Deep Phenotyping Reveals Distinct Immune Signatures Correlating with Prognostication, Treatment Responses, and MRD Status in Multiple Myeloma

 ,
,  ,
,  ,
,  , and
, and
Simple Summary
Abstract
1. Introduction
2. Patients and Methods
2.1. Patients
2.2. Next-Generation Flow Cytometry
2.3. Immune Profiling
2.4. Cytogenetics
2.5. Statistical Analysis
3. Results
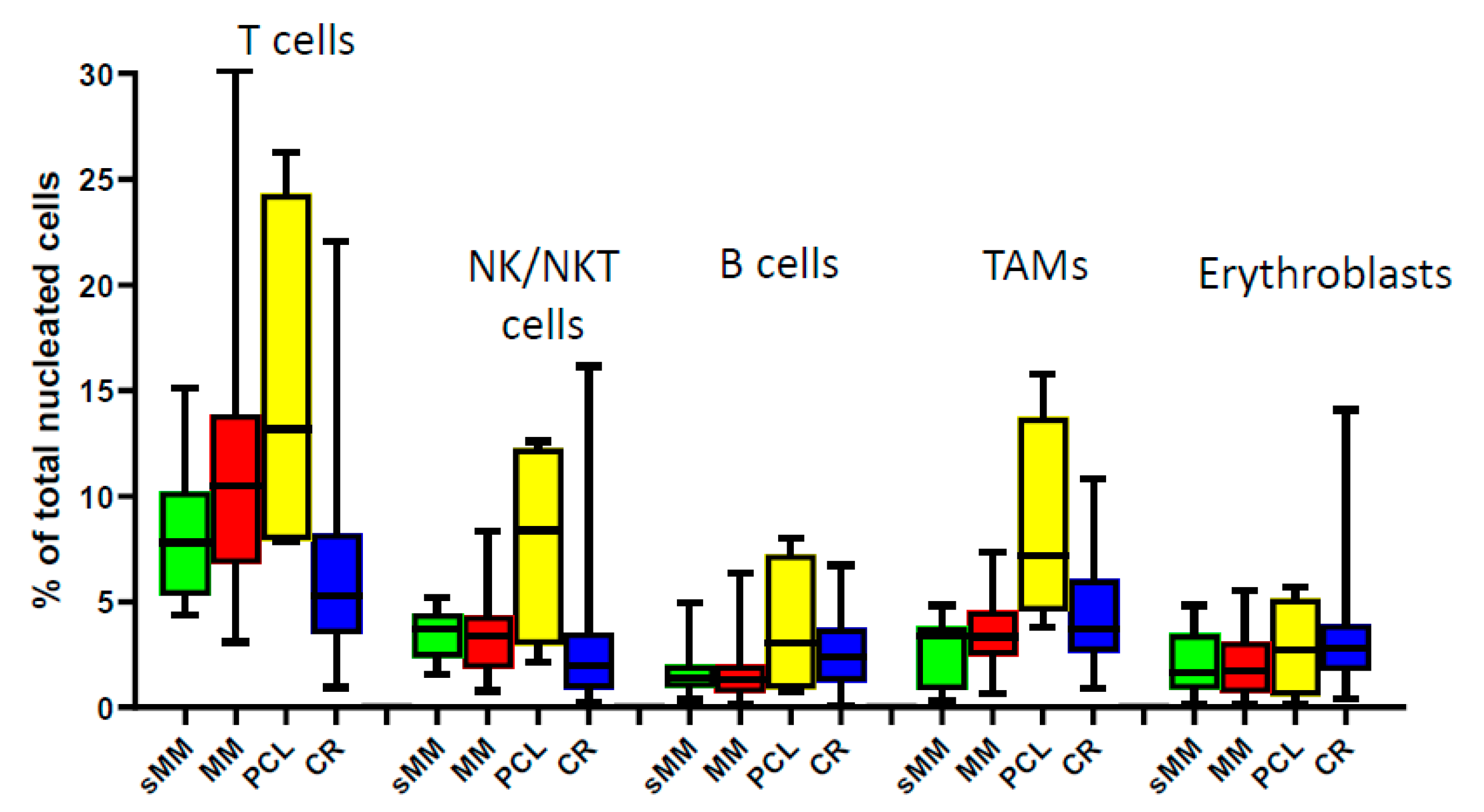
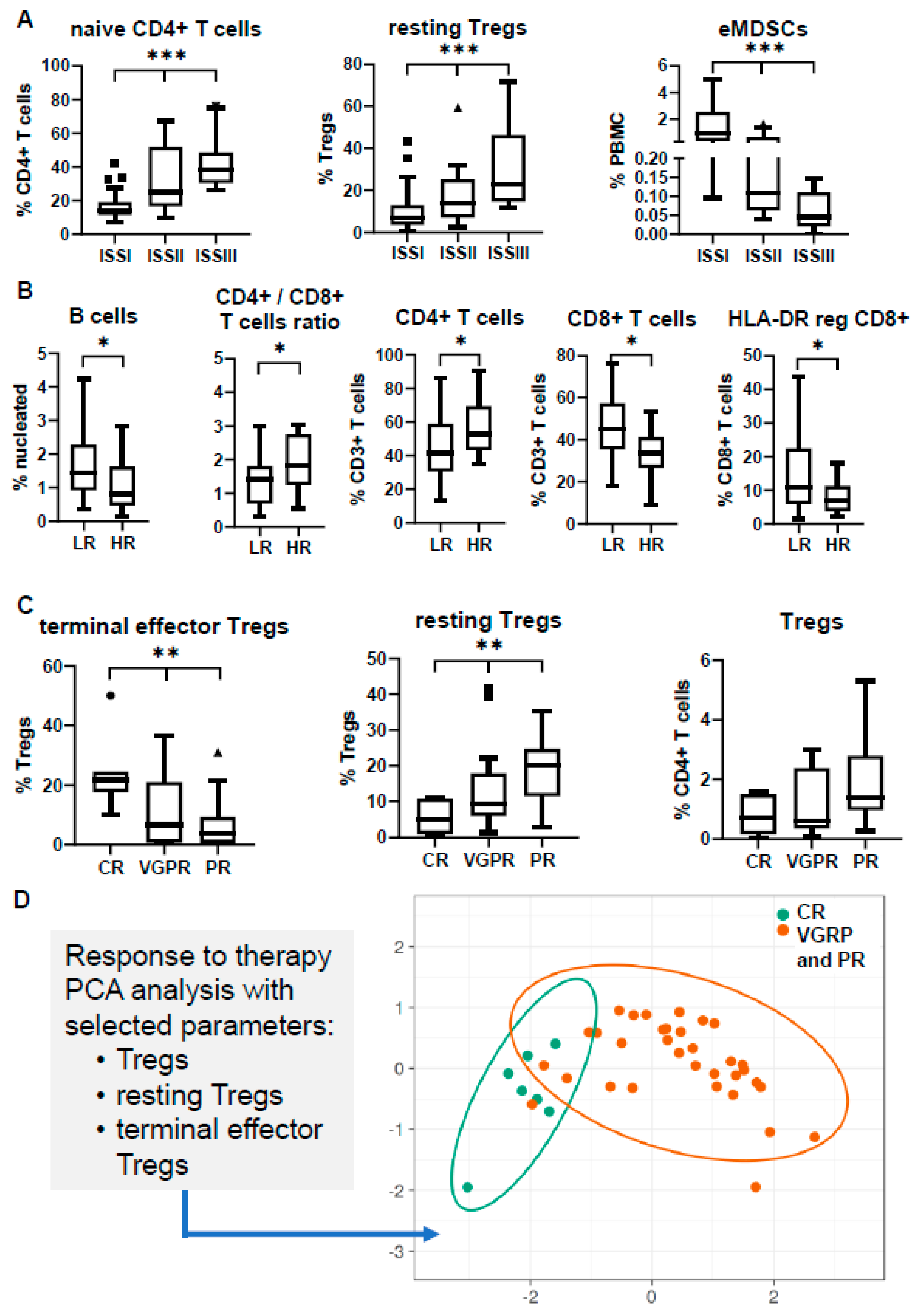
3.1. Composition of the BM Microenvironment at Different Stages of MM Progression
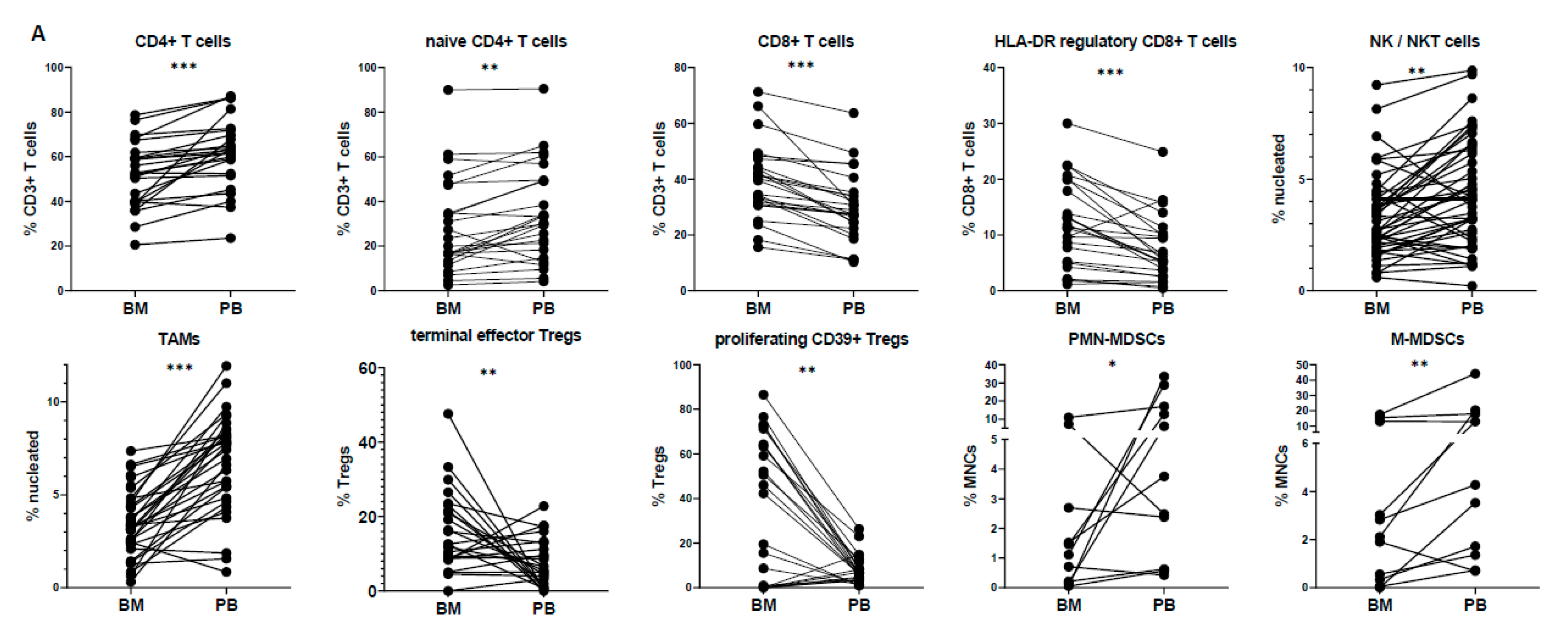
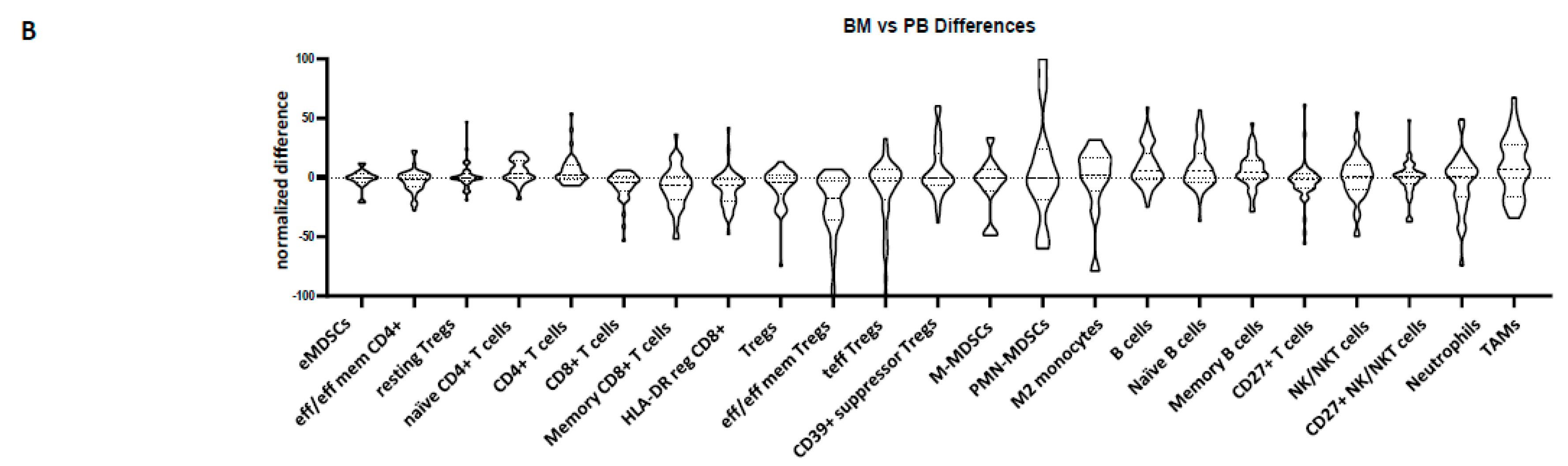
3.2. Peripheral Blood Cannot Reflect the Bone Marrow Microenvironment
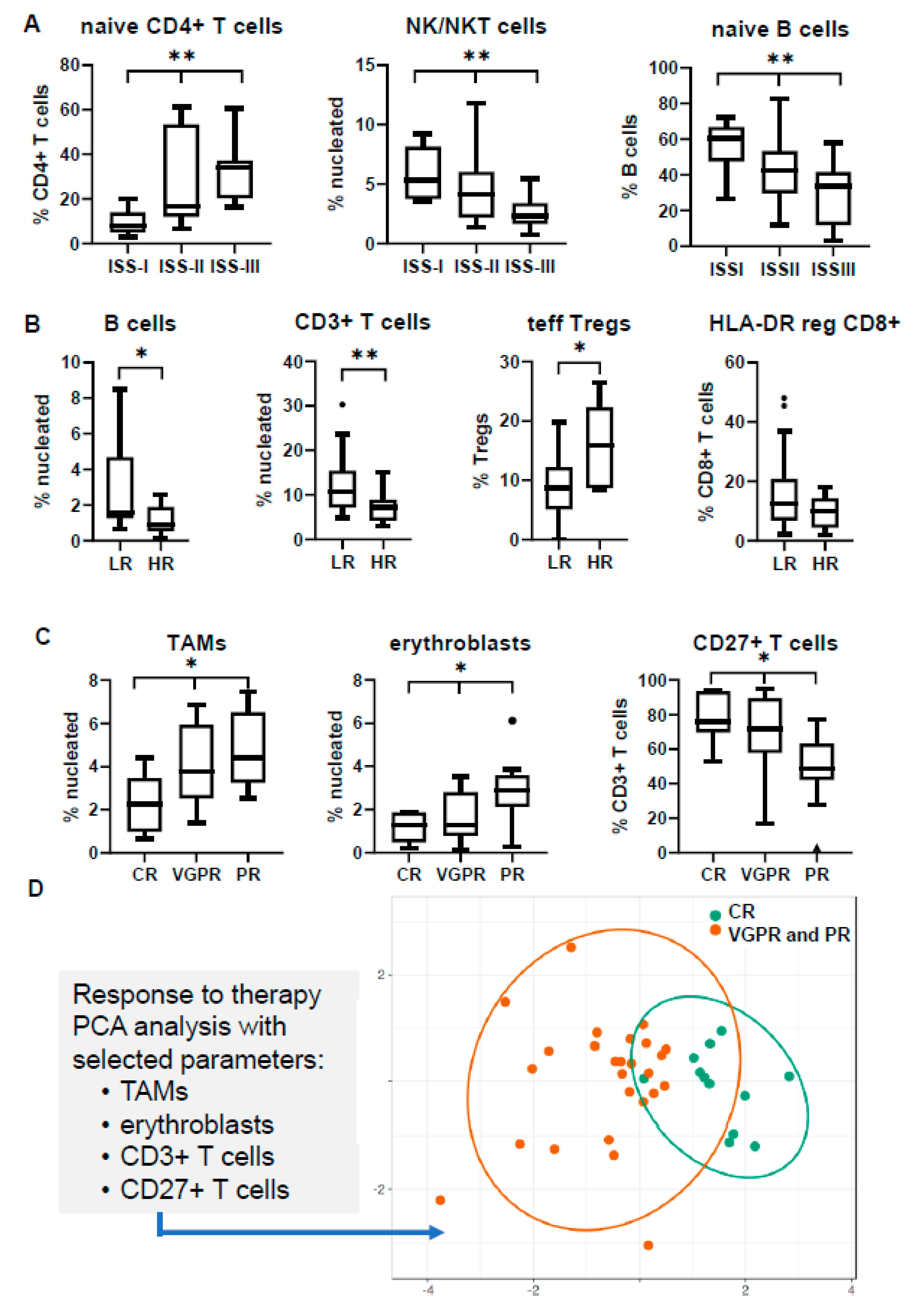
3.3. Immune Profiling May Differ in Separate Prognostic Groups
3.4. Immune Signatures May Predict Response to Induction Therapy
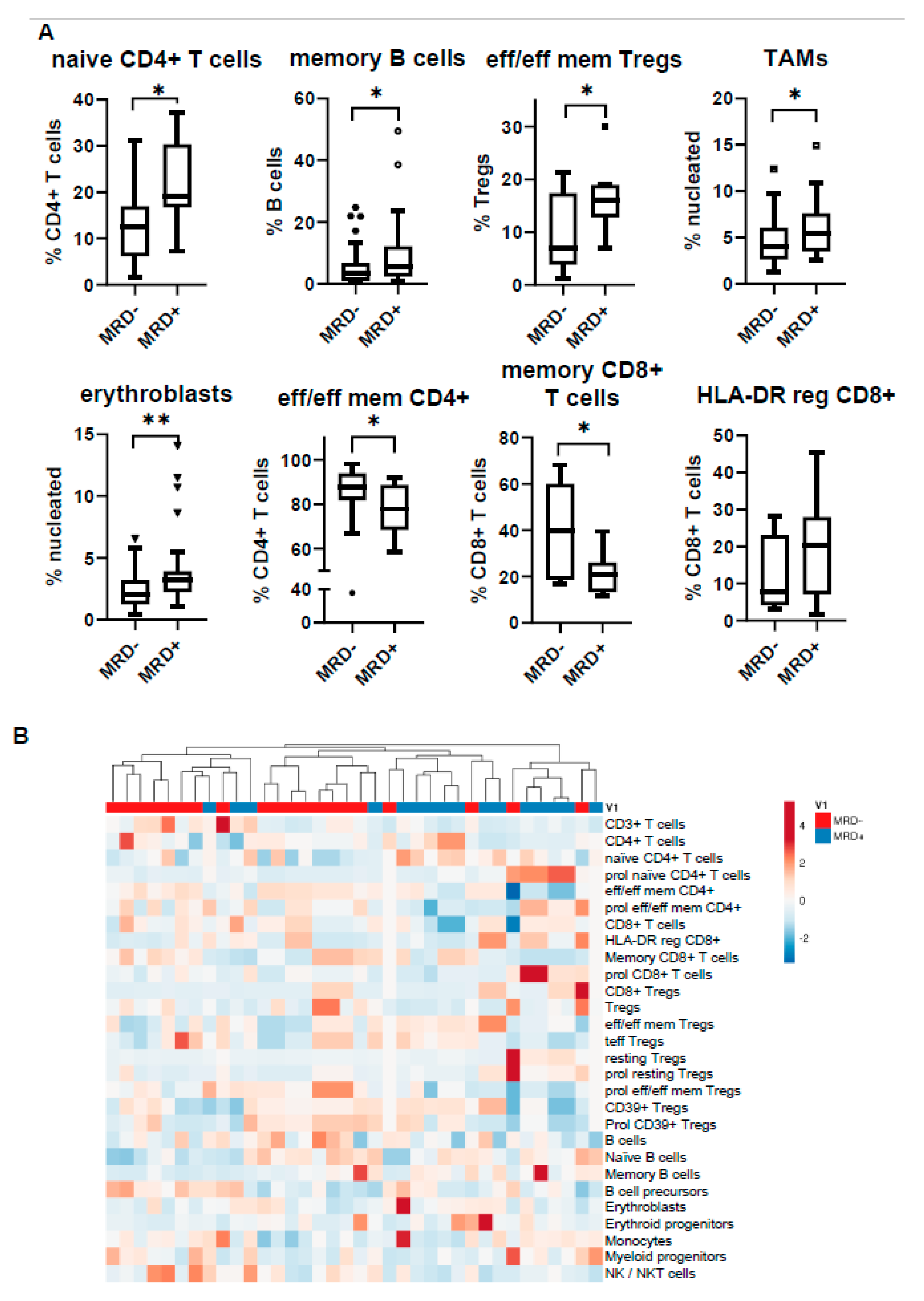
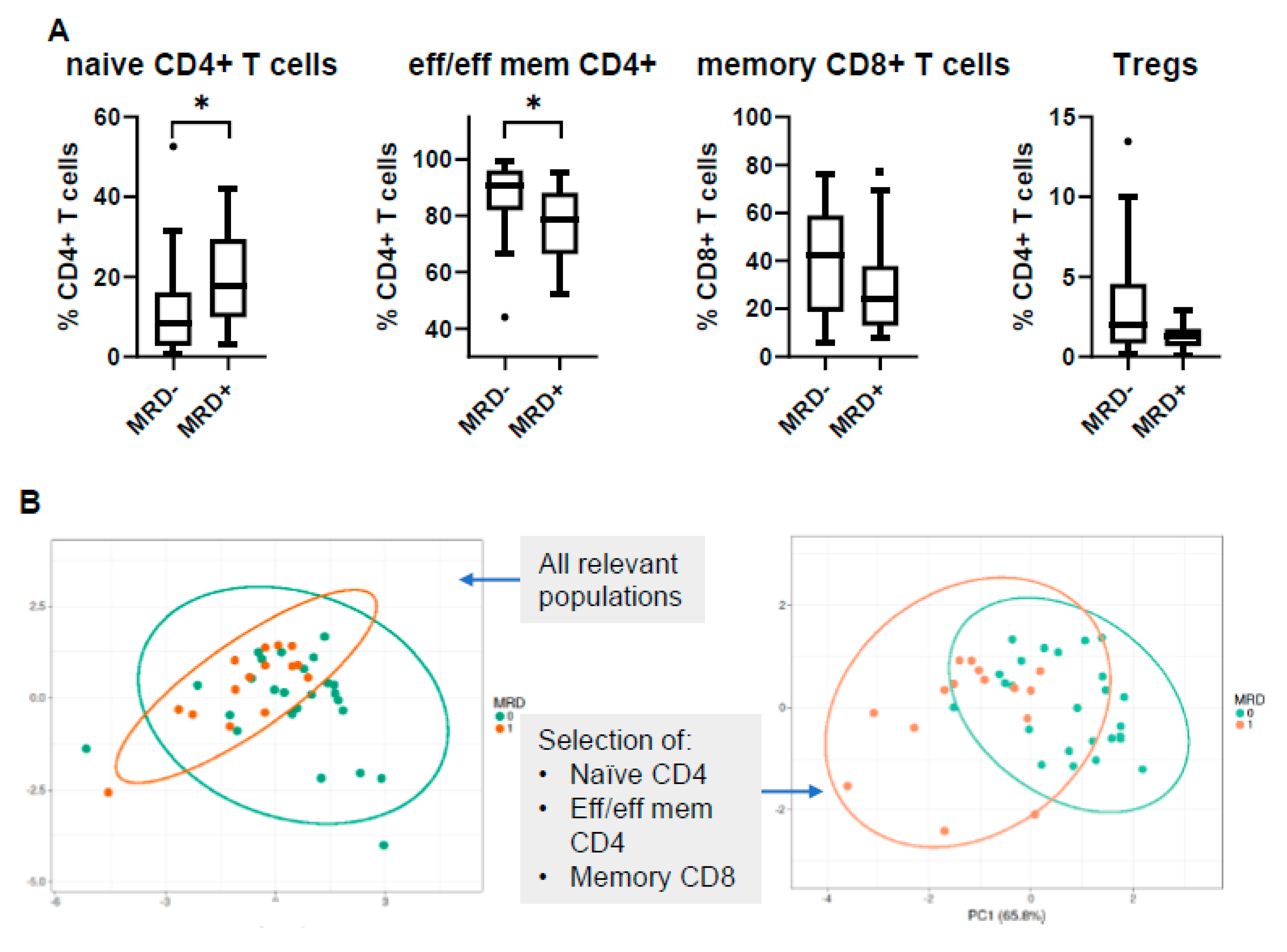
3.5. MRD Positivity Is Associated with a Distinct Immune Profile
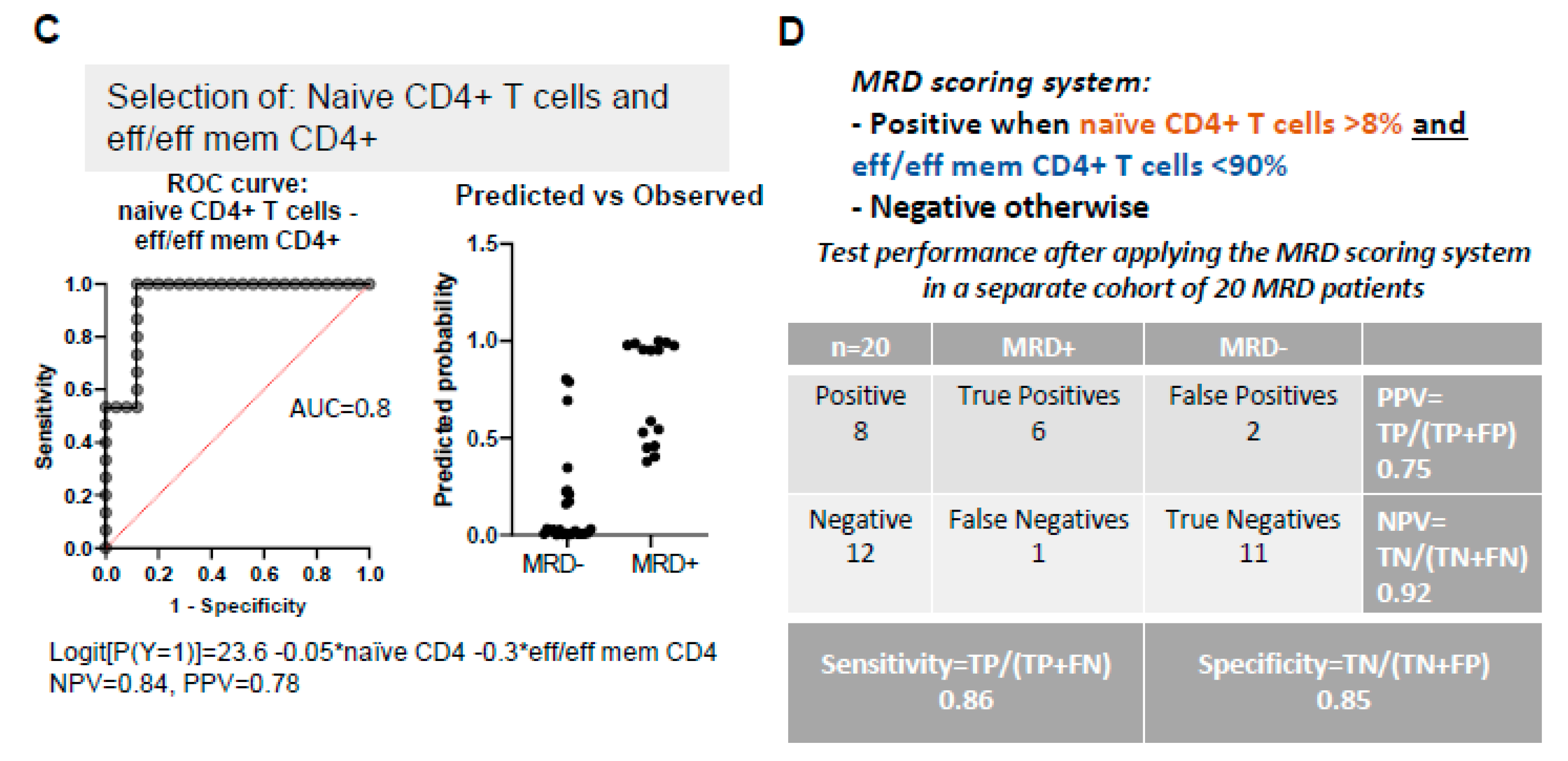
3.6. PB Signatures as Indicators for MRD Status
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- De Mel, S.; Lim, S.H.; Tung, M.L.; Chng, W.-J. Implications of Heterogeneity in Multiple Myeloma. BioMed Res. Int. 2014, 2014, 232546. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; Van Duin, M.; Sonneveld, P.; Mateos, M.-V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Primers 2017, 3, 17046. [Google Scholar] [CrossRef] [PubMed]
- Pawlyn, C.; Davies, F.E. Toward personalized treatment in multiple myeloma based on molecular characteristics. Blood 2019, 133, 660–675. [Google Scholar] [CrossRef] [PubMed]
- Mimura, N.; Hideshima, T.; Anderson, K.C. Novel therapeutic strategies for multiple myeloma. Exp. Hematol. 2015, 43, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Quach, H.; Ritchie, D.; Stewart, A.K.; Neeson, P.; Harrison, S.; Smyth, M.J.; Prince, H.M. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia 2010, 24, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Holstein, S.A.; McCarthy, P.L. Immunomodulatory Drugs in Multiple Myeloma: Mechanisms of action and clinical experience. Drugs 2017, 77, 505–520. [Google Scholar] [CrossRef]
- Madan, S.; Lacy, M.Q.; Dispenzieri, A.; Gertz, M.A.; Buadi, F.; Hayman, S.R.; Detweiler-Short, K.; Dingli, D.; Zeldenrust, S.; Lust, J.; et al. Efficacy of retreatment with immunomodulatory drugs (IMiDs) in patients receiving IMiDs for initial therapy of newly diagnosed multiple myeloma. Blood 2011, 118, 1763–1765. [Google Scholar] [CrossRef]
