibodies as Sensors of Rab Protein Conformation. Methods Enzymol. 2005, 403, 135–153. [Google Scholar] [CrossRef]
- Plamont, M.A.; Billon-Denis, E.; Maurin, S.; Gauron, C.; Pimenta, F.M.; Specht, C.G.; Shi, J.; Querard, J.; Pan, B.; Rossignol, J.; et al. Small fluorescence-activating and absorption-shifting tag for tunable protein imaging in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.J.; Jenkins, D.; Batt, D.; Mosher, R.; Isaacs, R.; Hu, T.; Capka, V.; Zhang, X.; Chen, D.; Tang, L.; et al. Abstract 1680: In vitro and in vivo activity of a highly potent and novel FGFR2/FGFR4 dual targeting antibody-drug conjugate. Cancer Res. 2015, 75, 1680. [Google Scholar] [CrossRef]
- Baskar, S.; Shivaprasad, N.; Zhu, Z.; Dimitrov, D.; Sigrist, M.; Sorensen, P.; Yohe, M.; Shern, J.; Maris, J.; Mackall, C.; et al. Abstract 2488: FGFR4 as a potential therapeutic target for monoclonal antibody based intervention in rhabdomyosarcoma. Cancer Res. 2015, 75, 2488. [Google Scholar] [CrossRef]
- Baskar, S. Abstract 4996: Targeting FGFR4 with monoclonal antibodies as therapeutic agents for the treatment of rhabdomyosarcoma. Cancer Res. 2016, 76, 4996. [Google Scholar] [CrossRef]
- Shivaprasad, N.; Xiong, Y.; Yohe, M.; Schneider, D.; Shern, J.; Baskar, S.; Dimitrov, D.; Sorenson, P.; Orentas, R.; Khan, J. Developing FGFR4 chimeric antigen receptor CAR T cell therapy against rhabdomyosarcoma. Mol. Ther. 2016, 24, S257–S258. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SdAb | kon1 * (1/M × s) | koff1 (1/s) | KD1 (M) | kon2 (1/M × s) | koff2 (1/s) | KD2 (M) | Rmax1 ** (RU) | Rmax2 (RU) |
|---|---|---|---|---|---|---|---|---|
| A8 | 3.14 × 105 | 1.32 × 10−9 | 4.22 × 10−15 | 6.67 × 104 | 2.45 × 10−3 | 3.68 × 10−8 | 25.6 | 18.1 |
| B1 | 1.11 × 106 | 1.18 × 10−6 | 1.06 × 10−12 | 2.16 × 105 | 9.92 × 10−4 | 4.60 × 10−9 | 26.1 | 17.5 |
| B5 | 1.84 × 106 | 5.66 × 10−4 | 3.08 × 10−10 | 1.73 × 105 | 3.75 × 10−8 | 2.16 × 10−13 | 23.1 | 10.3 |
| F8 | 5.45 × 104 | 1.04 × 10−6 | 1.91 × 10−11 | 1.35 × 106 | 5.57 × 10−3 | 4.14 × 10−9 | 83.0 | 86.4 |
| mCh | 2.60 × 103 | 5.11 × 10−3 | 1.96 × 10−6 | 2.32 × 103 | 5.05 × 10−3 | 2.18 × 10−6 | 20.7 | 20.7 |
| Liposomes Characteristics | L-A8 | L-B1 | L-B5 | L-F8 | L-mCh |
|---|---|---|---|---|---|
| Hydr. diameter (nm) | 126 | 127 | 129 | 136 | 122 |
| PDI | 0.129 | 0.108 | 0.093 | 0.122 | 0.025 |
| VCR conc (µg/mL) * | 308.4 | 253.8 | 254.6 | 270.3 | 319.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alijaj, N.; Moutel, S.; Gouveia, Z.L.; Gray, M.; Roveri, M.; Dzhumashev, D.; Weber, F.; Meier, G.; Luciani, P.; Rössler, J.K.; et al. Novel FGFR4-Targeting Single-Domain Antibodies for Multiple Targeted Therapies against Rhabdomyosarcoma. Cancers 2020, 12, 3313. https://doi.org/10.3390/cancers12113313
Alijaj N, Moutel S, Gouveia ZL, Gray M, Roveri M, Dzhumashev D, Weber F, Meier G, Luciani P, Rössler JK, et al. Novel FGFR4-Targeting Single-Domain Antibodies for Multiple Targeted Therapies against Rhabdomyosarcoma. Cancers. 2020; 12(11):3313. https://doi.org/10.3390/cancers12113313
Chicago/Turabian StyleAlijaj, Nagjie, Sandrine Moutel, Zelia L. Gouveia, Maxim Gray, Maurizio Roveri, Dzhangar Dzhumashev, Florian Weber, Gianmarco Meier, Paola Luciani, Jochen K. Rössler, and et al. 2020. "Novel FGFR4-Targeting Single-Domain Antibodies for Multiple Targeted Therapies against Rhabdomyosarcoma" Cancers 12, no. 11: 3313. https://doi.org/10.3390/cancers12113313
APA StyleAlijaj, N., Moutel, S., Gouveia, Z. L., Gray, M., Roveri, M., Dzhumashev, D., Weber, F., Meier, G., Luciani, P., Rössler, J. K., Schäfer, B. W., Perez, F., & Bernasconi, M. (2020). Novel FGFR4-Targeting Single-Domain Antibodies for Multiple Targeted Therapies against Rhabdomyosarcoma. Cancers, 12(11), 3313. https://doi.org/10.3390/cancers12113313