- Siegel, D.S.; Dimopoulos, M.A.; Ludwig, H.; Facon, T.; Goldschmidt, H.; Jakubowiak, A.; San-Miguel, J.; Obreja, M.; Blaedel, J.; Stewart, A.K. Improvement in overall survival with carfilzomib, lenalidomide, and dexamethasone in patients with relapsed or refractory multiple myeloma. J. Clin. Oncol. 2018, 36, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Gavriatopoulou, M.; Kastritis, E.; Ntanasis-Stathopoulos, I.; Fotiou, D.; Roussou, M.; Migkou, M.; Ziogas, D.C.; Kanellias, N.; Terpos, E.; Dimopoulos, M.A. The addition of IMiDs for patients with daratumumab-refractory multiple myeloma can overcome refractoriness to both agents. Blood 2018, 131, 464–467. [Google Scholar] [CrossRef]
- Mogollón, P.; Díaz-Tejedor, A.; Algarín, E.M.; Paíno, T.; Garayoa, M.; Ocio, E.M. Biological background of resistance to current standards of care in multiple myeloma. Cells 2019, 8, 1432. [Google Scholar] [CrossRef]
- Podar, K.; Chauhan, D.; Anderson, K.C. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia 2019, 23, 10–24. [Google Scholar] [CrossRef]
- Abe, M. Targeting the interplay between myeloma cells and the bone marrow microenvironment in myeloma. Int. J. Hematol. 2011, 94, 334–343. [Google Scholar] [CrossRef]
- Andrews, S.W.; Kabrah, S.; May, J.E.; Donaldson, C.; Morse, H.R. Multiple myeloma: The bone marrow microenvironment and its relation to treatment. Br. J. Biomed. Sci. 2013, 70, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone marrow microenvironment in multiple myeloma progression. J. Biomed. Biotechnol. 2012, 2012, 157496. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Moschetta, M.; Manier, S.; Glavey, S.; Görgün, G.T.; Roccaro, A.M.; Anderson, K.C.; Ghobrial, I.M. Targeting the bone marrow microenvironment in multiple myeloma. Immunol. Rev. 2015, 263, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Flores-Montero, J.; Sanoja-Flores, L.; Paiva, B.; Puig, N.; García-Sánchez, O.; Böttcher, S.; van der Velden, V.H.J.; Pérez-Morán, J.-J.; Vidriales, M.-B.; García-Sanz, R.; et al. Next Generation Flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia 2017, 31, 2094–2103. [Google Scholar] [CrossRef]
- Kostopoulos, I.V.; Paterakis, G.; Papadimitriou, K.; Pavlidis, D.; Tsitsilonis, O.E.; Papadhimitriou, S.I. Immunophenotypic analysis reveals heterogeneity and common biologic aspects in monoclonal B-cell lymphocytosis. Genes Chromosomes Cancer 2015, 54, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Sergentanis, T.N.; Kastritis, E.; Terpos, E.; Dimopoulos, M.A.; Psaltopoulou, T. Cytogenetics and Survival of Multiple Myeloma: Isolated and Combined Effects. Clin. Lymphoma Myeloma Leuk. 2016, 16, 335–340. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Gundesen, M.T.; Lund, T.; Moeller, H.E.H.; Abildgaard, N. Plasma cell leukemia: Definition, presentation, and treatment. Curr. Oncol. Rep. 2019, 21, 8. [Google Scholar] [CrossRef] [PubMed]
- Landgren, O.; Lu, S.X.; Hultcrantz, M. MRD Testing in Multiple Myeloma: The Main Future Driver for Modern Tailored Treatment. Semin. Hematol. 2018, 55, 44–50. [Google Scholar] [CrossRef]
- Kostopoulos, I.V.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Tsitsilonis, O.E.; Terpos, E. Minimal residual disease in multiple myeloma: Current landscape and future applications with immunotherapeutic approaches. Front. Oncol. 2020, 10, 860. [Google Scholar] [CrossRef]
- Landgren, O.; Devlin, S.; Boulad, M.; Mailankody, S. Role of MRD status in relation to clinical outcomes in newly diagnosed multiple myeloma patients: A meta-analysis. Bone Marrow Transplant. 2016, 51, 1565–1568. [Google Scholar] [CrossRef]
- Munshi, N.C.; Avet-Loiseau, H.; Rawstron, A.C.; Owen, R.G.; Child, J.A.; Thakurta, A.; Sherrington, P.; Samur, M.K.; Georgieva, A.; Anderson, K.C.; et al. Association of minimal residual disease with superior survival outcomes in patients with multiple myeloma: A meta-analysis. JAMA Oncol. 2017, 3, 28. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Zeldenrust, S.R.; Dingli, D.; Russell, S.J.; Lust, J.A.; et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008, 111, 2516–2520. [Google Scholar] [CrossRef]
- Costa, L.J.; Brill, I.K.; Omel, J.; Godby, K.; Kumar, S.K.; Brown, E.E. Recent trends in multiple myeloma incidence and survival by age, race, and ethnicity in the United States. Blood Adv. 2017, 1, 282–287. [Google Scholar] [CrossRef]
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A. Microenvironment drug resistance in multiple myeloma: Emerging new players. Oncotarget 2016, 7, 60698–60711. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Khong, T.; Ramachandran, M.; Chow, A.; Klarica, D.; Mai, L.; Walsh, S.; Broemeling, D.; Marziali, A.; Wiggin, M.; et al. Circulating tumour DNA analysis demonstrates spatial mutational heterogeneity that coincides with disease relapse in myeloma. Leukemia 2017, 31, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- de Larrea, C.F.; Kyle, R.A.; Durie, B.G.M.; Ludwig, H.; Usmani, S.; Vesole, D.H.; Hajek, R.; San Miguel, J.F.; Sezer, O.; Sonneveld, P.; et al. Plasma cell leukemia: Consensus statement on diagnostic requirements, response criteria and treatment recommendations by the International Myeloma Working Group. Leukemia 2013, 27, 780–791. [Google Scholar] [CrossRef]
- Brimnes, M.K.; Vangsted, A.J.; Knudsen, L.M.; Gimsing, P.; Gang, A.O.; Johnsen, H.E.; Svane, I.M. Increased level of both CD4+FOXP3+ regulatory T cells and CD14+HLA-DR−/low Myeloid-Derived Suppressor Cells and decreased level of dendritic cells in patients with multiple myeloma: Immune impairment in patients with multiple myeloma. Scand. J. Immunol. 2010, 72, 540–547. [Google Scholar] [CrossRef]
- Foglietta, M.; Castella, B.; Mariani, S.; Coscia, M.; Godio, L.; Ferracini, R.; Ruggeri, M.; Muccio, V.; Omede, P.; Palumbo, A.; et al. The bone marrow of myeloma patients is steadily inhabited by a normal-sized pool of functional regulatory T cells irrespectiveof the disease status. Haematologica 2014, 99, 1605–1610. [Google Scholar] [CrossRef]
- Görgün, G.T.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood 2013, 121, 2975–2987. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, J.; Brown, R.; Aklilu, E.; Yang, S.; Suen, H.; Hart, D.; Fromm, P.; Gibson, J.; Khoo, L.; Ho, P.J.; et al. Myeloma skews regulatory T and pro-inflammatory T helper 17 cell balance in favor of a suppressive state. Leuk. Lymphoma 2014, 55, 1090–1098. [Google Scholar] [CrossRef]
- Lad, D.; Huang, Q.; Hoeppli, R.; Garcia, R.; Xu, L.; Levings, M.; Song, K.; Broady, R. Evaluating the role of Tregs in the progression of multiple myeloma. Leuk. Lymphoma 2019, 60, 2134–2142. [Google Scholar] [CrossRef] [PubMed]
- Zelle-Rieser, C.; Thangavadivel, S.; Biedermann, R.; Brunner, A.; Stoitzner, P.; Willenbacher, E.; Greil, R.; Jöhrer, K. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J. Hematol. Oncol. 2016, 9, 116. [Google Scholar] [CrossRef]
- Pessoa de Magalhães, R.J.; Vidriales, M.-B.; Paiva, B.; Fernandez-Gimenez, C.; García-Sanz, R.; Mateos, M.-V.; Gutierrez, N.C.; Lecrevisse, Q.; Blanco, J.F.; Hernández, J.; et al. Analysis of the immune system of multiple myeloma patients achieving long-term disease control by multidimensional flow cytometry. Haematologica 2013, 98, 79–86. [Google Scholar] [CrossRef]
- Marsh-Wakefield, F.; Kruzins, A.; McGuire, H.M.; Yang, S.; Bryant, C.; St. Groth, B.F.; Nassif, N.; Byrne, S.N.; Gibson, J.; Brown, C.; et al. Mass cytometry discovers two discrete subsets of CD39−Treg which discriminate MGUS from multiple myeloma. Front. Immunol. 2019, 10, 1596. [Google Scholar] [CrossRef]
- San Miguel, J.F.; González, M.; Gascón, A.; Moro, M.J.; Hernández, J.M.; Ortega, F.; Jiménez, R.; Guerras, L.; Romero, M.; Casanova, F. Lymphoid subsets and prognostic factors in multiple myeloma. Cooperative Group for the Study of Monoclonal Gammopathies. Br. J. Haematol. 1992, 80, 305–309. [Google Scholar] [CrossRef]
- Koike, M.; Sekigawa, I.; Okada, M.; Matsumoto, M.; Iida, N.; Hashimoto, H.; Oshimi, K. Relationship between CD4(+)/CD8(+) T cell ratio and T cell activation in multiple myeloma: Reference to IL-16. Leuk. Res. 2002, 26, 705–711. [Google Scholar] [CrossRef]
- Dosani, T.; Carlsten, M.; Maric, I.; Landgren, O. The cellular immune system in myelomagenesis: NK cells and T cells in the development of MM and their uses in immunotherapies. Blood Cancer J. 2015, 5, e306. [Google Scholar] [CrossRef]
- Všianská, P.; Říhová, L.; Varmužová, T.; Suská, R.; Kryukov, F.; Mikulášová, A.; Kupská, R.; Penka, M.; Pour, L.; Adam, Z.; et al. Analysis of B-Cell subpopulations in monoclonal gammopathies. Clin. Lymphoma Myeloma Leuk. 2015, 15, e61–e71. [Google Scholar] [CrossRef]
- Pittari, G.; Vago, L.; Festuccia, M.; Bonini, C.; Mudawi, D.; Giaccone, L.; Bruno, B. Restoring natural killer cell immunity against multiple myeloma in the era of new drugs. Front. Immunol. 2017, 8, 1444. [Google Scholar] [CrossRef]
- Berardi, S.; Ria, R.; Reale, A.; De Luisi, A.; Catacchio, I.; Moschetta, M.; Vacca, A. Multiple myeloma macrophages: Pivotal players in the tumor microenvironment. J. Oncol. 2013, 2013, 1–6. [Google Scholar] [CrossRef]
- De Beule, N.; De Veirman, K.; Maes, K.; De Bruyne, E.; Menu, E.; Breckpot, K.; De Raeve, H.; Van Rampelbergh, R.; Van Ginderachter, J.A.; Schots, R.; et al. Tumour-associated macrophage-mediated survival of myeloma cells through STAT3 activation: TAM-mediated survival of myeloma cells. J. Pathol. 2017, 241, 534–546. [Google Scholar] [CrossRef]
- Panchabhai, S.; Kelemen, K.; Ahmann, G.; Sebastian, S.; Mantei, J.; Fonseca, R. Tumor-associated macrophages and extracellular matrix metalloproteinase inducer in prognosis of multiple myeloma. Leukemia 2016, 30, 951–954. [Google Scholar] [CrossRef]
- Botta, C.; Perez, C.; Puig, N.; Cedena, M.; Cordon, L.; Zabaleta, A.; Burgos, L.; Maia, C.; Goicoechea, I.; Rodriguez, S.; et al. Single-cell characterization of the multiple myeloma (MM) immune microenvironment identifies CD27-negative T cells as potential source of tumor-reactive lymphocytes. Blood 2019, 134, 506. [Google Scholar] [CrossRef]
- Bryant, C.; Suen, H.; Brown, R.; Yang, S.; Favaloro, J.; Aklilu, E.; Gibson, J.; Ho, P.J.; Iland, H.; Fromm, P.; et al. Long-term survival in multiple myeloma is associated with a distinct immunological profile, which includes proliferative cytotoxic T-cell clones and a favourable Treg/Th17 balance. Blood Cancer J. 2013, 3, e148. [Google Scholar] [CrossRef]
- Giannopoulos, K.; Kaminska, W.; Hus, I.; Dmoszynska, A. The frequency of T regulatory cells modulates the survival of multiple myeloma patients: Detailed characterisation of immune status in multiple myeloma. Br. J. Cancer 2012, 106, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Kotsakis, A.; Koinis, F.; Katsarou, A.; Gioulbasani, M.; Aggouraki, D.; Kentepozidis, N.; Georgoulias, V.; Vetsika, E.-K. Prognostic value of circulating regulatory T cell subsets in untreated non-small cell lung cancer patients. Sci. Rep. 2016, 6, 39247. [Google Scholar] [CrossRef]
- Perrot, A.; Lauwers-Cances, V.; Corre, J.; Robillard, N.; Hulin, C.; Chretien, M.-L.; Dejoie, T.; Maheo, S.; Stoppa, A.-M.; Pegourie, B.; et al. Minimal residual disease negativity using deep sequencing is a major prognostic factor in multiple myeloma. Blood 2018, 132, 2456–2464. [Google Scholar] [CrossRef]
- Paiva, B.; Puig, N.; Cedena, M.-T.; Rosiñol, L.; Cordón, L.; Vidriales, M.-B.; Burgos, L.; Flores-Montero, J.; Sanoja-Flores, L.; Lopez-Anglada, L.; et al. Measurable residual disease by Next-Generation flow cytometry in multiple myeloma. J. Clin. Oncol. 2020, 38, 784–792. [Google Scholar] [CrossRef]
- Paiva, B.; Cedena, M.-T.; Puig, N.; Arana, P.; Vidriales, M.-B.; Cordon, L.; Flores-Montero, J.; Gutierrez, N.C.; Martín-Ramos, M.-L.; Martinez-Lopez, J.; et al. Minimal residual disease monitoring and immune profiling in multiple myeloma in elderly patients. Blood 2016, 127, 3165–3174. [Google Scholar] [CrossRef]
- Terpos, E.; Kostopoulos, I.V.; Kastritis, E.; Ntanasis-Stathopoulos, I.; Migkou, M.; Rousakis, P.; Argyriou, A.T.; Kanellias, N.; Fotiou, D.; Eleutherakis-Papaiakovou, E.; et al. Impact of minimal residual disease detection by next-generation flow cytometry in multiple myeloma patients with sustained complete remission after frontline therapy. HemaSphere 2019, 3, e300. [Google Scholar] [CrossRef]
- Bhutani, M.; Foureau, D.; Zhang, Q.; Robinson, M.; Wynn, A.S.; Steuerwald, N.M.; Druhan, L.J.; Guo, F.; Rigby, K.; Turner, M.; et al. Peripheral immunotype correlates with minimal residual disease status and is modulated by Immunomodulatory drugs in multiple myeloma. Biol. Blood Marrow Transplant. 2019, 25, 459–465. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Parameters | Patients at Diagnosis (n = 53) |
|---|---|
| Age (years) | 66 (44–93) |
| Male sex (%) | 26 (39%) |
| Hemoglobin (g/dL) | 10.8 (8.0–14.7) * |
| Platelet counts (×109/L) | 243 (56–591) |
| Neutrophil counts/μL | 4513 (1000–13,000) |
| Serum Creatinine (mg/dL) | 1.63 (0.5–11.1) |
| Serum B2MG (mg/L) | 5.7 (0.3–26.4) |
| Serum LDH (U/L) | 167 (74–293) |
| Serum Calcium (mg/dL) | 9.6 (8.4–14.4) |
| BM infiltration (%) | 50.7 (0–90) |
| ISS stage | |
| I | 20/53 (38%) |
| II | 18/53 (34%) |
| III | 15/53 (28%) |
| FISH aberrations | |
| High risk | 22/53 (42%) |
| Low risk | 31/53 (58%) |
| Heavy chain | |
| IgA | 13/53 (25%) |
| IgG | 32/53 (60%) |
| IgD | 1/53 (2%) |
| Light chain only | 7/53 (13%) |
| Immune Subset | Expression of Markers |
|---|---|
| NGF MRD panel | |
| Plasma cells | CD38brCD138+ |
| B cells | CD19+CD45+ |
| Naïve B cells | CD19+CD27-CD38-/dimCD45+SSClow |
| B cell precursors | CD19+CD27-CD38brCD45dimSSClow |
| Memory B cells | CD19+CD27+CD38-/dimCD45+SSClow |
| T cells | CD19-CD45+CD56-SSClow |
| CD27+ T cells | CD19-CD45+CD56-CD27+SSClow |
| NK/NKT cells | CD19-CD45+ CD56-SSClow |
| CD27+ NK/NKT cells | CD19-CD45+CD56-CD27+SSClow |
| Neutrophils | CD45dimSSChigh |
| Myeloid progenitors | CD38+CD45dimCD117+SSChigh |
| Monocytes—TAMs | CD38+CD45+CD81+SSCint |
| Mast cells | CD45dimCD117br |
| Erythroblasts | CD38-CD45-SSClow |
| Erythroid progenitors | CD38-/dimCD45-/dimCD117+SSClow |
| T cell panel | |
| T regulatory cells (Tregs) | CD3+CD4+CD25+CD127lowFoxP3+ |
| Effector/effector memory Tregs (eff/eff mem Tregs) | CD3+CD4+CD25+CD127lowFoxP3+CD45RA-CD45RO+HLA-DR-CTLA4+ |
| Terminal effector Tregs (teff Tregs) | CD3+CD4+CD25+CD127lowFoxP3+CD45RA-CD45RO+HLA-DR+ CTLA4+ |
| Resting Tregs | CD3+CD4+CD25+CD127lowFoxP3+CD45RA-CD45RO+HLA-DR-CTLA4- |
| CD39+ suppressor Tregs (CD39 Tregs) | CD3+CD4+CD25+CD127lowFoxP3+CD45RA-CD45RO+CD39+ |
| CD4+ Τ cells | CD3+CD4+ |
| Naïve CD4+ Τ cells | CD3+CD4+CD45RA+CD45RO- |
| Effector/Effector memory CD4+T cells (eff/eff mem CD4+) | CD3+CD4+CD45RA-CD45RO+ |
| CD8+ T cells | CD3+CD8+ |
| CD8+ Tregs | CD3+CD8+CD25+FoxP3+ |
| Memory CD8+ T cells | CD3+CD8+CD45RO+ |
| HLA-DR regulatory CD8+ T cells (HLA-DR reg CD8+) | CD3+CD8+HLA-DR+ |
| MDSC panel | |
| Polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) | CD14-CD11b+CD15+SSChigh |
| Early myeloid-derived suppressor cells (eMDSCs) | Lin(CD3/CD14/CD15/CD19/CD56)-HLA-DR-CD33+ |
| Monocytic myeloid-derived suppressor cells (M-MDSCs) | CD11b-CD14+HLA-DRlow/-CD15- |
| M1 monocytes | Lin(CD3/CD14/CD15/CD19/CD56)-CD14+CD124- |
| M2 monocytes | Lin(CD3/CD14/CD15/CD19/CD56)-CD14+CD124+ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadimitriou, K.; Tsakirakis, N.; Malandrakis, P.; Vitsos, P.; Metousis, A.; Orologas-Stavrou, N.; Ntanasis-Stathopoulos, I.; Kanellias, N.; Eleutherakis-Papaiakovou, E.; Pothos, P.; et al. Deep Phenotyping Reveals Distinct Immune Signatures Correlating with Prognostication, Treatment Responses, and MRD Status in Multiple Myeloma. Cancers 2020, 12, 3245. https://doi.org/10.3390/cancers12113245
Papadimitriou K, Tsakirakis N, Malandrakis P, Vitsos P, Metousis A, Orologas-Stavrou N, Ntanasis-Stathopoulos I, Kanellias N, Eleutherakis-Papaiakovou E, Pothos P, et al. Deep Phenotyping Reveals Distinct Immune Signatures Correlating with Prognostication, Treatment Responses, and MRD Status in Multiple Myeloma. Cancers. 2020; 12(11):3245. https://doi.org/10.3390/cancers12113245
Chicago/Turabian StylePapadimitriou, Konstantinos, Nikolaos Tsakirakis, Panagiotis Malandrakis, Panagiotis Vitsos, Andreas Metousis, Nikolaos Orologas-Stavrou, Ioannis Ntanasis-Stathopoulos, Nikolaos Kanellias, Evangelos Eleutherakis-Papaiakovou, Panagiotis Pothos, and et al. 2020. "Deep Phenotyping Reveals Distinct Immune Signatures Correlating with Prognostication, Treatment Responses, and MRD Status in Multiple Myeloma" Cancers 12, no. 11: 3245. https://doi.org/10.3390/cancers12113245
APA StylePapadimitriou, K., Tsakirakis, N., Malandrakis, P., Vitsos, P., Metousis, A., Orologas-Stavrou, N., Ntanasis-Stathopoulos, I., Kanellias, N., Eleutherakis-Papaiakovou, E., Pothos, P., Fotiou, D., Gavriatopoulou, M., Kastritis, E., Dimopoulos, M.-A., Terpos, E., Tsitsilonis, O. E., & Kostopoulos, I. V. (2020). Deep Phenotyping Reveals Distinct Immune Signatures Correlating with Prognostication, Treatment Responses, and MRD Status in Multiple Myeloma. Cancers, 12(11), 3245. https://doi.org/10.3390/cancers12113245